

Abstract
Purpose
To determine the frequency and association with relapse-free survival (RFS) of MET and PIK3CA copy number elevations in early stage breast cancer.
Methods
Tumor DNA was extracted from 971 formalin-fixed paraffin-embedded early breast cancers for molecular inversion probes arrays. Data was segmented using the SNP-FASST2 segmentation algorithm. Copy number gains were called when copy number of each segment was greater than 2.3 or 1.7 respectively. RFS was estimated by Kaplan-Meier. Cox proportional hazards models were fit to determine independent associations of copy number with RFS.
Results
82 (8.44%) and 134 (13.8%) of tumors had MET or PIK3CA copy number elevation respectively, and 25.6% with MET copy number elevation had PIK3CA copy number elevation. Patients with either MET or PI3KCA high copy number tended to have poorer prognostic features (larger tumor size, higher grade, and hormone receptor negativity), Both, MET and PIK3CA high copy number were more likely to occur in triple negative disease (P=0.019 and <0.001, respectively). At a median follow-up of 7.4 years, there were 252 recurrences. Five-year RFS were 63.5%, and 83.1% for MET high copy number and MET normal/low copy number respectively, (P=0.06); and 73.1%, and 82.3% for PIK3CA high copy number and PIK3CA normal/low copy number respectively, (P=0.15). High copy number for either gene was not an independent predictor of RFS.
Conclusion
High copy number of MET or PIK3CA was associated with poorer prognostic features and triple negative disease. Co-amplification was frequent. Patients with high MET copy number tumors tended to have a worst RFS.
Keywords: MET, PIK3CA, gene copy number, breast cancer, prognosis
INTRODUCTION
The hepatocyte growth factor (HGF) and its receptor, the transmembrane tyrosine kinase MET interaction leads to formation of phosphorylated MET which activates downstream effectors such as PI3K (Phosphatidylinositol-3 kinase)/AKT, PLC γ (Phospholipase C γ), RAS-MAPK (mitogen-activated protein kinase), c-Src and STATs (signal transducer and activator of transcription) to promote cell proliferation, survival motility, and invasion, as well as morphogenic changes that in normal cells stimulate tissue repair and regeneration but are also co-opted during tumor growth.1-8 MET over-expression, with or without gene amplification, has been reported in a variety of human cancers including breast, lung, and gastrointestinal malignancies.9-11 Further, high levels of HGF and/or MET correlate with poor prognosis in several tumor types, including breast, ovarian, cervical, gastric, head and neck, and non-small cell lung cancer.10,12-15
The PI3K pathway plays a key role in cell growth, protein translation, autophagy, metabolism, and cell survival.16-18 Thus, tight regulation of the PI3K pathway is paramount to ensure that cellular inputs are integrated for appropriate cellular outcomes. The PI3K pathway is downstream of most growth factor tyrosine kinase receptors including MET, EGFR, HER2 and IGFR that have been implicated in breast cancer. PI3K signaling is deregulated through a variety of mechanisms, including overexpression or activation of TKR, activating mutations, gene amplification of PIK3CA and AKT isoforms, as well as loss of the negative regulators PTEN and INPP4B.19,20 Other forms of deregulation and aberrations of this pathway have been implicated not only in breast cancer development and progression, but also in resistance to targeted therapies directed to tyrosine kinase receptors and hormone receptors.20-23 As a result, multiple drugs targeting the PI3K pathway are in early clinical trials as mono or combination therapies in breast cancer.18,24
In breast cancer, reported aberrations in the PI3K pathway have been mostly restricted to detection of activating mutations and loss of suppressors, and data in MET receptor aberrations have been limited in the disease. The purpose of this study was to determine the frequency and association with relapse-free survival (RFS) of MET and PIK3CA copy number elevations and their interaction in a large cohort of early stage breast cancer.
MATERIALS AND METHODS
Patients and tumor samples
Adequate tumor DNA from formalin-fixed paraffin-embedded (FFPE) tissue blocks, clinical history, and follow-up data of 1,003 patients diagnosed with early breast cancer between 1985 and 1999 were identified from the Early Stage Breast Cancer Repository (ESBCR) at MD Anderson Cancer Center. Clinical information (including patient’s age, race/ethnicity, stage, tumor size, lymph node status, nuclear grade, hormone receptor (HR) status) and primary treatment (including surgery, radiotherapy therapy, chemotherapy, and endocrine therapy) was extracted from the medical records.
Molecular inversion probes and copy number
Tumor DNA was extracted from FFPE tissues and processed for copy number analyses. Briefly, 5-10 (5-μm) macrodissected tumor sections containing > 80% tumor cells per protocol were pooled and treated three times with proteinase K in ATL Tissue Lysis Buffer™ (Qiagen, Valencia, CA). Following lysis, samples were applied to uncoated Argylla Particles™ (Argylla Technologies, Tucson, AZ) and processed according to manufacturer recommendations (http://www.argylla.com). For 129 cases, DNA from non-tumor bearing lymph nodes, stored as FFPE, was isolated as an internal germline reference for the population. Tumor and normal DNA at 10 ng/μL was shipped to the Affymetrix™ MIP laboratory for copy number measurement. The laboratory was blinded to all sample and subject information including identity of duplicates. Data from the MIP high-density arrays are deposited at the National Center for Biotechnology Information (NCBIs) (GSE31424).
Nexus Copy Number v5.1 (BioDiscovery, El Segundo, CA) was used for processing the MIP data of these patients’ samples. Nexus Copy Number segmented the data using the SNP-FASST2 segmentation algorithm, and called copy number gains or losses when the estimated copy number of each segment was greater/less than 2.3/1.7 for MET and PIK3CA respectively. Each sample has, in general, a different set of segments. Common segments were derived in order to perform analyses, with a size of 77,487 of the union of all segment break-points for 971 samples. In order to reduce the dimensionality of the data, similar procedures were followed for the CGH regions. We clustered consecutive segments if no two segments within the cluster had different gain/loss calls for at least 97% of the samples. This simple criterion yielded 3378 segments with common breakpoints across all 971 samples.
Statistical Analysis
Patient characteristics were tabulated and described by their medians and ranges by copy number (high vs. normal/low) with a chi-square test or Wilcoxon’s rank sum test as appropriate. Relapse free-survival (RFS) was measured from the date of diagnosis to the date of first local/distant metastasis or last follow-up. Patients who died before experiencing a disease recurrence were considered censored at their date of death in the analysis. Survival outcomes were estimated according to the Kaplan-Meier product limit method. Using log rank statistics, groups were compared between high copy number and normal/low copy number groups for MET, PI3KCA and their co-amplifications as well as other important clinical variables. Three multivariate Cox proportional hazard models were developed. The first model incorporated MET, PIK3CA, copy number and their interaction. The second and third models incorporated either MET copy number or PIK3CA copy number and other prognostic clinical variables. Models were based on a backward selection procedure where all variables of interest were first included in a full model for screening and only variables with P<0.1 were retained. P-values less than 0.05 were considered statistically significant. Analysis was performed by using R 2.11.2. (R Development Core Team, 2010, Vienna, Austria).
RESULTS
Table 1 illustrates the patient and tumor characteristics as well as the therapy received by MET and PIK3CA copy number groups. Eighty-two (8.44%) were found to have elevated MET copy number, 134 (13.8%) had elevated PIK3CA copy number respectively, 25.6% of tumors with elevated MET copy number, also had elevated PIK3CA copy number, and 15.7% of tumors with elevated PIK3CA copy number, also had elevated MET copy number (Figure 1). Patients with tumors harboring either MET or PI3KCA high copy number tended to have more aggressive prognostic features including larger tumor size, higher tumor grade, and negative hormone receptors. There were no significant differences on adjuvant chemotherapy or radiation therapy However, more patients with normal/low copy number in either MET or PIK3CA received adjuvant endocrine therapy (P=0.003 and <0.000, respectively. Elevated MET or PIK3CA copy number were more likely to occur in triple negative disease (P=0.019 and <0.001, respectively).
Table 1.
Patient and Tumor Characteristics
| ALL | MET copy number | PI3KCA copy number | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | N=971 | % | High | Normal/Low | P-value | High | Normal/Lost | P-value | ||||
| N=82 | % | N=889 | % | N=134 | % | N=837 | ||||||
| Age at Diagnosis | ||||||||||||
| Min | 25 | -- | 28 | -- | 25 | -- | 25 | -- | 25 | -- | ||
| Median | 53 | -- | 52 | -- | 53.5 | -- | 51 | -- | 54 | -- | ||
| Max | 87 | -- | 86 | -- | 87 | -- | 0.3899 | 84 | -- | 87 | -- | 0.0387 |
| Tumor size | ||||||||||||
| <2cm | 566 | 58.29 | 34 | 41.46 | 532 | 59.84 | 62 | 46.27 | 504 | 60.22 | ||
| >=2cm | 369 | 38 | 46 | 56.1 | 323 | 36.33 | 0.0009 | 69 | 51.49 | 300 | 35.84 | 0.0012 |
| Nodal Status | ||||||||||||
| Positive | 383 | 39.44 | 34 | 41.46 | 349 | 39.26 | 38 | 28.36 | 345 | 41.22 | ||
| Negative | 565 | 58.19 | 47 | 57.32 | 518 | 58.27 | 0.8543 | 93 | 69.4 | 472 | 56.39 | 0.0057 |
| Grade | ||||||||||||
| 1 | 92 | 9.47 | 4 | 4.88 | 88 | 9.9 | 5 | 3.73 | 87 | 10.39 | ||
| 2 | 477 | 49.12 | 35 | 42.68 | 442 | 49.72 | 50 | 37.31 | 427 | 51.02 | ||
| 3 | 336 | 34.6 | 41 | 50 | 295 | 33.18 | 0.0153 | 72 | 53.73 | 264 | 31.54 | <0.0001 |
| Estrogen Receptor | ||||||||||||
| Negative | 293 | 30.18 | 38 | 46.34 | 255 | 28.68 | 71 | 52.99 | 222 | 26.52 | ||
| Positive | 666 | 68.59 | 44 | 53.66 | 622 | 69.97 | 0.0018 | 62 | 46.27 | 604 | 72.16 | <0.0001 |
| Progesterone Receptor | ||||||||||||
| Negative | 406 | 41.81 | 42 | 51.22 | 364 | 40.94 | 80 | 59.7 | 326 | 38.95 | ||
| Positive | 555 | 57.16 | 40 | 48.78 | 515 | 57.93 | 0.109 | 53 | 39.55 | 502 | 59.98 | <0.0001 |
| HER2 | ||||||||||||
| Negative | 764 | 78.68 | 68 | 82.93 | 696 | 78.29 | 102 | 76.12 | 662 | 79.09 | ||
| Positive | 207 | 21.32 | 14 | 17.07 | 193 | 21.71 | 0.4009 | 32 | 23.88 | 175 | 20.91 | 0.5051 |
| Breast Cancer Subtype | ||||||||||||
| HER2-positive | 207 | 21.32 | 14 | 17.07 | 193 | 21.71 | 32 | 23.88 | 175 | 20.91 | ||
| Hormone receptor-positive | 583 | 60.04 | 44 | 53.66 | 539 | 60.63 | 53 | 39.55 | 530 | 63.32 | ||
| Triple receptor negative | 173 | 17.82 | 24 | 29.27 | 149 | 16.76 | 0.0193 | 49 | 36.57 | 124 | 14.81 | <0.0001 |
| Menopausal Status | ||||||||||||
| Peri/pre-menopausal | 312 | 32.13 | 28 | 34.15 | 284 | 31.95 | 49 | 36.57 | 263 | 31.42 | ||
| Post-menopausal | 624 | 64.26 | 52 | 63.41 | 572 | 64.34 | 0.8363 | 80 | 59.7 | 544 | 64.99 | 0.2686 |
| Stage | ||||||||||||
| I | 304 | 31.31 | 18 | 21.95 | 286 | 32.17 | 32 | 23.88 | 272 | 32.5 | ||
| II | 662 | 68.18 | 64 | 78.05 | 598 | 67.27 | 0.0694 | 101 | 75.37 | 561 | 67.03 | 0.0599 |
| Histology | ||||||||||||
| Ductal | 898 | 92.48 | 78 | 95.12 | 820 | 92.24 | 128 | 95.52 | 770 | 92 | ||
| Other | 58 | 5.97 | 3 | 3.66 | 55 | 6.19 | 0.4914 | 2 | 1.49 | 56 | 6.69 | 0.0332 |
| MET | ||||||||||||
| High copy number | 82 | 8.44 | -- | -- | -- | -- | 21 | 15.67 | 61 | 7.29 | ||
| Normal/Low copy number | 889 | 91.56 | -- | -- | -- | -- | -- | 113 | 84.33 | 776 | 92.71 | 0.0021 |
| PIK3CA | ||||||||||||
| High copy number | 134 | 13.8 | 21 | 25.61 | 113 | 12.71 | -- | -- | -- | -- | ||
| Normal/Low copy number | 837 | 86.2 | 61 | 74.39 | 776 | 87.29 | 0.0021 | -- | -- | -- | -- | -- |
| Chemotherapy | ||||||||||||
| No | 480 | 49.43 | 38 | 46.34 | 442 | 49.72 | 62 | 46.27 | 418 | 49.94 | ||
| Anthracycline-based | 323 | 33.26 | 26 | 31.71 | 297 | 33.41 | 49 | 36.57 | 272 | 32.74 | ||
| Anthracycline and Taxane-based | 114 | 11.74 | 12 | 14.63 | 102 | 11.47 | 0.6496 | 14 | 10.45 | 100 | 11.95 | 0.5961 |
| Radiation Therapy | ||||||||||||
| Yes | 410 | 42.22 | 38 | 46.34 | 372 | 41.84 | 47 | 35.07 | 363 | 43.37 | ||
| No | 535 | 55.1 | 43 | 52.44 | 492 | 55.34 | 0.5805 | 84 | 62.69 | 451 | 53.88 | 0.0762 |
| Endocrine Therapy | ||||||||||||
| Yes | 422 | 43.46 | 23 | 28.05 | 399 | 44.88 | 36 | 26.87 | 386 | 46.12 | ||
| No | 522 | 53.76 | 58 | 70.73 | 464 | 52.19 | 0.003 | 95 | 70.9 | 427 | 51.02 | <0.0001 |
Figure 1.
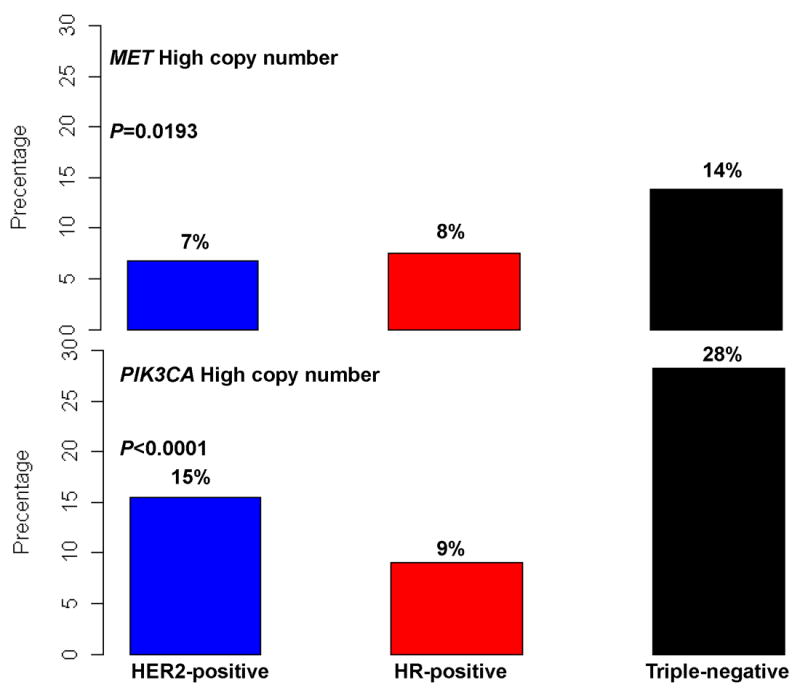
Distribution of MET and PIK3CA copy number by breast cancer subtype.
At a median follow-up of 7.5 years (range 0-21.1 years), there were 252 recurrences. Table 2 summarizes the 5-year RFS by MET and PIK3CA copy number and by other patient and tumor characteristics. Five-year RFS was 63.5%, and 83.1% for MET high copy number and MET normal/low copy number respectively, (P=0.06); and 73.1%, and 82.3% for PIK3CA high copy number and PIK3CA normal/low copy number respectively, (P=0.15) (Figures 2A and 2B). To evaluate the interaction of coordinate gene copy elevations in MET and PIK3CA, patients were classified into four groups: normal/low both PIK3CA and MET copy number, high both PIK3CA and MET copy number, MET high copy number and PIK3CA high copy number. No statistically significant difference in 5-year RFS estimates was found (P=0.137) (Figure 2C).
Table 2.
Five-year relapse-free survival estimates by copy number and patient and tumor characteristics
| 5 Year RFS Estimates | |||||
|---|---|---|---|---|---|
| N | Events | Survival | 95% CI | P-Value | |
| All | 971 | 252 | 81.00% | (78.5%, 83.6%) | |
| MET copy number | |||||
| High | 82 | 29 | 72.60% | (63.5%, 83.1%) | |
| Normal/Low | 889 | 223 | 81.80% | (79.3%, 84.5%) | 0.0637 |
| PI3KCA copy number | |||||
| High | 134 | 44 | 73.10% | (65.7%, 81.3%) | |
| Normal/Low | 837 | 208 | 82.30% | (79.7%, 85.1%) | 0.154 |
| Subtype | |||||
| HER2-positive | 207 | 62 | 76.30% | (70.5%, 82.5%) | |
| Hormone receptor-positive | 583 | 137 | 84.70% | (81.7%, 87.8%) | |
| Triple receptor-negative | 173 | 49 | 74.20% | (67.8%, 81.3%) | 0.071 |
| Age at Diagnosis | |||||
| <50 | 371 | 123 | 77.40% | (73.2%, 81.9%) | |
| >=50 | 595 | 129 | 83.30% | (80.3%, 86.5%) | 0.0003 |
| Tumor size | |||||
| <2cm | 566 | 116 | 86.50% | (83.6%, 89.5%) | |
| >=2cm | 369 | 123 | 73.90% | (69.4%, 78.7%) | <0.0001 |
| Nodal Status | |||||
| Positive | 383 | 113 | 86.60% | (83.7%, 89.5%) | |
| Negative | 565 | 130 | 73.90% | (69.5%, 78.6%) | <0.0001 |
| Grade | |||||
| 1 | 92 | 22 | 85.00% | (77.8%, 92.9%) | |
| 2 | 477 | 113 | 83.30% | (79.9%, 86.9%) | |
| 3 | 336 | 95 | 77.50% | (73.0%, 82.2%) | 0.249 |
| Menopausal Status | |||||
| Peri/pre-menopausal | 312 | 101 | 76.30% | (71.5%, 81.3%) | |
| Post-menopausal | 624 | 140 | 83.90% | (81.0%, 87.0%) | 0.0009 |
| Stage | |||||
| I | 304 | 43 | 91.90% | (88.8%, 95.1%) | |
| II | 662 | 209 | 76.00% | (72.7%, 79.5) | <0.0001 |
| Histology | |||||
| Ductal | 898 | 235 | 80.70% | (78.0%, 83.4) | |
| Other | 58 | 14 | 85.30% | (76.3%, 95.2%) | 0.826 |
| Chemotherapy | |||||
| No | 480 | 102 | 85.20% | (81.9%, 88.6%) | |
| Anthracycline-based | 323 | 110 | 75.50% | (70.9%, 80.5%) | |
| Anthracycline and Taxane-based | 114 | 24 | 81.60% | (74.6%, 89.2%) | 0.001 |
| Radiation Therapy | |||||
| Yes | 410 | 109 | 82.50% | (78.8%, 86.4%) | |
| No | 535 | 134 | 80.60% | (77.2%, 84.2%) | 0.645 |
| Endocrine Therapy | |||||
| Yes | 422 | 79 | 87.30% | (84%, 90.6%) | |
| No | 522 | 164 | 76.80% | (73.1%, 80.6%) | 0.0008 |
Figure 2.
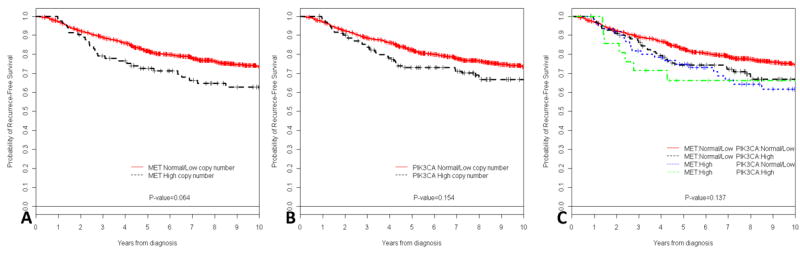
Kaplan-Meier relapse-free survival curves of all patients and MET (A) copy number, PIK3CA (B) copy number, and MET and PIK3CA copy number (C).
The Kaplan-Meier survival curves by MET and PIK3CA gene copy number and breast cancer subtype are presented in Figure 3. When looking at MET copy number, patients with HR-positive and high MET copy number breast cancer had a significant lower 5-year RFS compared with patients with HR-positive and normal/low MET copy number breast cancer (76.4% vs. 85.4%, P=0.034). There was a trend to worse 5-year RFS in patients with HER2-positive and high MET copy number breast cancer compared with patients with HER2-positive and normal/low MET copy number breast cancer (64.3% vs. 77.2%, P=0.061). No difference was seen in triple receptor-negative disease (P=0.80). When looking at PIK3CA copy number, there were no differences in 5-year RFS estimates by breast cancer subtype. Exploratory survival analysis to evaluate the interaction of high gene copy number in both MET and PIK3CA by breast cancer subtypes showed no statistically significant difference in 5-year RFS estimates (data not shown).
Figure 3.
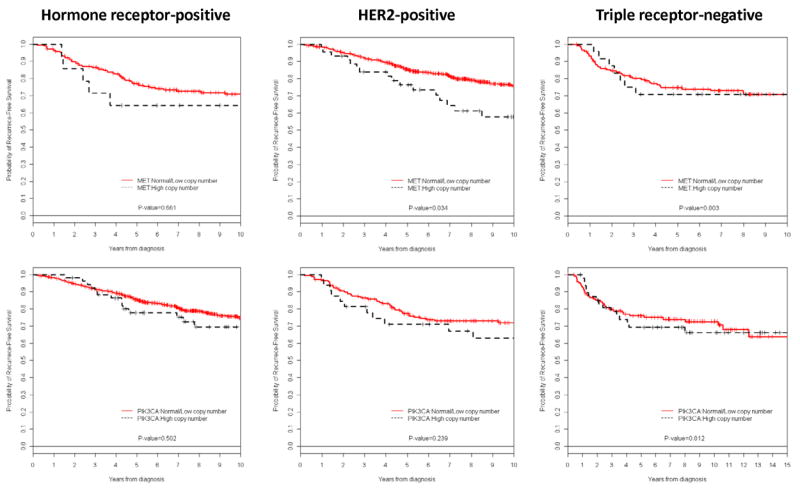
Kaplan-Meier relapse-free survival curves of patients by breast cancer subtype and MET or PIK3CA copy number.
Table 3 summarizes the multivariate models MET, PIK3CA, copy number and their interaction. The results were consistent with the RFS univariate analysis. Overall, patients with tumors harboring high MET copy number tended to be at higher risk to develop a recurrence compared to patients with tumors with normal/low MET copy number (HR:1.53, 95% CI:0.98-2.38, P=0.06). High PIK3CA copy number was not an independent predictor risk for recurrence (HR:1.3, 95% CI:0.91-1.86, P=0.147), nor was the interaction of both MET and PIK3CA high copy number (HR:0.7, 95% CI:0.28-1.77, P=0.458). When looking at patients with hormone receptor-positive breast cancer, patients with tumors with high MET copy number were more likely to develop recurrences (HR:1.86, 95% CI:1.07-3.25, P=0.029). In multivariate models including patient and tumor characteristics, MET or PIK3CA high copy number were not independent predictors of RFS after adjustment for age, stage, nodal status, tumor size and breast cancer subtype (HR:1.21, 95% CI:0.8-1.82, P=0.357, and HR:1.29, 95% CI:0.85-1.94, P=0.229).
Table 3.
Multivariate Cox proportional hazard model including MET, PIK3CA copy number and their interaction
| Hazard Ratio | 95% Confidence Intervals | P-value | |
|---|---|---|---|
| All Tumors | |||
| MET Normal/Low copy number | 1 | ||
| MET High Copy number | 1.53 | 0.98-2.38 | 0.06 |
| PIK3CA Normal/Low copy number | 1 | ||
| PIK3CA High Copy number | 1.3 | 0.91-1.86 | 0.147 |
| MET and PIK3CA high copy number | 0.7 | 0.28-1.77 | 0.458 |
| HER2-positive | |||
| MET Normal/Low copy number | 1 | ||
| MET High Copy number | 0.92 | 0.29-2.95 | 0.886 |
| PIK3CA Normal/Low copy number | 1 | ||
| PIK3CA High Copy number | 1.3 | 0.67-2.52 | 0.43 |
| MET and PIK3CA high copy number | 2.68 | 0.4-18.16 | 0.312 |
| Hormone receptor-positive | |||
| MET Normal/Low copy number | 1 | ||
| MET High Copy number | 1.86 | 1.07-3.25 | 0.0287 |
| PIK3CA Normal/Low copy number | 1 | ||
| PIK3CA High Copy number | 1.25 | 0.7-2.23 | 0.4418 |
| MET and PIK3CA high copy number | 0.59 | 0.12-2.89 | 0.515 |
| Triple receptor-negative | |||
| MET Normal/Low copy number | 1 | ||
| MET High copy number | 1.33 | 0.51-3.43 | 0.56 |
| PIK3CA Normal/Low copy number | 1 | ||
| PIK3CA High copy number | 1.17 | 0.6-2.3 | 0.645 |
| MET and PIK3CA high copy number | 0.61 | 0.12-2.95 | 0.535 |
DISCUSSION
The purpose of this study was to determine the frequency of MET and PIK3CA copy number in breast cancer and their associations with patient outcome. We found that 82 (8.44%) and 134 (13.8%) of tumors had elevated MET or PIK3CA copy number respectively, and 25.6% of tumors had both PIK3CA and MET high copy number and that high copy number of MET or PIK3CA was associated with poorer prognostic features and triple negative disease. Patients with tumors harboring elevated MET copy number tended to have worse 5-year RFS, (P=0.06). High copy number for either gene was not an independent predictor of RFS.
Deregulation of MET receptor activity occurs in a wide spectrum of human cancers, as a result of germline SNPs, somatic mutations, gene amplification, protein overexpression, and autocrine circuits driven by HGF.24 In breast cancer, reports have been small and restricted to assessment of protein overexpresion of the MET receptor as well as its ligand HGF in tumor tissues.13-15,26,27 The estimated rate of MET protein overexpression has been 20-30%,26,27 and as in several other tumor types, increased expression of MET receptor or its ligand HGF in breast cancer correlates with increased aggressiveness of disease and an overall poorer prognosis.14,25,26 Initial work assessing immunoreactive (ir)-HGF concentrations in tumor extracts of 258 primary human breast cancers using an enzyme-linked immunosorbent assay showed that ir-HGF level was correlated with large tumor size (P=0.05). Patients with high ir-HGF concentration had a significantly shorter relapse-free (P=0.001) and overall survival (P=0.001) rates. ir-HGF level was an independent predictor of relapse-free (P=0.041) and overall survival (P=0.036).14 In a smaller cohort of 91 tumors, high MET receptor expression by immunofluorescence in patients with positive and negative lymph nodes correlated with lower 5-year survival rate (P=0.008, and P=0.006 respectively).27 IA small study of 40 primary breast cancers detecting MET and HGF by immunoflourescence and immunohistochemistry showed that MET levels did not correlate with established poor prognostic factors. However, overexpression of MET correlated with disease progression (P=0.037). They also showed that when accounting for HER2 status, MET overexpression identified a subset of patients with adverse outcomes independently of HER2 positivity.26 Our study is the first one to report MET gene copy number, its distribution by tumor subtype and correlation with patient outcome. In our analysis, patients with high MET copy number had larger tumors, of higher grade, and negative hormone receptors. Two interesting findings should be highlighted: the higher proportion of triple-negative disease with this aberration, and the prognostic value of copy number in breast cancer, especially in hormone receptor-positive disease. This is important since we continue to need new therapeutic targets that activate signaling in triple receptor-negative breast cancer, and MET receptor inhibition could contribute to cell proliferation, survival and invasion. On the other hand, in our search for mechanisms of therapy resistance to endocrine therapy, MET signaling should be investigated. Lastly, we need to define the frequency of MET protein overexpression and its correlation with other aberrations such as gene amplification and activating mutations. Comprehensive work in our institution is on-going to answer this question.
In breast cancer, reported aberrations in the PI3K pathway have been mostly restricted to detection of activating mutations and loss of tumor suppressors and the data on PIK3CA gene amplification being limited.21,28,29 In a series of 92 primary breast cancers, investigators used quantitative real-time PCR to measure gene copy number and reported that 8.7% (8 of 92) of the tumors harbored a gain of PIK3CA gene copy number suggesting that gene amplification is not the main molecular mechanism in activating the PI3K-driven tumorigenesis pathway in breast cancer.30 In a second cohort of breast cancers, researchers reported that 10 of 161 tumors had PIK3CA gene amplification, and half of them also had an activation mutation in the gene suggesting that an additive effect of point mutation and copy number gain can contribute to oncogenesis.31 In our large cohort of early breast cancers, we also showed that 13.8% of all breast cancers have elevated PIK3CA copy number. However, assessing breast cancer subtypes, 28% of triple negative breast cancers had a high PIK3CA copy number suggesting that gene amplification in conjunction with loss PTEN and INPP4B may contribute to PI3K pathway activation in this subtype. However, basal breast cancers have a greater frequency of copy number aberrations, thus determining whether PIK3CA (or MET) amplification is a tumor “driver” remains to be determined. Previous to our report, the largest tumor set in which PIK3CA amplification was assessed, consisted of 292 invasive breast cancers, from which 209 were tested and 28 were found to be amplified (13.4%).32 In this work the investigators looked at other PI3K pathway aberrations and correlated them with breast cancer molecular subtype and outcome. Mutations and copy number gains were almost exclusive events with only 1 cancer encompassing both mutation and copy number gain of the PIK3CA gene. Neither mutations nor copy number gain were associated with clinicopathological parameters, breast cancer molecular subtype or outcome.32
Lastly we demonstrated that 26% of MET-amplified tumors also have PIK3CA amplification, a higher frequency than predicted by chance implying that coaberrations in the PI3K and MET pathways occur at sufficient frequency that could contribute to patient outcomes. Further studies including comprehensive analysis of large cohorts of breast cancers (i.e. TCGA- The Cancer Genome Atlas) to determine frequencies of mutations, copy number, methylation, translational changes in PI3K pathway-related genes and MET alone and in combination to determine the frequency of coaberrations in the pathways across multiple modalities are on-going in our group.
Acknowledgments
Funding: This work was supported in part by the Kleberg Center for Molecular Markers at MD Anderson Cancer Center, National Cancer Institute 1K23CA121994-01 (AMG), National Cancer Institute Breast Specialized Program for Research Excellence P50-CA116199 (GBM, MLB). National Cancer Institute through The University of Texas MD Anderson’s Cancer Center Support Grant (P30 CA016672). The Susan G. Komen Foundation FAS0703849 (AMG, GBM), and SAC100004 (to AMG, GRB).
Footnotes
Financial Disclosures: The authors have no financial disclosures.
References
- 1.Stoker M, Gherardi E, Perryman M, et al. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature. 1987;327:239–42. doi: 10.1038/327239a0. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura T, Nishizawa T, Hagiya M, et al. Molecular cloning and expression of human hepatocyte growth factor. Nature. 1989;342:440–3. doi: 10.1038/342440a0. [DOI] [PubMed] [Google Scholar]
- 3.Miyazawa K, Tsubouchi H, Naka D, et al. Molecular cloning and sequence analysis of cDNA for human hepatocyte growth factor. Biochem Biophys Res Commun. 1989;163:967–73. doi: 10.1016/0006-291x(89)92316-4. [DOI] [PubMed] [Google Scholar]
- 4.Zarnegar R, Michalopoulos G. Purification and biological characterization of human hepatopoietin A, a polypeptide growth factor for hepatocytes. Cancer Res. 1989;49:3314–20. [PubMed] [Google Scholar]
- 5.Gherardi E, Stoker M. Hepatocytes and scatter factor. Nature. 1990;346:228. doi: 10.1038/346228b0. [DOI] [PubMed] [Google Scholar]
- 6.Bottaro DP, Rubin JS, Faletto DL, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 7.Weidner KM, Arakaki N, Hartmann G, et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA. 1991;88:7001–5. doi: 10.1073/pnas.88.16.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma PC, Maulik G, Christensen J, et al. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–25. doi: 10.1023/a:1023768811842. [DOI] [PubMed] [Google Scholar]
- 9.Boix L, Rosa JL, Ventura F, et al. c-Met mRNA overexpression in human hepatocellular carcinoma. Hepatology. 1994;19:88–91. [PubMed] [Google Scholar]
- 10.Ichimura E, Maeshima A, Nakajima T, et al. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. 1996;87:1063–9. doi: 10.1111/j.1349-7006.1996.tb03111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edakuni G, Sasatomi E, Satoh T, et al. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int. 2001;51:172–8. doi: 10.1046/j.1440-1827.2001.01182.x. [DOI] [PubMed] [Google Scholar]
- 12.Ayhan A, Ertunc D, Tok EC, et al. Expression of the c-Met in advanced epithelial ovarian cancer and its prognostic significance. Int J Gynecol Cancer. 2005;15:618–23. doi: 10.1111/j.1525-1438.2005.00117.x. [DOI] [PubMed] [Google Scholar]
- 13.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109:863–867. doi: 10.1172/JCI15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita J, Ogawa M, Yamashita S, et al. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 1994;54:1630–3. [PubMed] [Google Scholar]
- 15.Parr C, Watkins G, Mansel RE, et al. The hepatocyte growth factor regulatory factors in human breast cancer. Clin Cancer Res. 2004;10:202–11. doi: 10.1158/1078-0432.ccr-0553-3. [DOI] [PubMed] [Google Scholar]
- 16.Hennessy BT, Smith DL, Ram PT, et al. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 17.Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27:2278–2287. doi: 10.1200/JCO.2008.20.0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal R, Carey M, Hennessy B, Mills GB. PI3K pathway-directed therapeutic strategies in cancer. Curr Opin Investig Drugs. 2010;11:615–28. [PubMed] [Google Scholar]
- 19.Gewinner C, Wang ZC, Richardson A, et al. Evidence that inositol polyphosphate 4- phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–25. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller TW, Perez-Torres M, Narasanna A, et al. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69:4192–201. doi: 10.1158/0008-5472.CAN-09-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Tenorio G, Stal O. Activation of AKT/PKB in breast cancer predicts a worse outcome among endocrine treated patients. Br J Cancer. 2002;86:540–5. doi: 10.1038/sj.bjc.6600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagata Y, Lan KH, Zhou X, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–27. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 23.Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12:395–402. doi: 10.1016/j.ccr.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 24.McAuliffe PF, Meric-Bernstam F, Mills GB, Gonzalez-Angulo AM. Deciphering the role of PI3K pathway in breast cancer biology and pathogenesis. Clin Breast Cancer. 2010;(Suppl 3):S59–65. doi: 10.3816/CBC.2010.s.013. [DOI] [PubMed] [Google Scholar]
- 25.Eder JP, Vande Woude GF, Boemer SA, LoRusso PM. Novel Therapeutic Inhibitors of the c-Met signaling Pathway in Cancer. Clin Cancer Res. 2009;15:2207–14. doi: 10.1158/1078-0432.CCR-08-1306. [DOI] [PubMed] [Google Scholar]
- 26.Lengyel E, Prechtel D, Resau JH, et al. c-Met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of Her2/neu. Int J Cancer. 2004;113:678–682. doi: 10.1002/ijc.20598. [DOI] [PubMed] [Google Scholar]
- 27.Ghoussoub RAD, Dillon DA, D’Aquilla T, et al. Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer. 1998;92:1523–20. doi: 10.1002/(sici)1097-0142(19980415)82:8<1513::aid-cncr13>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 28.Saal LH, Johansson P, Holm K, et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc Natl Acad Sci U S A. 2007;104:7564–7569. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–6091. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu G, Xing M, Mambo E, et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7:R609–16. doi: 10.1186/bcr1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kadota M, Sato M, Duncan B, et al. Identification of novel gene amplifications in breast cancer and coexistence of gene amplification with an activating mutation of PIK3CA. Cancer Res. 2009;69:7357–65. doi: 10.1158/0008-5472.CAN-09-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.López-Knowles E, O’Toole SA, McNeil CM, et al. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int J Cancer. 2010;126:1121–31. doi: 10.1002/ijc.24831. [DOI] [PubMed] [Google Scholar]