

Abstract
p150, product of the SALL2 gene, is a binding partner of the polyoma virus large T antigen and a putative tumor suppressor. p150 binds to the nuclease hypersensitive element of the c-MYC promoter and represses c-MYC transcription. Overexpression of p150 in human ovarian surface epithelial cells leads to decreased expression, and downregulation to increased expression, of c-MYC. c-MYC is repressed upon restoration of p150 to ovarian carcinoma cells. Induction of apoptosis by etoposide results in recruitment of p150 to the c-MYC promoter and to repression of c-MYC. Analysis of data in The Cancer Genome Atlas shows negative correlations between SALL2 and c-MYC expression in four common solid tumor types.
Introduction
Oncogenic DNA viruses target tumor suppressor genes of their hosts for inactivation ostensibly to promote cell cycle progression and to block apoptosis as essential steps in virus replication. These functions are also critical for cell transformation and tumor induction by these viruses [1]. The highly oncogenic mouse polyoma virus (Py) stands apart from SV40 and other DNA tumor viruses in failing to target p53 for inactivation or destruction. A screen was previously designed to identify tumor suppressors or other host factors with which Py must interact in order to replicate. The product of the SALL2 gene (p150) was identified as a Py large T binding protein using this screen [2]. p150 binds at the C-terminus of Py large T in a region bearing no homology to SV40 large T. p150 blocks Py DNA replication and binding of p150 by Py large T overcomes this inhibition. A Py mutant unable to bind p150 is unable to replicate and fails to induce a broad spectrum of tumors in the mouse [2]. Targeting of p150 by the human papilloma virus HPV16 E6 oncoprotein has recently been reported [3].
Evolutionarily conserved SALL genes encode multi-zinc finger transcription factors that function in embryonic development in invertebrate and vertebrate species [4] including man [5], [6], [7], [8]. Unique within this family, SALL2 has been implicated as a possible tumor suppressor [9], [10]. SALL2 has growth arrest and proapoptotic functions that overlap with those of p53. Specifically, p150 binds to the p21Cip1/Waf1 promoter in regions adjacent to the known p53 binding sites and transactivates p21Cip1/Waf1 in the absence of p53 [9]. p150 also binds to the BAX promoter and activates transcription [9], [11]. Restoration of p150 to human ovarian carcinoma cells deficient in p150 expression results in partial inhibition of tumor growth in SCID mice accompanied by elevated levels of p21Cip1/Waf1 and BAX [9]. SALL2 has also been recognized and characterized as a ‘quiescence factor’, essential for arresting growth of human fibroblasts under conditions of serum deprivation [12]. When treated with SALL2-siRNA, serum-deprived cells fail to arrest [12]. Upon restoration of serum, p150 is rapidly degraded as cells re-enter the cell cycle. Factors that regulate SALL2 expression at the transcriptional and posttranslational levels as a function of growth conditions have been identified [13].
The DNA sequence specificity of binding by p150 has recently been investigated [11]. The optimal consensus sequence for binding in vitro is the heptanucleotide GGG(T/C)GGG. p150 binds to GC-rich elements related to this sequence present in the promoter regions of p21Cip1/Waf1 and BAX [11]. The present investigation was undertaken to determine whether the protooncogene c-MYC may come under negative regulation by p150, consistent with the frequent overexpression of c-MYC in many forms of cancer and with the action of p150 as a putative tumor suppressor.
Materials and Methods
Cells and transfections
HOSE cells were telomerase-immortalized, HPV E6-transformed human ovarian surface epithelial cells [14], [15]. Ovarian carcinoma-derived RMUGS cells which are deficient in p150 [9] were obtained from the American Type Culture Collection. Cells were grown in DMEM with 10% fetal bovine serum. pcDNA 3.1 (Invitrogen) and pcDNA3.1-p150 were transfected with lipofectamine 2000 (Invitrogen) into HOSE cells for 48 hours. Negative control siRNA (Invitrogen, #45-2001) and SALL2 siRNA (Invitrogen # HSS 109406) were transfected with oligofectamine (Invitrogen) for 48 hours.
Antisera
Polyclonal antisera were raised in rabbits against the N-terminus (amino acids 1–550) and the C-terminus (amino acids 717–1005) of human p150 purified as GST fusions [11]. Antisera were purified by flow through over a GST column and binding to Staph A agarose beads. c-MYC epitope antibody was from Santa Cruz ; it recognizes the 62 kDa c-Myc2 protein referred to as c-MYC in this study. The anti-GAPDH was from Calbiochem.
Western blots
Immunoblotting was carried out on cell extracts using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). Intensity values were determined with LI-COR Odyssey software (Li-COR Biosciences).
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described [16] with slight modifications. For binding of endogenous p150 to the c-MYC promoter in HOSE cells, Staph A-agarose beads coated with N- or C-terminal p150 antibody or normal rabbit sera were used. Immunoselected chromatin fragments were subjected to PCR (32 cycles) with a primer set (5′-CTCTCTTACTCTGTTTACATCCTAG and 5′-CTGGAATTACTACAGCGAGTTAG) for c-MYC amplification. Band intensities of the amplified DNA were revealed on a 1% agarose gel. ChIP assays were performed on HOSE cells with or without prior si-RNA SALL2 knockdown. Control- or SALL2-siRNA was introduced into HOSE cells. At 48 hours post transfection, sonicated chromatin was immunoprecipitated with N-terminal p150 antibody. qRT-PCR assays were performed with primer sets (c-MYC promoter, 5′-GCGTAGTTAATTCATGCGGCTCTC and 5′- CCCACGCCCTCTGCTTTGGGAACC; aldolase A promoter, 5′- CGGTCTGTTCGTTGCACAGAGTAG and 5′-GTTGAGGCAGTAGACAGAGAAAGC). The threshold values of each sample were normalized with those of corresponding 1% input control sample. To determine the effects of etoposide on p150-binding to the c-MYC promoter, HOSE cells were transfected with p150 expression vector for 48 hours and incubated for an additional 22 hours with etoposide (40 µM ) to induce apoptosis. ChIP was carried out with anti-N-terminal p150 antibody. Immunopurified chromatin fragments were amplified by RT-PCR.
Quantitative RT-PCR
Total RNA was isolated using RNeasy® kit (Qiagen), reversed transcribed using QuantiTect Reverse Transcription Kit (Qiagen) and quantitated by RT-PCR using specific primers: SALL2 F: 5′-CACGAATCCGAGAGGAGCTCTC, R: 5′-CACCATTACAGGAGGGTCAGTAG; c-Myc F: 5′-GCTCCACCTCCAGCTTGTAC, R: 5′CGAGCTGCTGTCGTTGAGAG; Aldolase A F: 5′-CGCAGAAGGGGTCCTGGTGA, R: 5′-CAGCTCCTTCTTCTGCTCCGGGGT. qRT-PCR was carried out on a Roche LightCycler 480 using SYBR Green Master Mix. The data were analyzed by the comparative CT (ΔΔCT) method and quantitated relative to the aldolase A gene and normalized to the controls.
Gel shift assays for binding of p150 to the c-MYC promoter
Oligonucleotides containing the NHE III1 of the human c-MYC promoter (5′-TGGGGAGGGTGGGGAGGGTGGGGAAGG), the p150 consensus binding sequence (CS) (5′-GGATCACTGGGTGGGAATCACGCT) and Oct1 binding site (5′-TGTCGAATGCAAATCACTAGAA) and their complementary sequences were purchased from IDT (Coralville, Iowa). Annealed complementary fragments were radiolabeled with [γ-32P] dATP. Binding reactions were carried out in the presence of 100 ng purified GST or GST tagged p150 in reaction buffer containing 10 mM HEPES (pH 7.5), 25 nM KCl, 2.5 mM MgCl, 5 µl ZnCl, 3% glycerol, 2 µg BSA and 200 ng Poly(dA-dT). For specific and nonspecific competition, 50 molar excess unlabeled oligonucleotides were added to the mixture.
Luciferase reporter assay
Reporter plasmids myc-Luc and myc-Luc-ΔNHE with and without the NHE III1 respectively were constructed using two primer sets: myc-Luc, 5′-ATAGATCTCTCTTACTCTGTTTACATCCTAGAGC and 5′-ATAAGCTTCCGGGAGGGGCGCTTATGGGGAGGG; myc-Luc-ΔNHE, 5′- ATAAGCTTCCTCAGCCGTCCAGACCCTCGCATT and 5′- ATAAGCTTGGAGACTCAGCCGGGCAGCCGAGCAC. Amplified fragments were cloned into pGL3 and the constructs verified by sequencing. The vectors myc-Luc and myc-Luc-ΔNHE and pRL-TK were introduced into HOSE cells pretreated with control siRNA or SALL2-siRNA oligonucleotides. For a dose-dependent reporter assay, RMUGS cells were transfected with myc-Luc, pRL-CMV and pcDNA or pcDNA-p150. After 48 hours of incubation at 37°C, the transfected cells were subjected to luciferase activity assays (Dual-Luciferase Reporter System; Promega, Madison, WI). Firefly luciferase values were normalized to those of Renilla luciferase.
Correlations between SALL2 and c-MYC expression levels in tumors
Gene expression data from The Cancer Genome Atlas (TCGA) were accessed and downloaded from TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/). Cancer types selected for correlation analysis were those having more than 100 samples with Level 3 Expression-Gene data (https://tcga-data.nci.nih.gov/tcga/tcgaDataType.jsp) and ones known to arise from tissues that normally express SALL2 based on data in the mouse [2] and human ovarian epithelial cells [9]. This resulted in selection of ovarian serous cystadenocarcinoma (OVCA), glioblastoma multiforme (GBM), lung squamous cell carcinoma (LUSC) and breast invasive carcinoma (BRCA). For all data sets, tumor specimen-specific SALL2 and MYC expression levels were retrieved, compiled, and entered into the SAS JMP Pro 9.0.0 software package. Scatter plots of SALL2 by MYC were generated using the software's “Fit Y by X” command. Pearson correlation coefficients and associated p values were calculated using the default Restricted Maximum Likelihood (REML) method.
Results and Discussion
p150 binds to the NHE III1 of the c-MYC promoter
Several approaches have been used to determine whether p150 binds to the c-MYC promoter region and regulates its expression. Initial focus was on the GC-rich nuclease-hypersensitive element (NHE III1) as a key regulator of c-MYC expression. NHE III1 has tandem repeats of the optimal consensus binding sequence established for p150 [11] (Figure 1A). The NHE III1 can adopt a four stranded ‘quadruplex’ secondary structure known to be targeted by zinc-finger transcription factors and to be associated with silencing of c-MYC transcription [17], [18]. A recombinant GST fusion protein (GST-p150) was expressed and purified from 293 cells [11]. GST-p150 was used in a gel shift assay with radioactive NHE III1 (Figure 1B). GST-p150 clearly binds to the NHE III1 as well as to the consensus sequence (CS) oligonucleotide as a control. A 50-fold molar excess of unlabeled NHE III1 or CS oligonucleotide competed for binding by the labeled NHE III1 while the unrelated Oct1 oligonucleotide did not.
Figure 1. GST-p150 binds to the c-MYC promoter region in vitro and in vivo.
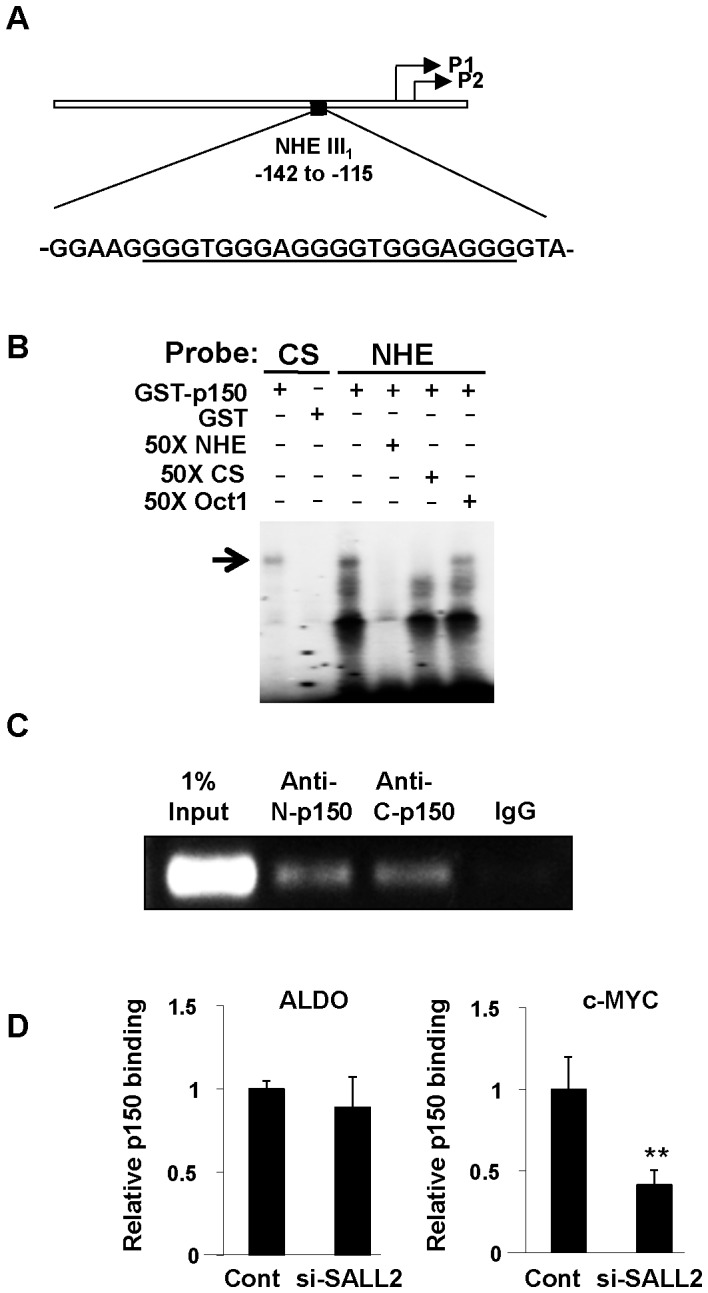
(A) The GC-rich nuclease hypersensitive element NHE III1 of the human c-MYC promoter. Consensus binding sites for p150 are underlined. Approximate positions of transcriptional start sites are shown (P1, P2). (B) An electrophoretic mobility shift assay shows binding of GST-p150 to the labeled NHE III1 and to the consensus binding sequence (CS) GGGTGGG- as control (<$>\raster(75%)="rg1"<$>). 50× molar excess of unlabeled NHE III1 and CS compete for binding while the unrelated Oct1 olionucleotide does not. (C) Chromatin immunoprecipitation assays with antibodies to the N- and C-terminal portions of p150 show binding of endogenous p150 to the NHE III1 region of the c-MYC promoter in HOSE cells. (D) SALL2-knockdown leads to reduced binding of p150 to the c-MYC promoter. HOSE cells were treated with SALL2 siRNA or control oligonucleotide and followed by chromatin immunoprecipitation and quantitative RT-PCR assays. The threshold cycle values of immunoprecipitates were normalized to those of 1% input control samples. The human aldolase A promoter region was included as a negative control. Estimates of binding were based on triplicate samples. ** denotes p<0.01 based on Student t test run on HOSE cells treated with SALL2-siRNA or control RNA.
To determine whether p150 binds to the c-MYC promoter in vivo, chromatin immunoprecipitation was carried out using established human ovarian surface epithelial cells (HOSE). Binding of endogenous p150 to a region of the c-MYC promoter encompassing the NHE III1 was evident using either of two polyclonal antibodies to the human protein (Figure 1C). SALL2 knockdown resulted in a decreased level of p150 binding to the c-MYC promoter region (Figure 1D).
p150 negatively regulates c-MYC expression
To investigate whether binding of p150 to the c-MYC promoter results in activation or repression of c-MYC, levels of SALL2 expression were manipulated in HOSE cells and in RMUGS, an ovarian carcinoma-derived cell line lacking expression of p150 [9]. Overexpression of p150 in HOSE cells led to reduced levels of c-MYC RNA and a commensurate decrease in levels of the protein (Figure 2A). si-RNA targeting of SALL2 was previously shown to promote G1→S progression in these cells [9]. This treatment also led to increases in c-MYC mRNA (Figure 2B, left panel) and protein (Figure 2B, right panel).
Figure 2. p150 represses c-MYC expression.

(A) Left - overexpression of p150 results in reduced c-MYC expression in HOSE cells shown by quantitative RT PCR. Right – immunoblotting for c-MYC. (B) Left - siRNA-knockdown of endogenous SALL2 in HOSE cells leads to increased c-MYC expression by quantitative RT PCR. Right - immunoblotting for c-MYC. (C) Left - Human c-MYC promoter region and luciferase reporters used in promoter activity assays. The NHE region (−142 to −100) including p150 consensus binding sites (underlined) was deleted in the reporter myc-Luc-ΔNHE. Right - siRNA to SALL2 in HOSE cells results in increased expression of myc-Luc but has no effect on myc-Luc-ΔNHE. (D) Expression of exogenous p150 in p150-deficient RMUGS ovarian cancer cells leads to decreased expression of the reporter myc-Luc. All histograms are based on triplicate determinations. * and ** denote p<0.05 and p<0.01, respectively, comparing means of experimental and control.
HOSE cells pretreated with si-SALL2 or control RNA oligonucleotide were transfected with luciferase reporters with and without the NHE III1 (Figure 2C, left) and a pRL-TK-Renilla control vector for normalization. si-SALL2 RNA gave rise to increased expression of the NHE III1 reporter (Figure 2C, right). The ability of p150 to repress c-MYC expression was dependent on retention of the NHE III1 as the reporter plasmid lacking NHE III1 showed no response to si-SALL2 (Figure 2C, right). When RMUGS cells lacking p150 were transfected with increasing amounts of pcDNA- p150 expression vector, signals from the myc -Luc reporter were progressively reduced (Figure 2D).
These results demonstrate that p150 binds to the NHE III1 of the c-MYC promoter in vitro and in vivo and represses c-MYC transcription. The degree of transcriptional repression of c-MYC by p150 is relatively modest. Expression of c-MYC is tightly regulated, with small and often transient increases leading to cellular proliferation [19], [20]. Consistent with these results are earlier findings that downregulation of SALL2 stimulates growth of HOSE cells and that restoration of SALL2 in ovarian carcinoma cells results in suppression of tumor growth [9].
An apoptotic signal leads to increased binding of p150 to the c-MYC promoter
c-MYC can regulate apoptosis either positively or negatively depending on cell type and physiological conditions [21]. HOSE cells were treated with etoposide (40 µM for 22 hours) to induce apoptosis. A fluorimetric assay for caspase-3 was used to confirm the apoptotic response (Figure 3, left panel). Etoposide-treated cells were assayed for SALL2 and c-MYC mRNA. SALL2 was slightly induced and c-MYC strongly repressed following etoposide treatment (Figure 3, middle panel). The strong reduction in c-MYC RNA is most likely due in large part to p53 which is known to repress c-MYC [22], [23]. HOSE cells show a robust p53 response to etoposide (Figure S1). Chromatin immunoprecipitation with anti- p150 followed by qRT-PCR showed that etoposide treatment led to increased binding of p150 to the c-MYC promoter (Figure 3, right panel). Thus, recruitment of p150 to the c-MYC promoter occurs as part of a response to an apoptotic stimulus in HOSE cells. In other cell systems, modest increases in c-MYC expression lead to a proliferative response, while high levels of expression have been linked to tumor suppression and apoptosis [24], [25]. Negative regulation of c-MYC by SALL2 may function to maintain c-MYC levels within a normal range, avoiding thresholds that lead to overproliferation or apoptosis.
Figure 3. An apoptotic signal leads to increased binding of p150 to the c-MYC promoter.

(A) Left - Etoposide-treated HOSE cells were monitored for apoptosis by caspase-3 activation. Middle - Quantitative RT-PCR shows a moderate increase in SALL2 and a large decrease in c-MYC expression following etoposide treatment. Right - ChIP followed by qRT-PCR shows increased binding of p150 to the c-MYC promoter in etoposide-treated cells. All histograms are based on triplicate determinations. ** denotes p<0.01 comparing means of experimental and control.
SALL2 and c-MYC expression in tumors
Overexpression of c-MYC due to increased transcription or DNA amplification accompanies the development of many forms of human cancer including ovarian [26], [27], [28], [29]. Transcriptional repression of c-MYC coupled with activation of p21Cip1/Waf1 and BAX by p150 constitute powerful growth inhibitory and proapoptotic functions consistent with action of SALL2 as a tumor suppressor. To determine whether overexpression of c-MYC is correlated with reduced expression of SALL2 in tumors, levels of SALL2 and c-MYC expression in The Cancer Genome Atlas were accessed. Four tumor types were selected for analysis based on their derivations from normal tissues known to express p150 in the mouse [2] and on high levels of expression in normal human ovarian surface epithelial cells (HOSE). These are ovarian serous cystadenocarcinoma (OVCA), glioblastoma multiforme (GBM), breast invasive carcinoma (BRCA), and lung squamous cell carcinoma (LUSC). These tumor types exhibit highly significant negative correlations between SALL2 and c-MYC expression. Inverse correlations were of similar magnitude, with Pearson coefficients (r) ranging from −.195 for GBM to −.273 for LUSC (Figure 4). Acute myeloid leukemia (AML) was analyzed as a control as there is currently no evidence for SALL2 expression in normal spleen [2] or other lymphoid tissue. For AML the negative correlation is not significant (r = −0.065, n = 197, p = .3677). Taken together, these results suggest that repression of c-MYC by SALL2 may contribute to a role of the latter in suppressing these forms of cancer. They point as well to the need for further understanding of the molecular functions of SALL2.
Figure 4. SALL2 by c-MYC expression scatterplots for cancer types in The Cancer Genome Atlas.

(A) OVCA - ovarian serous cystadenocarcinoma. (B) GBM – glioblastoma multiforme. (C ) BRCA – breast invasive carcinoma. (D) LUSC – lung squamous cell carcinoma. n = number of samples; r = Pearson correlation coefficient; p = p-value of the correlation. The lines shown are those of best fit.
Supporting Information
Etoposide induces a strong p53 response in HOSE cells. HOSE cells were treated with either DMSO or etoposide (40 µM) for 7 hours. Cells were lysed in PBS with 1% NP40, protease and phosphatase inhibitors and subjected to a western analysis. Anti-p150, GAPDH and phospho-specific p53 antibodies were used.
(TIF)
Funding Statement
This work was supported by a grant from the National Cancer Institute RO1 CA 092520 to TB, NIH grant R01 GM077122 to CL, and grants P50 CA105009 and K08 CA108748, the Ovarian Cancer Research Fund, the Robert and Debra First Fund, and the Dana-Farber Women's Executive Council Genetic Fingerprinting Award to RD. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Howley PM, Livingston DM (2009) Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384: 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li D, Dower K, Ma Y, Tian Y, Benjamin TL (2001) A tumor host range selection procedure identifies p150(sal2) as a target of polyoma virus large T antigen. Proc Natl Acad Sci U S A 98: 14619–14624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Parroche P, Touka M, Mansour M, Bouvard V, Thepot A, et al. (2011) Human papillomavirus type 16 E6 inhibits p21(WAF1) transcription independently of p53 by inactivating p150(Sal2). Virology 417: 443–448. [DOI] [PubMed] [Google Scholar]
- 4. Sweetman D, Munsterberg A (2006) The vertebrate spalt genes in development and disease. Dev Biol 293: 285–293. [DOI] [PubMed] [Google Scholar]
- 5. Kohlhase J, Chitayat D, Kotzot D, Ceylaner S, Froster UG, et al. (2005) SALL4 mutations in Okihiro syndrome (Duane-radial ray syndrome), acro-renal-ocular syndrome, and related disorders. Hum Mutat 26: 176–183. [DOI] [PubMed] [Google Scholar]
- 6. Kohlhase J, Taschner PE, Burfeind P, Pasche B, Newman B, et al. (1999) Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am J Hum Genet 64: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al-Baradie R, Yamada K, St Hilaire C, Chan WM, Andrews C, et al. (2002) Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet 71: 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiefer SM, Ohlemiller KK, Yang J, McDill BW, Kohlhase J, et al. (2003) Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum Mol Genet 12: 2221–2227. [DOI] [PubMed] [Google Scholar]
- 9. Li D, Tian Y, Ma Y, Benjamin T (2004) p150(Sal2) is a p53-independent regulator of p21(WAF1/CIP). Mol Cell Biol 24: 3885–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma Y, Li D, Chai L, Luciani AM, Ford D, et al. (2001) Cloning and characterization of two promoters for the human HSAL2 gene and their transcriptional repression by the Wilms tumor suppressor gene product. J Biol Chem 276: 48223–48230. [DOI] [PubMed] [Google Scholar]
- 11. Gu H, Li D, Sung CK, Yim H, Troke P, et al. (2011) DNA-binding and regulatory properties of the transcription factor and putative tumor suppressor p150(Sal2). Biochim Biophys Acta 1809: 276–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu H, Adler AS, Segal E, Chang HY (2007) A transcriptional program mediating entry into cellular quiescence. PLoS Genet 3: e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sung CK, Dahl J, Yim H, Rodig S, Benjamin TL (2011) Transcriptional and post-translational regulation of the quiescence factor and putative tumor suppressor p150(Sal2). FASEB J 25: 1275–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clauss A, Ng V, Liu J, Piao H, Russo M, et al. (2010) Overexpression of elafin in ovarian carcinoma is driven by genomic gains and activation of the nuclear factor kappaB pathway and is associated with poor overall survival. Neoplasia 12: 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drapkin R, von Horsten HH, Lin Y, Mok SC, Crum CP, et al. (2005) Human epididymis protein 4 (HE4) is a secreted glycoprotein that is overexpressed by serous and endometrioid ovarian carcinomas. Cancer Res 65: 2162–2169. [DOI] [PubMed] [Google Scholar]
- 16. Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185. [DOI] [PubMed] [Google Scholar]
- 17. Gonzalez V, Hurley LH (2010) The c-MYC NHE III(1): function and regulation. Annu Rev Pharmacol Toxicol 50: 111–129. [DOI] [PubMed] [Google Scholar]
- 18. Yang D, Hurley LH (2006) Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids 25: 951–968. [DOI] [PubMed] [Google Scholar]
- 19. Shichiri M, Hanson KD, Sedivy JM (1993) Effects of c-myc expression on proliferation, quiescence, and the G0 to G1 transition in nontransformed cells. Cell Growth Differ 4: 93–104. [PubMed] [Google Scholar]
- 20. Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, et al. (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773. [DOI] [PubMed] [Google Scholar]
- 21. Hoffman B, Liebermann DA (2008) Apoptotic signaling by c-MYC. Oncogene 27: 6462–6472. [DOI] [PubMed] [Google Scholar]
- 22. Ho JS, Ma W, Mao DY, Benchimol S (2005) p53-Dependent transcriptional repression of c-myc is required for G1 cell cycle arrest. Mol Cell Biol 25: 7423–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sachdeva M, Zhu S, Wu F, Wu H, Walia V, et al. (2009) p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci U S A 106: 3207–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, et al. (2008) Distinct thresholds govern Myc's biological output in vivo. Cancer Cell 14: 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soucek L, Evan GI (2010) The ups and downs of Myc biology. Curr Opin Genet Dev 20: 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Darcy KM, Brady WE, Blancato JK, Dickson RB, Hoskins WJ, et al. (2009) Prognostic relevance of c-MYC gene amplification and polysomy for chromosome 8 in suboptimally-resected, advanced stage epithelial ovarian cancers: a Gynecologic Oncology Group study. Gynecol Oncol 114: 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haverty PM, Hon LS, Kaminker JS, Chant J, Zhang Z (2009) High-resolution analysis of copy number alterations and associated expression changes in ovarian tumors. BMC Med Genomics 2: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gorringe KL, George J, Anglesio MS, Ramakrishna M, Etemadmoghadam D, et al. (2010) Copy number analysis identifies novel interactions between genomic loci in ovarian cancer. PLoS One 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nowee ME, Snijders AM, Rockx DA, de Wit RM, Kosma VM, et al. (2007) DNA profiling of primary serous ovarian and fallopian tube carcinomas with array comparative genomic hybridization and multiplex ligation-dependent probe amplification. J Pathol 213: 46–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Etoposide induces a strong p53 response in HOSE cells. HOSE cells were treated with either DMSO or etoposide (40 µM) for 7 hours. Cells were lysed in PBS with 1% NP40, protease and phosphatase inhibitors and subjected to a western analysis. Anti-p150, GAPDH and phospho-specific p53 antibodies were used.
(TIF)