

Severe traumatic brain injury is a significant risk factor for development of epilepsy that often becomes clinically apparent after a latent period of months to years (Salazar et al, 1985; Annegers et al, 1998). Unfortunately, a large percentage of patients with symptomatic epilepsies due to trauma or other etiologies are not seizure free on appropriate pharmacological antiseizure treatment (Kwan and Brodie, 2000). The high incidence of epileptogenesis following severe injury in war time (Salazar et al., 1985) and the expected marked increase due to the conflicts in Iraq and Afghanistan, emphasize the importance of developing prophylactic strategies that might be applied during the latent period between injury and the onset of seizures (Garga and Lowenstein, 2006). To date, multiple clinical trials aimed at prevention of epilepsy after traumatic brain injury (TBI) with older antiseizure agents (Glötzner et al, 1983; Young et al, 1983a,b; Temkin et al. 1990, 1999), glucocorticoids (Watson et al, 2004) and magnesium (Temkin et al. 2007) have been unsuccessful (see Temkin 2001, 2009 for reviews).
In previous experiments, we used the partial cortical isolation (“undercut”or “UC” ) model of chronic posttraumatic epileptogenesis (Echlin, 1959; Sharpless and Halpern, 1962) to assess cellular mechanisms that might underlie injury-induced focal cortical hyperexcitability (Tseng and Prince, 1993; Salin et al, 1995; Hoffman et al, 1994; Li and Prince, 2002; Jin et al 2006; Faria and Prince, 2010; Jin et al, 2011; reviewed in Graber and Prince, 2006; Prince et al, 2009; Li et al, 2010). Rodent in vitro slices from the undercut (UC) cortex generate spontaneous and evoked epileptiform discharges after a latent period of ~7–14 days (Tseng and Prince, 2003; Hoffman et al, 1994; Salin et al, 1995). Focal suppression of cortical activity with tetrodotoxin (TTX) during a critical period of 3 days after injury reduces subsequent epileptogenesis in this model, providing a proof in principle that prophylaxis of posttraumatic epileptogenesis is possible (Graber and Prince 1999, 2004). The mechanisms for this effect have not been fully explored; however, recent data indicate that TTX treatment reduces immunocytochemical evidence for axonal and terminal sprouting (Prince et al, 2009).
Enhanced excitatory connectivity, is a prominent pathophysiological alteration in injured epileptogenic neocortex (Salin et al, 1995; Li and Prince 2002; Jin et al., 2006) and hippocampus (Tauck and Nadler 1985, McKinney et al 1997, Yang et al 1995), as well as in human epileptogenic temporal lobes (Babb et al, 1991, 1992; Masukawa et al, 1992; Isokawa et al 1993; Marco and DeFelipe, 1997). Layer V excitatory pyramidal (Pyr) neurons in UC neocortex develop elaborate sprouting of axonal arbors with increased branching and a higher density of boutons on their axons (Salin et al. 1995), and whole cell recordings from these cells show an increase in the frequency of spontaneous (s) and miniature (m) excitatory postsynaptic currents (EPSCs) (Li and Prince, 2002). Experiments in which laser scanning photostimulation of caged glutamate has been used to map excitatory circuitry have also revealed a more extensive functional excitatory connectivity in the UC cortex (Jin et al. 2006). These findings have led to the hypothesis that an intervention to decrease new excitatory synapse formation after injury might be one approach to antiepileptogenesis in the undercut model of posttraumatic epileptogenesis. Formation of new synaptic connections is an important event during development in cortical structures (Sutor and Luhmann, 1995, Sur and Leamey, 2001) that appears to be recapitulated after injury (Salin et al, 1995; Carmichael and Chesselet, 1992). A potential approach to limiting excessive excitatory synapse formation, and possibly epileptogenesis following injury, was suggested by recent results that have shown a prominent role for thrombospondins (TSPs), and perhaps other astrocyte-secreted proteins, in excitatory synapse formation during development (Christopherson et al 2005; Eroglu et al, 2009). The receptor for TSPs is the calcium channel α2δ1 subunit (Gee et al., 1996; Field et al., 2006), which is significantly upregulated by nerve and brain injury (Luo et al 2001, 2002 ) and isalso the receptor for the antiepileptic/antiallodynic drugs gabapentin(GBP) (Eroglu et al, 2009; Gee et al., 1996) and pregabalin. GBP also has potentialneuroprotective properties (Rothstein and Kuncl, 1995). High affinity binding of GBP inhibits TSP-induced excitatory synapse formation during development both in vitro and in vivo, providing it is administered within a ~5 days of formation of a new synapse; already existing synapses are unaffected (Eroglu et al., 2009). These developmental results lead to the hypothesis that GBP treatment might also block or limit excitatory synaptogenesis following injury, and reduce posttraumatic hyperexcitability in this model. We therefore treated animals with GBP via ip injections or sc via infusion pumps following injury and used the expression of epileptiform activity in in vitro slices, recordings of sEPSCs and mEPSCs in layer V Pyr neurons, and anatomical measures of excitatory synapses to assess drug effects. Results suggest that GBP administered after cortical injury limits excitatory synapse formation, reduces the frequency of excitatory postsynaptic currents (EPSCs), decreases the incidence of epileptiform discharges in neocortical slices and has neuroprotective effects. Portions of these results were published in an abstract (Li et al., 2009).
Materials and Methods
All experiments were carried out according to protocols approved by the Stanford Institutional Animal Care and Use Committee.
Preparation of partial cortical isolations
Using previously described methods (Graber and Prince, 2006), partial isolations of sensorimotor cortex (UCs) were made in 141 male Sprague-Dawley rats at postnatal ages (P)30 ±1 day. Animals were deeply anesthetized with ketamine (Phoenix, St. Joseph, MO, 80 mg/kg ip) and xylazine (Rompun Lloyd Lab, Shenandoah Iowa, 8 mg/kg ip), the scalp incised and retracted, and a 3 × 5-mm bone window centered on the coronal suture removed with dura intact. A 30-gauge needle, bent 90° 2 mm from the tip, was inserted parasagittally ~1–2 mm from the interhemispheric sulcus, advanced under direct vision tangentially just beneath the pial vessels, and lowered 2 mm. The needle was rotated ~180° to produce a contiguous white matter lesion, elevated to just beneath the pia to make a second transcortical cut, and removed. An additional transcortical lesion was placed ~2 mm lateral and parallel to the initial parasagittal cut without needle rotation. The skull opening was covered with sterile Saran Wrap®, and the skin sutured. Animals were treated with 0.02 – 0.03 mg/kg buprenorphine, s.c. and recovered uneventfully. Groups of UC rats were subsequently re-anaesthetized and used for in vitro slice, immunohistochemical or Western blot experiments as below.
Protocols for GBP administration
Undercut littermates were randomly treated with GBP (USP) or saline beginning within 1h following surgery using two protocols (Fig. 1A). Group 1 had i.p injections of saline or GBP, 100mg/kg/day 3x/day for 2 days (Grp 1 of Fig. 1A). Group 2 had a continuous subcutaneous infusion of saline or saline containing GBP via implanted Alzet pumps at an estimated dose of ~120 mg/kg/day for 13–15 days, taking increase in body weight over the dose period into account. In vitro slices for electrophysiological recordings were obtained 13–15 days after the UC in both groups, i.e. on the last day of treatment in group 2 and 11–13 days after the end of GBP treatment in group 1 (Methods below).
Figure 1. Treatment with GBP decreases the incidence of evoked epileptiform discharges in UC cortex.

A: GBP treatment protocols. Group (Grp)1: GBP 100mg/kg or saline i.p 3x/day for 2 days (gray arrows/lines) beginning within 1h of placement of UC lesion (time 0), followed by slice experiment (dashed line) on day 15 after UC. Grp2: GBP ~10 mg/day or saline via Alzet pump infusion sc followed by slice experiment on day 15.
B: Representative traces of epileptiform field potentials evoked at threshold by 2 consecutive stimuli delivered on column at the white matter/layer VI border from a slice of saline-treated UC rat. Acute GBP application in vitro (400μM x 2h) failed to block such responses (n = 4 slices; not shown).
C: Representative responses to stimuli as in B in a slice from UC GBP-treated rat of Grp 2 in which epileptiform responses are not evoked.
D: Percentage of slices in which stimuli evoked epileptiform responses similar to those in B, in groups 1 and 2 of A. Numbers in bars: number of rats in saline and GBP groups. 4–5 slices assayed/animal (average 4.3). Data are expressed as mean ± SEM. *: p < 0.05.
Neocortical slice preparation and field potential recordings
UC rats or naïve control littermates were deeply anesthetized with sodium pentobarbital (55 mg/kg i.p.), brains removed and blocked and ~350 μm thick coronal sensorimotor cortical slices cut with a vibrotome (Leica VT1000s) as previously described (Li and Prince 2002). Slices were incubated for at least 1 hour in artificial cerebral spinal fluid (ACSF) containing (in mM): 126 NaCl, 5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, 10 glucose, and 26 NaHCO3; pH 7.4 when gassed with 95% O2/5% CO2 at 32°C. Slices were placed in a modified interface recording chamber (34 – 36°C) where they were perfused with ACSF at a rate of 2 ml/min. The partially isolated cortical area was easily identified under a dissecting microscope and evoked field potentials recorded in layer V with an ACSF-filled glass pipette. Focal extracellular 50 μsec single and paired square-wave pulses (10 and 30 msec interpulse interval) were delivered at 0.1 Hz through a concentric bipolar electrode (Fine Surgical Instruments Inc. Hempstead, New York) placed at the layer VI/white matter junction on column with the recording electrode. Stimulus intensity was adjusted to be threshold for eliciting a short latency field potential response. Recordings were obtained from 4–5 slices/rat in a total of 49 rats. Stimulating and recording electrode pairs were moved together across the slice to up to 10 sites to be certain that “hot spots” for evoking epileptiform responses were not missed in slices when epileptiform responses were not initially evoked (Graber and Prince, 2004). Stimuli in epileptogenic slices elicited interictal epileptiform events that are easily distinguished from other responses on the basis of their variable and often long latency and duration, their occurrence at a threshold as all-or-none responses, and their polyphasic contours with peaks often associated with bursts of extracellular action potentials (Prince and Tseng, 1993; Hoffman et al, 2004). Similar events occurred spontaneously in some slices. A blinded observer viewed the traces offline and verified the classification of slices as epileptogenic vs. non-epileptogenic. Evoked field potentials were further assessed using a coastline burst index (Korn et al, 1987), a rough measure of response intensity.
Whole cell patch clamp recordings
Slices (300 μm) were obtained, incubated as above and transferred one at a time to a recording chamber on the stage of a compound microscope where they were minimally submerged and perfused at a rate of 3 ml/min with the above ACSF containing 2.5 mM KCl. Patch electrodes pulled from borosilicate glass tubing (1.5 mm OD) had impedances of 3–5 MΩ when filled with (in mM): 120 K-gluconate, 10 KCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 EGTA, 0.5% biocytin and pH 7.3 adjusted with KOH, 285–295 mosM.
Whole cell current- and voltage-clamp recordings of spontaneous (s)EPSCs and miniature (m)EPSCs were obtained from layer V Pyr cells that were identified with infrared video microscopy and differential interference contrast optics (Zeiss Axioskop) as neurons with large somata, a single emerging dendrite oriented toward the pial surface, and an adapting firing pattern to depolarizing current pulses. DNQX (6,7-dinitroquinoxaline-2,3-dione; Ascent Scientific) 10 μM) was used in these experiments to identify spontaneous events as EPSCs. In experiments where mEPSCs were analyzed, ACSF containing 1 μM tetrodotoxin (TTX, Ascent Scientific) was used.
Recordings were made with a Multiclamp 700A amplifier, sampled at 10 kHz, filtered at 2 kHz with a Digidata 1320A digitizer, and analyzed using Clampfit (Axon Instruments Inc. Foster City, CA), Mini Analysis (Synaptosoft, Decatur, GA), Origin 6.1 (OriginLab, Northampton, MA), Excel (Microsoft) and Prism (GraphPad software). Only recordings with a stable access resistance < 25 MΩ that varied <15% during the recording were accepted for analysis. All experiments were carried out at room temperature (~22°C).
Immunohistochemistry
Animals were deeply anesthetized with Beuthanasia-D (Schering-Plough Animal Heath Corp, Union ) (0.28mg/kg) and perfused intracardially with heparinized saline and 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were removed, post-fixed overnight at 4° C and cryoprotected in 30% sucrose in PBS at 4° C. Sections (25–40 μm) were prepared on a Heidelberg Microm (Micron International GmbH, Walldorf, Germany). Sections from GBP- and saline-treated UC rats were processed together and primary antibodies were omitted in some sections as an additional control. Sections were processed with 10% normal goat serum (Jackson Immunoresearch Labs Inc. West Grove, PA) and 0.2% Triton X-100 for 60 min and exposed overnight (4° C) to the primary antibodies against: neurofilament heavy chain 200 kD (monoclonal; 1: 1000, Epitomics Inc. CA), anti-postsynaptic density 95 KDa protein (PSD95) (monoclonal;1: 200, Sigma); anti-vesicular glutamate transporter 1 (VGLUT1; polyclonal; 1:1000; kindly provided by Dr. R. Reimer, Stanford University School of Medicine); mouse anti-vesicular glutamate transporter 1 (VGLUT1; 1:500, Millipore, CA); pan neuronal marker NeuN (1:100, Millipore, CA); and glial fibrillary acidic protein (GFAP; 1:1000, Epitomics Inc. CA; Millipore, CA ). After several washes, sections were incubated for 2h at room temperature with fluorescein-labeled secondary antibodies: Alexa Fluors 594 goat anti-mouse IgG and Alexa Fluors 488 goat anti-rabbit IgG (Millipore, CA), goat anti- mouse Dylight 649, and goat anti-rabbit Dylight 594, (Jackson Immunoresearch Labs Inc. West Grove, PA). Double or triple immunofluorescence was assessed with a laser confocal microscope (Talamasca 2P). Indirect assessment of putative excitatory synapses in layer V was obtained by a blinded observer who measured the density of sites of close apposition between PSD-95 and VGLUT1 puncta (colocalized VGLUT1/PSD-95 puncta; Fig. 5) from non-adjacent sections in UC and naive littermates.
Figure 5. Chronic GBP treatment decreases posttraumatic synaptogenesis.
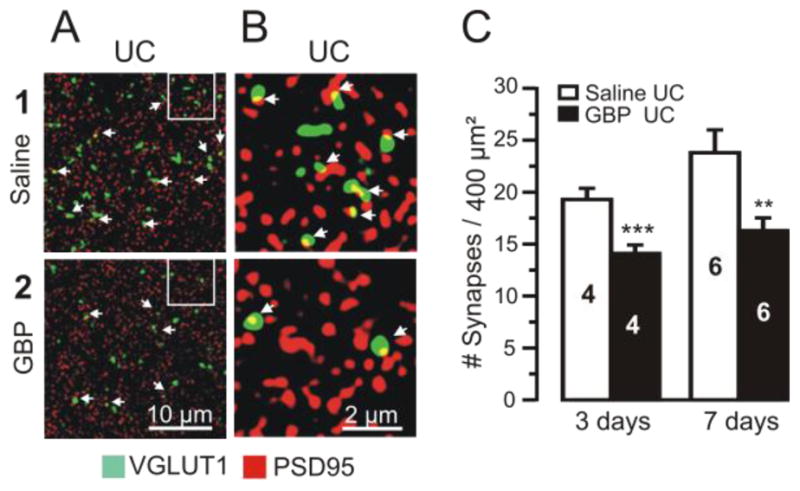
Confocal images of dual VGLUT1-IR (green) and PSD95-IR (red) from layer V of saline-treated (upper images) and GBP-treated rats (lower images).
A1–2: Images of UC sections from rats treated with saline (A1) and GBP (100 mg/kg/3x/d for 7d), (A2). Arrows in A–B point to sites of close apposition of VGLUT1- and PSD95-IR (yellow; presumed synapses).
B1–2: Images from marked segments of A1, A2 at 5x higher magnification. Calibrations in A2 for A1–2 and in B2 for B1–2.
C: Blinded counts of profiles of VGLUT1/ PSD95 colocalizations in UC cortical sections from UC rats treated with saline x3d (left graph, white bar) or GBP (100 mg/kg/3x/d x3d) (black bar). Right graph: 7d GBP treatment at same dose. Numbers in bars: number of animals. 2–3 sections analyzed/animal and 3 images from each section. Both GBP treatment durations resulted in a significant decrease in synaptic density compared to UC saline controls. ***: p < 0.001; **: p < 0.01). Data are expressed as mean ± SEM.
Western Blots
Fresh full thickness cortical brain tissue was dissected from naïve, UC and contralateral homotopic cortical areas, and sonicated in homogenization buffer: 12.5% 0.5 M Tris HCl, 10% glycerol, 2% SDS and protease inhibitor cocktail (Thermo Scientific, Rockford, IL). Homogenates were centrifuged at 14,000g for 15 min at 4° C, and supernatant collected. Samples of protein (15–20 μg) from saline and GBP-treated brain tissue were separated by 4–15% Tris-HCl running gel and transferred to Amersham Hybond-P transfer membranes (GE Healthcare). Membranes were blocked with 5% nonfat milk in TBST buffer for 1 h at room temperature, and incubated overnight at 4° C with primary antibodies (1:25,000 for rabbit anti-GFAP, Epitomics, Inc., Burlingame, CA; 1:1000 for mouse monoclonal anti-human thrombospondin-1 (TSP1), R&D Systems and 1:10,000 for mouse monoclonal anti-β-actin, Sigma). After several washes, the membranes were incubated with secondary antibodies at dilutions of 1:10,000–20,000 for 1.5 hrs, room temperature. After further extensive washing, the immunoreactive bands were detected with ECL plus Western blot detection system reagents (GE healthcare). Quantification of optical absorbency of Western blots was performed by using ImageJ software (NIH). Relative expression of specific protein was normalized and calculated as the OD of specific protein/OD of β-actin.
Fluoro-Jade C (FJC) histochemistry
Sections from isolated neocortical regions from 6 UC GBP-treated and 6 saline treated rats were processed for FJC histochemistry according to the modified manufacture’s protocol (Millipore, CA). Slides were immersed in a 1% sodium hydroxide in 80% alcohol for 5 min, followed by 70% alcohol, 30% alcohol and distilled water, each for 2 min, transferred into a 0.06% potassium permanganate solution and agitated for 15 min. After rinsing in distilled water for 2 min, the slides were stained with 0.0001% FJC dye (Millipore, CA) dissolved in 0.1% acetic acid vehicle, for 30 min in the dark and washed in distilled water three times, each for 1 min. The slides were air dried overnight, dehydrated in ethanol (95% twice and 100% twice), cleared in xylene and coversliped with D.P.X. The FJC stained sections were examined under a fluorescence microscope (Nikon Eclipse E800, Japan). For multiple labeling studies, sections were blocked with 3% BSA (Jackson Immunoresearch Labs Inc. West Grove, PA) and 0.2% Triton X-100, exposed overnight (4° C) to antibodies (as above) against NeuN (1: 100 and GFAP (1:1000), washed three times and incubated for 2h at room temperature with fluorescein-labeled secondary antibodies. After several PBS washes, sections were mounted, dried overnight, rehydrated in distilled water for 2 min, transferred into 0.06% potassium permanganate for 5 min, rinsed in distilled water for 2 min, stained with 0.0001% FJC dye for 15 min, washed three times in distilled water (1 min each), air dried, cleared and coversliped with D.P.X. Sections were coded, and blind counts of FJC reactive cells were obtained from an average of 3 fields/section in 2–3 non-adjacent sections from UC-GBP and saline-treated UC animals using ImageJ (NIH).
Statistical analyses
Data are expressed as mean ± S.E.M unless otherwise noted. Statistical significance (p < 0.05) of differences between saline-treated and GBP-treated UC groups, were measured using two-tailed Student t-tests or one way ANOVA.
Results
In vivo GBP treatment decreases the incidence of evoked epileptiform discharges in cortical slices containing UC lesions
Animals with UC lesions were treated with GBP or saline immediately after injury, using 2 different protocols (Fig. 1A) and the incidence of evoked epileptiform discharges in UC cortical slices was assessed with field potential recordings. Experiments in Group 2 of Fig. 1 were done initially. The larger number of experiments in saline vs. GBP-treated animals allowed us to verify the high incidence of epileptiform activity found in slices from undercut cortex in previous experiments (Graber and Prince, 1999; Hoffman et al, 1994). Chronic in vivo treatment with GBP (100 mg/kg 3x/d ip or 120 mg/kg/d sc by pump x 14 days) resulted in a large (69%) reduction in the incidence of epileptogenic slices from UC cortex (Fig. 1A, Group 2; 3/17 epileptogenic slices from 4 GBP-treated rats vs. 21/43 slices from 10 saline-treated animals; Fig. 1B–C; 1D; p < 0.04). A significant reduction in epileptogenic slices was also found in animals of Group 1 that had been treated for 2 days with GBP injections followed by a slice experiment 11–13 days later (Fig. 1A,D; 11/42 epileptogenic slices from 10 GBP rats vs. 11/21 in 5 saline-treated controls, 53% reduction; p < 0.05). There was no significant difference in the percentage of hyperexcitable slices between the short (2d, group 1) and long treatment groups (14d, group 2), and results remained highly significant when data from the 2 groups were combined (not shown). Analyses of the coastline burst index (Korn et al, 1987) revealed no significant differences between the epileptiform potentials evoked in GBP and saline-treated groups (not shown). Additional experiments will be required to determine effects of varying the onset and length of treatment following injury and the duration of the apparent antiepileptogenic effect once GBP is discontinued (see Discussion). We considered the possibility that GBP might acutely decrease spontaneous epileptiform activity known to occur in UC rats in vivo (e.g. Graber and Prince, 2006) and in some way influence development of chronic cortical hyperexcitability, or cause a persistent but acute effect in Group 2. However, acute bath application of GBP (400 μM) in vitro for 2 h did not affect the incidence or electrographic features of evoked epileptiform bursts in slices from 4 saline-treated UC rats. These results indicate that even brief GBP treatment given shortly after lesion placement in vivo has a long-lasting effect to decrease hyperexcitability in slices from chronically injured neocortex
Neuroprotective effects of GBP
One explanation for the reduction of epileptogenic discharges in UC cortices by GBP would be a neuroprotective effect, as has been reported after traumatic brain and spinal cord injury (Calabresi et al., 2000; Cunningham et al., 2004; Emmez, et al., 2010; Rothstein and Kuncl, 1995; Striano and Striano, 2008). We used the density of flurojade C (FJC)-stained neurons in sections cut through the chronically UC cortex as a marker of cell damage/death (Schmued et al. 2005). Previous reports showed that neuronal death detected by FJC after injury reaches a peak at ~3 days and lasts up to 14 days (Wang et al, 2008). We therefore used a day 7 time point. Analysis of images from confocal sections from UC regions in GBP vs. saline-treated UC rats showed that GBP reduced the incidence of FJC-positive cells in layers V and VI by 41%, 7 days after injury (GBP: 19.4 ± 2.3 FJC positive neurons/350 μm2, n = 18 sections, 6 rats; saline: 32.9 ± 2.9, n = 18 sections, 6 rats, p < 0.001; Fig 2A–C). To help identify cell subtypes of FJC-positive profiles, we reacted FJC stained sections with antibodies for pan-neuronal marker NeuN and astrocyte marker GFAP. A number of FJC positive profiles co-labeled for NeuN but not for GFAP (Fig. 2D). GBP treatment also significantly reduced expression of 200 kD neurofilament immunoreactivity (IR) in somata and proximal dendrites of layer V neurons (arrows in Fig 2E; Fig. 2F–G). The alteration in neurofilament expression by GBP may be a result of a reduction in neuronal injury and/or reduced activity in the injured cortex (e.g. Fig 1 of Prince et al, 2009).
Figure 2. Neuroprotective effects of chronic GBP treatment.

A–B: Representative confocal images of Fluoro-Jade C (FJC) staining from UC cortices of rats treated for 2d with saline (A) or GBP (B). Sections obtained 7 days after cortical injury. Arrows: FJC positive profiles.
C: Density of FJC positive cells/350×350 μm2 in layer V of UC cortex is decreased by GBP treatment. Graph shows data from 6 saline- and 6 GBP-treated UC rats. Counts done from 6–9 images and 2–3 sections/rat. ***P < 0.001. Error bars: SEM.
D: FJC positive profiles were neuronal. Confocal image from section triple labeled for FJC (green), GFAP-IR (red) and NeuN (blue). FJC did not colocalize with GFAP (large arrow), however a number of profiles were reactive for NeuN and FJC (small arrows). Calibration bar in B for A,B and in F for E,F.
E–F: 200KDa neurofilament-IR sections through UC from rat treated with saline (E) and GBP (F).
G: Graph shows significant decrease in density of neurofilament IR (#cells/700×700 μm2) in GBP-treated rats. Numbers in columns: # rats. *: p<0.05
Activation of astrocytes and increased GFAP-IR is a common response to neural injury (Ide et al, 1996; Cervos-Navarro and Lafuente, 1991). GFAP-IR was substantially increased at the edges of UC lesions in comparison to contralateral or naïve cortices (Fig. 3A, saline UC vs. naive) and GBP treatment reduced GFAP-IR in the injured area (Fig. 3A, GBP UC vs. Saline UC). Western blot analysis confirmed the significant increase in GFAP protein expression in UC compared to intact cortex (Fig. 3C, Saline1, Saline2) that was partially suppressed in GBP-treated animals (Fig. 3C, GBP1, GBP2). When normalized to β-actin, GFAP protein expression in GBP-treated UC lesions decreased by 41% at 7 days after injury, compared to saline treatment (Fig. 3D, right graph; GBP 0.51± 0.09 arbitrary OD units, n = 4 rats, vs. saline 0.86 ± 0.08, n = 4, p < 0.05); and by 35% 3 days after injury (left graph: GBP 0.41± 0.03, n = 6, vs. saline 0.63 ± 0.09, n = 6; p < 0.05). The pan-neuronal marker NeuN was used to verify the cortical depth of GFAP-IR in different rats (Fig. 3B, lower panels).
Figure 3. Increased GFAP expression in UC cortex is reduced toward naïve levels by chronic GBP treatment.

A: Representative images of GFAP-IR close to the UC lesion (dashed white lines) in sections from saline-treated (Saline UC, n = 6 rats), GBP-treated (GBP UC, 100mg/kg 3x/d x 7d, n = 6) and naive control cortex (n = 2). Pial surface up and to the left. Sections processed together and imaged using the same parameters. Increased GFAP-IR along cuts in Saline-UC is partially decreased toward naïve control level in GBP UC section. Lower images: NeuN-IR in sections adjacent to those in upper GFAP row to show laminar position of GFAP-IR. Roman numerals: cortical laminae. Calibration: 200 μm for all images
B: Western immunoblots of GFAP protein expression from UC cortex in 2 saline-treated rats (Saline 1 and Saline 2) and 2 UC rats treated with GBP (GBP1 and GBP2; 100 mg/kg 3xd x 7d). Cl: contralateral homotypic cortex. GFAP expression is increased in UC vs. contralateral cortex, and decreased in blots from GBP-treated vs. saline-treated UC animals.
C: Optical density of GFAP immunoblots normalized to the expression of β actin. Graphs show a significant decrease in GFAP expression in UC cortex for 3 and 7d GBP- vs. saline-treated UC rats. GBP dose/d as in B. *P < 0.05 for both comparisons. Data are expressed as mean ± SEM. Numbers of rats in bars.
Following injury to the mature cortex, reactive astrocytes transiently re-express thrombospondins 1 and 2 (TSPs1/2) (Lin et al., 2003) that may promote synapse formation (Christopherson et al, 2005; Eroglu et al., 2009). Such effects are reduced in TSP1/2 knockout animals (Liauw et al., 2008). There were significant increases in TSP1 measured by Western blot and TSP2 immunoreactivity in injured cortex 3 days after the undercut (Fig 4A–D), associated with increased GFAP (Fig 4D). However, this increase in TSPs 1/2 was not observed at day 7, 10 or 14 after injury (data not shown). When normalized to β-actin, TSP1 protein expression in UC lesions of GBP-treated rats 3 days after injury decreased by 55% (Fig. 4B; GBP 0.67± 0.09, n = 6, vs. saline 1.48 ± 0.28, n = 6; p < 0.05). Large increases in the expression of the TSP/GBP receptor α2δ-1-IR were also present 7 days after the UC lesion (c.f. Fig. 4E and F), as reported following spinal nerve injury, (Luo et al, 2001). Taken together, these results show that alterations which would be expected to promote new synapse formation after injury, such as enhanced expression of GFAP, TSPs and α2δ-1, are present in the partially isolated neocortex early after lesion placement. The reductions in FJC stained neurons, neurofilament-IR, GFAP and TSPs that result from GBP treatment are consistent with a significant neuroprotective effect by the drug.
Figure 4. Alterations in TSP1/2 and α2δ-1 expression in UC cortex.

A: Representative immunoblots of TSP1 protein expression from UC and contralateral homotopic cortex (Cl) 3 d after lesion in GBP treated (GBP UC) and saline treated rats (Saline UC). GBP dose: 100 mg/kg 3xd x 3d beginning on day of UC. TSP1 expression is increased in UC vs. contralateral cortex (wells, 1 vs. 2 and 3 vs. 4), and decreased in blots from GBP-treated vs. saline-treated UC animals (wells 1 vs. 3).
B: Optical density (OD) of TSP1 immunoblots normalized to the expression of β-actin. Graphs show a significant increase in TSP 1 in UC vs. contralateral cortex in both saline and GBP treated animals. TSP1 expression in the UC is reduced in GBP vs. saline treated UC cortex. Data obtained 3 days after UC.*P < 0.05. Data are expressed as mean ± SEM. Numbers of rats in bars.
C–D: Dual IR for GFAP and TSP2 in area of UC (D) vs. contralateral cortex (C) 3 days after UC lesion. Dashed lines here and in F: UC lesions. Pial surface up in D and up-right in F. Calibration in C for C,D and in E for E–F.
E–F: Increased α2δ-1-IR in area of the undercut 7 days after lesion (F) compared to contralateral cortex (E).
Chronic GBP treatment after injury inhibits synapse formation
GBP decreases excitatory synapse formation during development (Eroglu et al., 2009), and we hypothesized that it might also limit the enhanced excitatory connectivity that occurs following injury (Salin et al, 1995; Carmichael and Chesselet, 2002; Staley and Dudek, 2006; Jin et al, 2006). Such an action could, in part, account for reductions in epileptogenesis after neocortical trauma (Fig 1D). Sections containing cortical UCs from rats treated chronically with GBP or saline were double-immunolabeled for presynaptic and postsynaptic markers of excitatory synapses, VGLUT1 and PSD95, respectively. Profiles in which there was close apposition of pre- and postsynaptic markers (Fig. 5A–B, arrows) were presumed to represent synaptic contacts (Stevens et al, 2007; Eroglu et al., 2009). Blind counts of close appositions of VGLUT1- and PSD95-immunoreactvity showed that GBP treatments for 3 days and 7days following injury significantly decreased the density of presumed excitatory synapses (Fig. 5C. left graph: GBP 3days, 14.1 ± 0.82 profiles/20 μm2, n = 4 vs. saline, 19.3 ± 1.07, n = 4, p<0.001; 5C, right graph: GBP 7days, 16.3 ± 1.22 profiles/20 μm2, (n=6) vs. saline, 23.8 ± 2.2, (n=6), p < 0.01). Thus, similar to results of GBP treatment during brain development and effects on barrel plasticity following deafferentation (Eroglu et al, 2009), GBP significantly reduces excitatory synapse formation following cortical injury.
Chronic GBP treatment decreases the frequency of spontaneous and miniature EPSCs on layer V pyramidal neurons in undercut cortical slices
One functional consequence of a reduction in the density of excitatory synapses in the UC cortex resulting from GBP treatment might be a decrease in the frequency of excitatory postsynaptic currents in slices from drug vs. saline-treated UC rats. We therefore obtained whole cell recordings of sEPSCs and mEPSCs from layer V Pyr neurons in brain slices from rats treated with GBP (120 mg/kg/day s.c., 10–14d via pump) or saline following the injury. Spontaneous excitatory currents were completely blocked when the perfusate contained 10 μM DNQX (not shown), indicating that they were mediated by activation of AMPA/KA receptors. Representative recordings of sEPSCs from saline- and GBP-treated rats are shown in Fig. 6A. The frequency of excitatory currents in UC slices was lower than we previously reported for layer V Pyr neurons (Li and Prince, 2002), perhaps due to differences in bath temperature (32°C in earlier experiments and room temperature here). Nonetheless, neurons from the GBP-treated animals had a ~33% lower sEPSC frequency than those of saline-treated UC controls (saline: 5.08 ± 0.36 Hz, n = 19 neurons from 8 rats vs. GBP: 3.4 ± 0.44 Hz, n = 21 neurons from 8 rats; p < 0.001; Fig. 6A and left graph in 6B). sEPSC amplitudes were similar between these two groups (saline: 17.4 ± 1.6 pA vs. GBP: 16.7 ± 0.8 pA, p = >0.6, Fig 6B, right graph). The frequency of mEPSCs recorded in 1 μM TTX was also reduced in slices from GBP-treated vs. saline-treated UC animals (saline: 2.3 ± 0.2 Hz, n = 10 cells vs. GBP: 1.7 ± 0.1 Hz, n = 11, p < 0.05) without effect on mEPSC amplitude (saline: 14.6 ± 0.5 pA vs. GBP: 14.2 ± 0.5 pA, p = >0.6). No significant differences were found in sEPSC rise time, decay time and half width for GBP versus saline treatment (10–90% rise time: saline 2.5 ± 0.2 ms vs. GBP 2.6 ±0.1 ms; p=>0.6; decay time: saline 3.1 ± 0.2 ms vs. GBP 3.2±0.2 ms, p=>0.8; half width: saline 2.5 ± 0.3 ms vs. GBP 2.5 ± 0.2 ms, p=>0.9.). These functional results are consistent with the decreased density of excitatory synapses estimated from the dual VGLUT1/PSD95 close appositions in the above immunocytochemical experiments.
Figure 6. Chronic GBP treatment decreased the frequency of excitatory postsynaptic currents in layer V pyramidal neurons of undercut cortex.
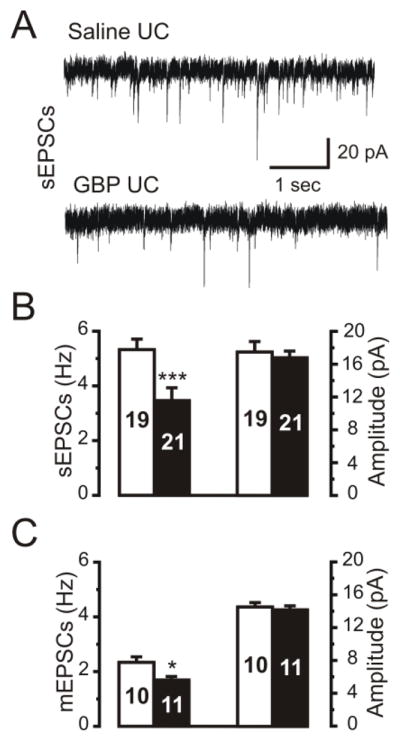
A: Representative recordings of spontaneous (s)EPSCs in layer V pyramidal (Pyr) neurons in slices from saline- (upper trace) and GBP-treated (100 mg/kg/3x/d ip x 14d.; lower trace) UC cortex. Holding potential: −65mV. Estimated ECl: −55 mV.
B: Group data for frequency and amplitude of spontaneous EPSCs from layer V Pyr neurons in slices from saline- (white bars) and GBP-treated (100 mg/kg/3x/d ip x 14d) UC cortex (black bars). GBP treatment decreased the frequency of sEPSCs, but not amplitude. Numbers in bars: number of cells here and in C. ***: P<0.001.
C: Frequency but not amplitude of mEPSCs was reduced in GBP-treated (black bars) but not saline-treated rats (white bars). *: P<0.05.
Discussion
Following injury to the mature cortex, reactive astrocytes re-express TSPs 1/2 that may promote axonal sprouting and synapse formation (Lin et al., 2003; Liauw et al., 2008). The extent to which such effects are adaptive or maladaptive is unknown, however, they are dependent on TSP actions as they are reduced in TSP1/2 knockout animals (Liauw et al., 2008). In addition, the α2δ-1 receptor is markedly up-regulated in UC cortex (Fig. 4) as it is in spinal nerve injury (Luo et al., 2001; Luo et al., 2002; reviewed in Maneuf et al., 2006 and Field et al., 2006), thus, providing increased targets for TSP actions. These and earlier results (Eroglu et al 2009) lead to the hypothesis that GBP might be an effective antiepileptogenic agent by blocking the interaction of TSPs with their α2δ-1 receptor following injury, reducing new aberrant synapse formation and perhaps indirectly decreasing axonal sprouting. These experiments were designed to test the potential effects of GBP in a model of injury-induced epileptogenesis.
We have previously shown that ~ 58–75 % of sensorimotor cortical slices, from areas of chronic partial cortical isolations/UCs placed 10–14 days earlier, generate spontaneous and evoked epileptiform responses consisting of variable latency and duration, all-or-none, prolonged polyphasic field potentials that originate in layer V and spread to other lamina and across the slice (Hoffman et al. 1994; Prince and Tseng 1993; Salin et al, 1995; Graber and Prince, 1999; reviewed in Graber and Prince, 2006). In these previous experiments and the current study, at least 1 slice from each “undercut” animal generated epileptiform events. In whole cell patch clamp recordings from UC slices, such epileptiform potentials are associated with an increase in frequency of spontaneous and evoked EPSCs (Li and Prince, 2002), and more widespread excitatory network connections onto layer V pyramidal neurons (Jin et al., 2006). We found a similar incidence of hyperexcitability in field potential recordings in in vitro slices from saline-treated UC rats in this study, in spite of differences in experimental conditions (e.g. temperature, [K+]o).
GBP treatment reduces posttraumatic hyperexcitability
The principal findings of these experiments are that high dose, brief GBP treatments, given for 2d, beginning immediately after brain injury, effectively reduce the incidence of epileptiform discharges assessed 14 days later in brain slices from the UC cortex (Group1 of Fig. 1). A similar reduction in the incidence of evoked bursts was present the day following 2 weeks of GBP treatment (Group 2 of Fig. 1). GBP has a short half life of ~1.7h after iv injection in rat (Radulovic et al, 1995) making it highly unlikely to influence the incidence of epileptogenic activity via an acute antiepileptic action in either protocol group, as the in vitro slice experiments were delayed by 4–8 hours or 13–14 days after the drug was discontinued in Groups 2 and 1, respectively (Fig. 1A and Methods). We have not thoroughly explored the effects of more prolonged treatments, or longer intervals between drug administration and measurements of electrophysiological or anatomical variables. There may be a critical period for GBP administration, as was the case in developmental experiments, so that newly formed synapses may be unaffected if drug treatment is delayed for a number of days (Eroglu et al, 2009). Such experiments will be important, as the signals that initiate sprouting and new synapse formation may persist in the injured cortex after GBP is discontinued, and other pathophysiological alterations with a different temporal onset and duration, e.g. decreases in GABAergic inhibition or alterations in neurotransmitter transporters, will presumably not be affected by GBP and may induce hyperexcitability even when new excitatory synapse formation is limited by the drug. These will be significant considerations in interpreting effects of GBP or other drugs on epileptogenesis in in vivo preclinical trials. In this context, it is important to emphasize that prophylactic actions of GBP on injury-induced epileptiform activity in vitro are not necessarily predictive of favorable effects on epileptogenesis and development of seizures in vivo. Although electrographic ictal episodes and associated behavioral alterations have been shown in vivo in implanted rodents and cats with partial cortical isolations (Graber and Prince, 2006; Nita et al, 2007), the incidence and frequency of seizures is not known.
Neuroprotective effects of GBP
The present results suggest that both neuroprotective effects and a decrease in injury-induced synapse formation by GBP may have a role in the antiepileptogenic effect. Neuronal injury and astrogliosis in the UC region (Figs. 2 and 3) may promote epileptogenesis by initiating reorganization of cortical circuits and formation of new excitatory connections (McKinney et al, 1997; Carmichael and Chesselet, 2002; Salin et al, 2005; Jin et al, 2006), and through alterations in glial function (Stewart et al, 2010; Ortinski et al, 2010). Our results show that GBP, administered for 2–3d beginning on the day of the UC lesion, decreased the density of FJC-positive profiles (Fig. 2A–C) that are known to localize with injured neurons (Eisch et al., 1998; Schmued et al., 1997, 2005; Wang et al., 2008; Fig 2C). This attenuation of neuronal markers of injury was accompanied by immunocytochemical and Western blot results showing that GBP also reduced gliosis (Fig. 3). Previously described enhanced neurofilament-IR in UC cortex (Prince et al, 2009), a marker for axonal injury and sprouting (Yaghmai and Povlishock, 1992; Christman et al, 1997; Chuckowree and Vickers, 2003), was also decreased in layer V Pyr neurons by GBP (Fig. 2E–G). Similar neuroprotective effects of GBP have been reported in other model systems (Baydas et al., 2005; Calabresi et al., 2000; Cunningham et al., 2004; Comi et al., 2008; Kim et al., 2009). For example, GBP given immediately following carotid ligation in mice reduced acute reactive seizures and brain atrophy (Comi et al. 2008), and also decreased markers of glial and neuronal abnormalities following hyperglycemic brain injury (Baydas et al. 2005). Although mechanisms for these effects are uncertain, reduction of glial-activated inflammatory mediators (Mototh et al, 2000) and reduced oxidative stress have been suggested (Baydas et al., 2005).
GBP has multiple other actions including effects on ion channels (Liu et al., 2006, Vega-Hernandez and Felex, 2002), as well as on ligand gated NMDA and GABAB receptors (Bertrand et al., 2003, Kim et al., 2009, Ng et al., 2001). GBP decreases trafficking of voltage gated calcium channels to the membrane and their density (Hendrich et al., 2008; Bauer et al., 2009; Davies et al., 2007) and reduces Ca++ currents in neurons, perhaps through actions involving presynaptic N- or P/Q-type Ca++ channels (van Hooft et al., 2002; Vega-Hernandez and Felix, 2002; Fink et al., 2000; Bertrand et al., 2003; Bayer et al., 2004; Li et al., 2006; Kato and Bredt, 2007), resulting in reductions in release of glutamate and other transmitters (Cunningham et al., 2004; (Bayer et al., 2004). Such acute effects may be related to anticonvulsant or to the neuroprotective effects of GBP, however, we cannot rule out the possibility that they also alter the processes important in the early stages of epileptogenesis. As mentioned above, acute residual antiepileptic effects of GBP could not have contributed to results with the dosing protocols used. Further, prolonged bath applications of high concentrations of GBP (200–400 μM x 2h) did not block the evoked epileptiform discharges in UC cortex (not shown). In contrast, in vivo administration of GBP for a few days significantly decreased the frequencies, but not amplitudes of sEPSCs and mEPSCs recorded from layer V pyramidal neurons (Fig. 6).
Effects of GBP on excitatory synapse formation
Previous anatomical (Salin et al, 1995) and functional results (Li et al, 2002; Jin et al, 2006) are compatible with increases in excitatory connectivity in the UC cortex. Because GBP treatment has been shown to limit excitatory synapse formation during development (Eroglu et al, 2009), we hypothesized that it might also limit injury-induced synaptogenesis in the UC cortex. Results showed a significant decrease in the density of close appositions of pre- and postsynaptic markers for excitatory synapses in the injured cortex from animals treated with GBP vs., saline, an effect similar to that reported in naïve neocortex of mice treated with GBP in the first week of life (Eroglu et al, 2009). In addition, our data show that the GBP suppression of synapse formation in the UC can be detected at the earliest time point examined, 3 days after the lesion (Fig. 5C). The electrophysiological data showing decreases in mEPSC and sEPSC frequency in the GBP treated animals are compatible with the above anatomical changes. Thus, at least one mechanism for the anti-epileptogenic effect of GBP may be attributed to the prevention of new synapse formation. Because the drug was administered beginning on the day of injury, it is unclear whether a similar effect would occur with delayed treatment (e.g. Eroglu et. al. 2009).
Acute effects of GBP mentioned above do not appear to contribute to suppression of evoked epileptiform bursts in slices because of the short half life of GBP in rats (Vollmer et al, 1986) and the effects seen long after the treatment in some experiments (e.g. Group 1 of Fig. 1A,D). However, such effects may be related to the neuroprotective actions of GBP found in models of spinal cord, retinal and brain ischemia (Lagreze et al., 2001; Traa et al., 2008; Kale et al., 2011) and in our experiments when there is a short interval between injury and treatment (Figs 2,3).
GPB is a widely available clinical pharmaceutical agent used for treatment of pain, epilepsy and anxiety (Rothstein and Kuncl, 1995). Current results raise the possibility of potential effects as an antiepileptogenic agent following TBI. Obviously a number of important issues remain to be addressed in additional experiments before consideration of such use, including drug dosage, the optimal timing and duration of treatment, efficacy in in vivo preclinical trials, and potential adverse effects on adaptive new connectivity that may occur hand in hand with maladaptive effects of sprouting and new synapse formation (Dancause et al, 2005; Lee et al, 2004).
Highlights.
Gabapentin (GBP) decreases injury-induced astrocytosis and is neuroprotective.
GBP given following cortical injury inhibits synapse formation and decreases excitatory synaptic activity.
In vivo GBP treatment reduces evoked epileptiform discharges in slices from injured cortex.
GBP is a potential antiepileptogenic agent
Acknowledgments
The work was supported by a grant from the Citizens United for Research in Epilepsy (CURE) and NIH grant NS12151 from the NINDS. We thank Dr. Richard Reimer for providing vGluR1 antibody, and Isabel Parada for providing images of Figure 4C.D.E.F. Cagla Eroglu provided valuable advice during the course of these experiments.
Footnotes
Statement of interest
All authors assert that there is no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med. 1998;338:20–24. doi: 10.1056/NEJM199801013380104. [DOI] [PubMed] [Google Scholar]
- Babb TL, Kupfer WR, Pretorius JK, Crandall PH, Levesque MF. Synaptic reorganization by mossy fibers in human epileptic fascia dentata. Neuroscience. 1991;42:351–363. doi: 10.1016/0306-4522(91)90380-7. [DOI] [PubMed] [Google Scholar]
- Babb TL, Pretorius JK, Kupfer WR, Mathern GW, Crandall PH, Levesque MF. Aberrant synaptic reorganization in human epileptic hippocampus: evidence for feedforward excitation. Dendron. 1992;1:7–25. [Google Scholar]
- Bauer CS, Nieto-Rostro M, Rahman W, Tran-Van-Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez-Alacid L, Millar NS, Dickenson AH, Lujan R, Dolphin AC. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J Neurosci. 2009;29(13):4076–88. doi: 10.1523/JNEUROSCI.0356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baydas G, Sonkaya E, Tuzcu M, Yasar A, Donder E. Novel role for gabapentin in neuroprotection of central nervous system in streptozotocine-induced diabetic rats. Acta Pharmacologica Sinica. 2005;26 (4):417–422. doi: 10.1111/j.1745-7254.2005.00072.x. [DOI] [PubMed] [Google Scholar]
- Bayer K, Ahmadi S, Zeilhofer HU. Gabapentin may inhibit synaptic transmission in the mouse spinal cord dorsal horn through a preferential block of P/Q-type Ca2+ channels. Neuropharmacology. 2004;46(5):743–9. doi: 10.1016/j.neuropharm.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Nouel D, Morin F, Nagy F, Lacaille JC. Gabapentin actions on Kir3 currents and N-type Ca2+ channels via GABAB receptors in hippocampal pyramidal cells. Synapse. 2003;50(2):95–109. doi: 10.1002/syn.10247. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Picconi B, Saulle E, Centonze D, Hainsworth AH, Bernardi G. Is pharmacological neuroprotection dependent on reduced glutamate release? Stroke. 2000;31(3):766–72. doi: 10.1161/01.str.31.3.766. [DOI] [PubMed] [Google Scholar]
- Carmichael ST, Chesselet MF. Synchronous neuronal activity is a signal for axonal sprouting after cortical lesions in the adult. J Neurosci. 2002;22:6062–6070. doi: 10.1523/JNEUROSCI.22-14-06062.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervos-Navarro J, Lafuente JV. Traumatic brain injuries: structural changes. J Neurol Sci. 1991;103(Suppl):S3–14. doi: 10.1016/0022-510x(91)90002-o. [DOI] [PubMed] [Google Scholar]
- Christman CW, Salvant JB, Jr, Walker SA, Povlishock JT. Characterization of a prolonged regenerative attempt by diffusely injured axons following traumatic brain injury in adult cat: a light and electron microscopic immunocytochemical study. Acta Neuropathol (Berl) 1997;94:329–337. doi: 10.1007/s004010050715. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chuckowree JA, Vickers JC. Cytoskeletal and morphological alterations underlying axonal sprouting after localized transection of cortical neuron Axons in vitro. J Neurosci. 2003;23:3715–3725. doi: 10.1523/JNEUROSCI.23-09-03715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi AM, Traa BS, Mulholland JD, Kadam SD, Johnston MV. Pediatr Res. 2008;64(1):81–5. doi: 10.1203/PDR.0b013e318174e70e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20(6):1566–76. doi: 10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, Stowe AM, Nudo RJ. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies A, Hendrich J, Van Minh AT, Wratten J, Douglas L, Dolphin AC. Functional biology of the alpha(2)delta subunits of voltage-gated calcium channels. Trends Pharmacol Sci. 2007;28(5):220–8. doi: 10.1016/j.tips.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Echlin FA. The supersensitivity of chronically “isolated” cerebral cortex as a mechanisms of focol epilepsy. Electroenceph Clin Neurophysiology. 1959;11:697–722. doi: 10.1016/0013-4694(59)90110-5. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Schmued LC, Marshall JF. Characterizing Cortical Neuron Injury With Fluoro-Jade Labeling After a Neurotoxic Regimen of Methamphetamine. Synapse. 1998;30:329–333. doi: 10.1002/(SICI)1098-2396(199811)30:3<329::AID-SYN10>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Emmez H, Börcek AÖ, Kaymaz M, Kaymaz F, Durdağ E, Civi S, Gulbahar O, Aykol S, Paşaoğlu A. Neuroprotective effects of gabapentin in experimental spinal cord injury. World Neurosurg. 2010;73(6):729–34. doi: 10.1016/j.wneu.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O’Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–92. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. J Cell Commun Signal. 2009;3:167–176. doi: 10.1007/s12079-009-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria LC, Prince DA. Presynaptic inhibitory terminals are functionally abnormal in a rat model of posttraumatic epilepsy. J Neurophysiol. 2010;104(1):280–90. doi: 10.1152/jn.00351.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MJ, Cox PJ, Stott E, Melrose H, Offord J, Su TZ, Bramwell S, Corradini L, England S, Winks J, Kinloch RA, Hendrich J, Dolphin AC, Webb T, Williams D. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci U S A. 2006;103:17537–17542. doi: 10.1073/pnas.0409066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K, Meder W, Dooley DJ, Gothert M. Inhibition of neuronal Ca(2+) influx by gabapentin and subsequent reduction of neurotransmitter release from rat neocortical slices. Br J Pharmacol. 2000;130:900–906. doi: 10.1038/sj.bjp.0703380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garga N, Lowenstein DH. Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 2006;6:1–5. doi: 10.1111/j.1535-7511.2005.00083.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- Glötzner FL, Haubitz I, Miltner F, Kapp G, Pflughaupt KW. Seizure prevention using carbamazepine following severe brain injuries. Neurochirurgia. 1983;26:66–79. doi: 10.1055/s-2008-1053615. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. Tetrodotoxin prevents posttraumatic epileptogenesis in rats. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. A critical period for prevention of posttraumatic neocortical hyperexcitability in rats. Ann Neurol. 2004;55:860–70. doi: 10.1002/ana.20124. [DOI] [PubMed] [Google Scholar]
- Graber KD, Prince DA. Chronic partial cortical isolation. In: Pitkanen A, Schwartzkroin P, Moshe S, editors. Models of Seizures and Epilepsy. Elsevier; San Diego: 2006. pp. 477–493. [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. Pharmacological disruption of calcium channel trafficking by the alpha2delta ligand gabapentin. Proc Natl Acad Sci U S A. 2008;105(9):3628–33. doi: 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman SN, Salin PA, Prince DA. Chronic neocortical epileptogenesis in vitro. J Neurophysiol. 1994;71:1762–1773. doi: 10.1152/jn.1994.71.5.1762. [DOI] [PubMed] [Google Scholar]
- Ide CF, Scripter JL, Coltman BW, Dotson RS, Snyder DC, Jelaso A. Cellular and molecular correlates to plasticity during recovery from injury in the developing mammalian brain. Prog Brain Res. 1996;108:365–377. doi: 10.1016/s0079-6123(08)62552-2. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Levesque MF, Babb TL, Engel J., Jr Single mossy fiber axonal systems of human dentate granule cells studied in hippocampal slices from patients with temporal lobe epilepsy. J Neurosci. 1993;13:1511–1522. doi: 10.1523/JNEUROSCI.13-04-01511.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Prince DA, Huguenard JR. Enhanced excitatory synaptic connectivity in layer v pyramidal neurons of chronically injured epileptogenic neocortex in rats. J Neurosci. 2006;26:4891–4900. doi: 10.1523/JNEUROSCI.4361-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Huguenard JR, Prince DA. Reorganization of inhibitory synaptic circuits in rodent chronically injured epileptogenic neocortex. Cereb Cortex. 2011;21(5):1094–104. doi: 10.1093/cercor/bhq181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale A, Börcek AÖ, Emmez H, Yildirim Z, Durdağ E, Lortlar N, Kurt G, Doğulu F, Kılıç N. Neuroprotective effects of gabapentin on spinal cord ischemia-reperfusion injury in rabbits. J Neurosurg Spine. 2011;15(3):228–37. doi: 10.3171/2011.4.SPINE10583. [DOI] [PubMed] [Google Scholar]
- Kato AS, Bredt DS. Pharmacological regulation of ion channels by auxiliary subunits. Curr Opin Drug Discov Devel. 2007;10(5):565–72. [PubMed] [Google Scholar]
- Kim YS, Chang HK, Lee JW, Sung YH, Kim SE, Shin MS, Yi JW, Park JH, Kim H, Kim CJ. Protective Effect of Gabapentin on N-Methyl-D-aspartate–Induced Excitotoxicity in Rat Hippocampal CA1 Neurons. J Pharmacol Sci. 2009;109:144–147. doi: 10.1254/jphs.08067sc. [DOI] [PubMed] [Google Scholar]
- Korn H, Burnod Y, Faber DS. Spontaneous quantal currents in a central neuron match predictions from binomial analysis of evoked responses. Proc Natl Acad Sci U S A. 1987;84(16):5981–5. doi: 10.1073/pnas.84.16.5981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9. doi: 10.1056/NEJM200002033420503. [DOI] [PubMed] [Google Scholar]
- Lagrèze WA, Müller-Velten R, Feuerstein TJ. The neuroprotective properties of gabapentin-lactam. Graefes Arch Clin Exp Ophthalmol. 2001;239(11):845–9. doi: 10.1007/s00417-001-0383-5. [DOI] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28(10):1722–32. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Lee JK, Kim JE, Sivula M, Strittmatter SM. Nogo receptor antagonism promotes stroke recovery by enhancing axonal plasticity. J Neurosci. 2004;24:6209–6217. doi: 10.1523/JNEUROSCI.1643-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A, Gross J, Gold MS, Dickenson AH, Feng G, Luo ZD. Calcium channel alpha2delta1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 2006;125(1–2):20–34. doi: 10.1016/j.pain.2006.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Prince DA. Synaptic activity in chronically injured, epileptogenic sensory-motor neocortex. J Neurophysiol. 2002;88:2–12. doi: 10.1152/jn.00507.2001. [DOI] [PubMed] [Google Scholar]
- Li H, Graber KD, Prince DA. Gabapentin Prevents Posttraumatic Epileptogenesis. Annual Epilepsy Society Abstract. 2009:3.054. [Google Scholar]
- Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34(1):177–86. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- Liu Y, Qin N, Reitz T, Wang Y, Flores CM. Inhibition of the rat brain sodium channel Nav1.2 after prolonged exposure to gabapentin. Epilepsy Research. 2006;70:263–268. doi: 10.1016/j.eplepsyres.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel alpha 2 delta-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Luo ZD, Lee K. Alpha2delta and the mechanism of action of gabapentin in the treatment of pain. Semin Cell Dev Biol. 2006;17(5):565–70. doi: 10.1016/j.semcdb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Marco P, DeFelipe J. Altered synaptic circuitry in the human temporal neocortex removed from epileptic patients. Exp Brain Res. 1997;114:1–10. doi: 10.1007/pl00005608. [DOI] [PubMed] [Google Scholar]
- Masukawa LM, Uruno K, Sperling M, O’Connor MJ, Burdette LJ. The functional relationship between antidromically evoked field responses of the dentate gyrus and mossy fiber reorganization in temporal lobe epileptic patients. Brain Res. 1992;579:119–127. doi: 10.1016/0006-8993(92)90750-4. [DOI] [PubMed] [Google Scholar]
- Matoth I, Pinto F, Sicsic C, Brenner T. Inhibitory effect of carbamazepine on inflammatory mediators produced by stimulated glial cells. Neurosci Res. 2000;38(2):209–12. doi: 10.1016/s0168-0102(00)00127-9. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- Ng GY, Bertrand S, Sullivan R, Ethier N, Wang J, Yergey J, Belley M, Trimble L, Bateman K, Alder L, Smith A, McKernan R, Metters K, O’Neill GP, Lacaille JC, Hebert TE. Gamma-aminobutyric acid type B receptors with specific heterodimer composition and postsynaptic actions in hippocampal neurons are targets of anticonvulsant gabapentin action. Mol Pharmacol. 2001;59:144–152. [PubMed] [Google Scholar]
- Nita DA, Cisse Y, Timofeev I, Steriade M. Waking-sleep modulation of paroxysmal activities induced by partial cortical deafferentation. Cereb Cortex. 2007;17:272–283. doi: 10.1093/cercor/bhj145. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince DA, Tseng GF. Epileptogenesis in chronically injured cortex: in vitro studies. J Neurophysiol. 1993;69:1276–1291. doi: 10.1152/jn.1993.69.4.1276. [DOI] [PubMed] [Google Scholar]
- Prince DA, Parada I, Scalise K, Graber K, Jin X, Shen F. Epilepsy following cortical injury: cellular and molecular mechanisms as targets for potential prophylaxis. Epilepsia. 2009;(Supp 2):30–40. doi: 10.1111/j.1528-1167.2008.02008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic LL, Türck D, von Hodenberg A, Vollmer KO, McNally WP, DeHart PD, Hanson BJ, Bockbrader HN, Chang T. Disposition of gabapentin (neurontin) in mice, rats, dogs, and monkeys. Drug Metab Dispos. 1995;23(4):441–8. [PubMed] [Google Scholar]
- Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985;35:1406–1414. doi: 10.1212/wnl.35.10.1406. [DOI] [PubMed] [Google Scholar]
- Salin P, Tseng GF, Hoffman S, Parada I, Prince DA. Axonal sprouting in layer V pyramidal neurons of chronically injured cerebral cortex. J Neurosci. 1995;15:8234–8245. doi: 10.1523/JNEUROSCI.15-12-08234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless SK, Halpern LM. The electrical excitability of chronically isolated cortex studied by means of permanently implanted electrodes. Electroencephalogr Clin Neurophysiol. 1962;14:244–55. doi: 10.1016/0013-4694(62)90034-2. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker W., Jr Fluoro-Jade: A novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res. 1997;751:37–46. doi: 10.1016/s0006-8993(96)01387-x. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Stowers CC, Scallet AC, Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Dudek FE. Interictal spikes and epileptogenesis. Epilepsy Curr. 2006;6(6):199–202. doi: 10.1111/j.1535-7511.2006.00145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, D’Ambrosio R. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. 2010;104:3345–3360. doi: 10.1152/jn.00398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Striano P, Striano S. Gabapentin: A Ca2+ channel alpha2-delta ligand far beyond epilepsy therapy. Drugs Today. 2008;44(5):353–168. doi: 10.1358/dot.2008.44.5.1186403. [DOI] [PubMed] [Google Scholar]
- Sur M, Leamey CA. Development and plasticity of cortical areas and networks. Nat Rev Neurosci. 2001;2:251–262. doi: 10.1038/35067562. [DOI] [PubMed] [Google Scholar]
- Sutor B, Luhmann HJ. Development of excitatory and inhibitory postsynaptic potentials in the rat neocortex. Perspect Dev Neurobiol. 1995;2:409–419. [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–1022. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: meta-analysis of controlled trials. Epilepsia. 2001;42:515–524. doi: 10.1046/j.1528-1157.2001.28900.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10–3. doi: 10.1111/j.1528-1167.2008.02005.x. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Anderson GD, Winn HR, Ellenbogen RG, Britz GW, Schuster J, Lucas T, Newell DW, Mansfield PN, Machamer JE, Barber J, Dikmen SS. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29–38. doi: 10.1016/S1474-4422(06)70630-5. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Anderson GD, Wilensky AJ, Holmes MD, Cohen W, Newell DW, Nelson P, Awan A, Winn HR. Valproate therapy for prevention of posttraumatic seizures: a randomized trial. J Neurosurg. 1999;91:593–600. doi: 10.3171/jns.1999.91.4.0593. [DOI] [PubMed] [Google Scholar]
- Temkin NR, Dikmen SS, Wilensky AJ, Keihm J, Chabal S, Winn HR. A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N Engl J Med. 1990;323:497–502. doi: 10.1056/NEJM199008233230801. [DOI] [PubMed] [Google Scholar]
- Traa BS, Mulholland JD, Kadam SD, Johnston MV, Comi AM. Gabapentin neuroprotection and seizure suppression in immature mouse brain ischemia. Pediatr Res. 2008;64(1):81–5. doi: 10.1203/PDR.0b013e318174e70e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng GF, Prince DA. Heterogeneity of rat corticospinal neurons. J Comp Neurol. 1993;335(1):92–108. doi: 10.1002/cne.903350107. [DOI] [PubMed] [Google Scholar]
- van Hooft JA, Dougherty JJ, Endeman D, Nichols RA, Wadman WJ. Gabapentin inhibits presynaptic Ca(2+) influx and synaptic transmission in rat hippocampus and neocortex. Eur J Pharmacol. 2002;449(3):221–28. doi: 10.1016/s0014-2999(02)02044-7. [DOI] [PubMed] [Google Scholar]
- Vega HA, Felix R. Down-Regulation of N-Type Voltage-Activated Ca2+ Channels by Gabapentin. Cellular and Molecular Neurobiology. 2002;22 (2):185–190. doi: 10.1023/a:1019865822069. [DOI] [PubMed] [Google Scholar]
- Vollmer KO, von Hodenberg A, Kölle EU. Pharmacokinetics and metabolism of gabapentin in rat, dog and man. Arzneimittelforschung. 1986;36(5):830–9. [PubMed] [Google Scholar]
- Wang L, Liua Y-H, Huang Y-G, Chen L-W. Time-course of neuronal death in the mouse pilocarpine model of chronic epilepsy using Fluoro-Jade C staining. Brain Res. 2008;1241:157–167. doi: 10.1016/j.brainres.2008.07.097. [DOI] [PubMed] [Google Scholar]
- Watson NF, Barber JK, Doherty MJ, Miller JW, Temkin NR. Does glucocorticoid administration prevent late seizures after head injury? Epilepsia. 2004;45(6):690–4. doi: 10.1111/j.0013-9580.2004.59403.x. [DOI] [PubMed] [Google Scholar]
- Yaghmai A, Povlishock J. Traumatically induced reactive change as visualized through the use of monoclonal antibodies targeted to neurofilament subunits. J Neuropathol Exp Neurol. 1992;51:158–176. doi: 10.1097/00005072-199203000-00006. [DOI] [PubMed] [Google Scholar]
- Yang K, Mu XS, Xue JJ, Perez-Polo JR, Hayes RL. Regional and temporal profiles of c-fos and nerve growth factor mRNA expression in rat brain after lateral cortical impact injury. J Neurosci Res. 1995;42:571–578. doi: 10.1002/jnr.490420415. [DOI] [PubMed] [Google Scholar]
- Young B, Rapp RP, Norton JA, Haack D, Tibbs PA, Bean JR. Failure of prophylactically administered phenytoin to prevent late posttraumatic seizures. J Neurosurg. 1983a;58:236–24. doi: 10.3171/jns.1983.58.2.0236. [DOI] [PubMed] [Google Scholar]
- Young B, Rapp RP, Norton JA, Haack D, Walsh JW. Failure of prophylactically administered phenytoin to prevent post-traumatic seizures in children. Childs Brain. 1983b;10:185–192. doi: 10.1159/000120113. [DOI] [PubMed] [Google Scholar]