

Abstract
Little is known about the reorganization capacity of water molecules at the active sites of enzymes and how this couples to the catalytic reaction. Here, we study the dynamics of water molecules at the active site of a highly proficient enzyme, Δ5-3-ketosteroid isomerase (KSI), during a light-activated mimic of its catalytic cycle. Photo-excitation of a nitrile containing photo-acid, coumarin183 (C183), mimics the change in charge density that occurs at the active site of KSI during the first step of the catalytic reaction. The nitrile of C183 is exposed to water when bound to the KSI active site, and we used time-resolved vibrational spectroscopy as a site-specific probe to study the solvation dynamics of water molecules in the vicinity of the nitrile. We observed that water molecules at the active site of KSI are highly rigid, during the light-activated catalytic cycle, compared to the solvation dynamics observed in bulk water. Based upon this result we hypothesize that rigid water dipoles at the active site might help in the maintenance of the pre-organized electrostatic environment required for efficient catalysis. The results also demonstrate the utility of nitrile probes in measuring the dynamics of local (H-bonded) water molecules in contrast to the commonly used fluorescence methods which measure the average behavior of primary and subsequent spheres of solvation.
Keywords: Vibrational Stokes shift, hydration dynamics, Nitrile probe, and enzyme active site dynamics
Introduction
Understanding the dynamical properties of water molecules surrounding a solute is of fundamental importance in chemical and biological systems 1–6. For a chemical reaction in aqueous solution, reorganization of water molecules around the reactants often controls or modulates the rate of the reaction 1,2. Most of the chemical reactions in biological systems occur at the active site of enzymes, which often contain ordered and disordered water molecules 4,7–11. Although the activity of enzymes depends upon the solvation environment around the active site11–13, very little is known about the dynamical properties and reorganization capacity of active site water molecules and whether water dynamics is coupled to the catalytic reaction co-ordinate 14–16.
NMR and fluorescence up-conversion spectroscopy have been widely used to study the dynamics of water molecules near the surface of proteins. In NMR methods, measurement of magnetic relaxation dispersion of the quadrupolar 2H and 17O nuclei in the water molecule 17 and intermolecular nuclear Overhauser effects between the water and the solute 18 are usually used to study hydration dynamics. These experiments are suitable only for measuring slow water dynamics 19 and the relationship of these observables to function is not clear. Dynamic motions of water molecules at the protein-water interface in the neighborhood of a fluorescent probe localized on the protein surface contribute to time-dependent fluorescence Stokes shifts20,21, which can be probed with femtosecond resolution using fluorescence up-conversion spectroscopy 2,20–22. While significant variations in the solvation response at different sites on the surface of a protein can be observed, it is difficult to parse the experimental results between the solvation response of the protein in the vicinity of the fluorescent probe solvating water molecules, and a specific connection with function is rarely available 23.
In contrast, vibrational dynamic Stokes shift measurements 24–30 have the ability to reveal site-specific information on solvent dynamics if vibrations localized in one section of the macromolecular system are probed. In this method, the solute is photo-excited into an electronic excited state and the time-dependent changes in the IR spectrum of a specific vibrational mode of the solute is measured, as the vibrational mode responds to changes in electrostatic environment due to the dynamic rearrangement of the solvent 24,26,29. This approach has been applied to study the dynamics of hydrogen bonds (H-bonds) between an H-bond donor solute and an H-bond acceptor solvent 29,30. For example, time-dependent changes in the stretching frequency of the carbonyl bond (-C=O) have been used to obtain site-specific information on dynamics of H-bonds between coumarin and phenol molecules 24,25.
Vibrational modes other than those of the carbonyl group, such as hydroxyl (-OH) 31,32 and nitrile (-C≡N) 33–35 stretching modes, have also been developed as local probes for the ultrafast dielectric response. Among these, the -C≡N stretching mode is particularly attractive because it absorbs in an uncluttered part of the vibrational spectrum. For example, the ultrafast frequency shifts of a -C≡N vibration has been used to study the solvent dependence of electron transfer rates between a -C≡N containing photo-acid and a base 34. To date only a few studies have employed this strategy to study local solvation in macromolecular systems like proteins, though we now have methods to systematically introduce nitrile probes at many sites in a protein35–41.
In this work we have carried out vibrational dynamic Stokes shift measurements on a -C≡N containing photo-acid, coumarin183 (C183), bound at the active site of a highly proficient enzyme, Δ5-3-ketosteroid isomerase (KSI) (Fig. 1) to study how the water molecules which are H-bonded to the -C≡N in the substrate pocket of the enzyme respond to a light-activated charge perturbation which mimics the movement of charges during its catalytic cycle. KSI catalyzes the isomerization of 3-oxo-Δ5-steroids to their Δ4-conjugated isomers 42–45 (Fig. 2A), and increases the rate of the isomerization reaction by 1011-fold over the uncatalyzed reaction. We have previously shown that the fluorescent photo-acid C183 (Fig. 1 and 2B) binds at the active site of KSI (dissociation constant ~ 1.3 μM, 46,47 similar to other transition-state analogues), and its photo excitation mimics the changes in electrostatic environment which occur during the first step of the catalytic cycle of the enzyme 46,47. The catalytic reaction of KSI starts with the abstraction of the alpha proton of the steroid substrate by the negatively charged Asp40 residue of the enzyme (Fig. 2A), which leads to the formation of a dienolate intermediate. The network of hydrogen bonds to the active site residues (Asp103, Tyr 16, Tyr57, and Tyr 32) of the enzyme, termed the oxyanion hole, stabilizes the negative charge on the oxygen atom of the dienolate intermediate. Hence, during the progress of the reaction from the reactant state to the intermediate state, negative charge moves toward the oxyanion hole. Conversely, negative charge moves away from the oxyanion hole while proceeding from the intermediate state to the reactant state (Fig. 2A). From the perspective of electrostatics, the first step of the catalytic reaction is characterized by this flow of charge. Because the light activation of enzyme-bound C183 relocates the charge density away from the oxyanion hole (Fig. 2B), and creates an electronic difference dipole (|Δμ⃗excitation|) of ~ 14.2 Debye 47 which is similar in magnitude to the |Δμ⃗reaction| of ~ 20.2 Debye created during the first step of KSI catalysis 47 (though the direction of the dipole appears to be rotated by approximately 45° between the two systems46,47), it mimics the movement of charge density during the first step of the KSI catalytic cycle 47. C183 contains a -C≡N moiety at structural position 3 located toward the solvent exposed side of the KSI active site when bound to the enzyme (Fig. 2B) 46,47. In this study we have utilized this nitrile vibrational probe to study the dynamical behavior of water molecules present in the substrate pocket of KSI where the locally-probed solvation response can be related to the catalytic function of the enzyme.
Figure 1.
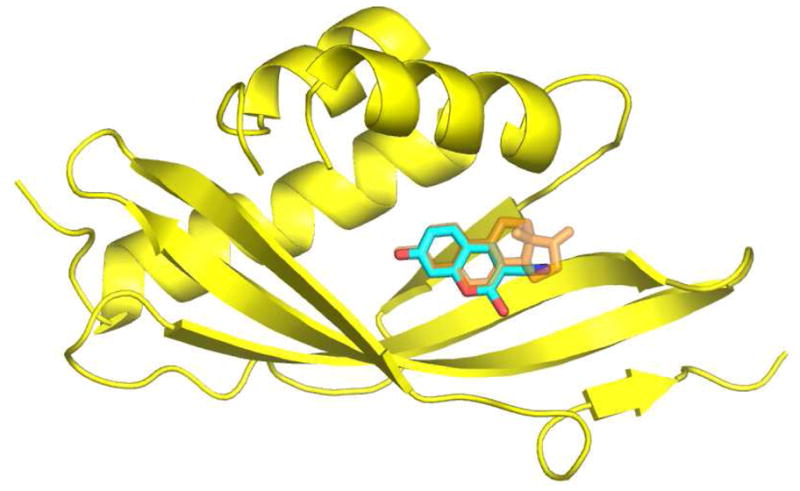
Structural model of KSI (yellow) bound to equilenin (orange) (PDB file: 1OH0). C183 (cyan blue) is modeled into this structure, aligned with the naphthol rings of equilenin. A β-strand and a loop consisting of residues 36–46 has been removed for clarity. The structural model was created using the program PyMOL (http://www.pymol.org).
Figure 2.

Comparison of the enzymatic reaction of KSI with photocycle of C183. (A) The movement of electronic charge during the enzymatic reaction of KSI is compared to the movement of electronic charge during the photo-excitation of C183 (B). The red arrow in panels (A) and (B) show the direction of movement of electronic charge when moving from the KSI-intermediate complex to the KSI-reactant complex. An electronic difference dipole (|Δμ⃗reaction|) of ~ 20.2 Debye is created during the first step of KSI catalysis which is similar in magnitude to |Δμ⃗excitation| ~ 14.2 Debye created during photo-excitation of enzyme-bound C183 46,47. The red circle in panel B highlights the nitrile of C183 which faces the solvent exposed side of KSI active site and H-bonded to water and whose IR spectrum is used to probe dynamics in the substrate pocket of KSI.
Material and Methods
Buffers
The buffers used in this study are pH 5 buffer (40 mM citrate, 1 mM EDTA), pH 7.2 buffer (40 mM potassium phosphate, 1 mM EDTA), and pH 9.0 buffer (40 mM CHES (N-cyclohexyl-2-aminoethanesulfonic acid), 1 mM EDTA).
Protein expression and purification
All experiments in the present work used KSI from Pseudomonas putida with the common mutation, D40N, to enhance binding 46. The protein was expressed and purified according to a previously published protocol 48. The fluorescence and circular dichroism spectra of the mutant protein are similar to the wild-type KSI in both the native and unfolded states 47 which indicates that D40N mutation does not perturb the gross tertiary and secondary structure of KSI. The D40N form of KSI mimics the charge distribution of the enzyme active site in the intermediate state (protonated base), and is not the active form of the enzyme.
UV-Vis absorption and steady-state fluorescence
The UV-Vis absorption spectra of free and enzyme bound C183 were measured with a Perkin Elmer Lambda 25 spectrophotometer. The concentrations of C183 and KSI were respectively 5 μM and 50 μM. Steady-state fluorescence spectra of the unbound and enzyme bound C183 were measured with a Perkin Elmer LS 55 fluorescence spectrometer, with excitation set to 408 nm.
Steady-state FT-IR experiments
All the steady state IR-spectra were obtained on a Bruker Vertex 70 FTIR spectrometer (Billerica, MA) equipped with an InSb detector and processed as detailed previously 47. Peak positions were determined using a second derivative-based method built into the OPUS FT-IR software (Bruker Photonics, Billerica, MA). The concentration of free and enzyme bound C183 was ~ 2mM for FTIR experiments.
UV-pump IR-probe experiments
We use sub-picosecond duration 400 nm pulses to excite C183, and broadband mid-IR pulses to probe the transient spectra of the nitrile stretch of C183. We generated pump and probe pulses with a regeneratively amplified Ti:sapphire laser system (Spitfire, Spectra Physics) with a 1 kHz repetition rate and 35 fs FWHM pulse duration at 800 nm. We produced 400 nm pulses with a 7 nm FWHM spectral width by frequency doubling the amplifier output with a 0.1 mm thick BBO crystal, and sent ~ 5mW to excite the electronic transition of C183. The pump beam spot size was ~ 200 μm in diameter. Tunable mid-infrared pulses were generated by difference frequency generation of the near-infrared signal and idler from an optical parametric amplifier (OPA800CF, Spectra Physics). These mid-IR pulses have duration of 60 fs FWHM with a spectral width of about 300 cm−1. The cross-correlation of the pump and probe pulses is less than 200 fs FWHM. We measured the pump-probe signal with the mid-IR polarization both parallel and perpendicular to the 400 nm pump pulse. Isotropic signals were calculated with this data and used for further analysis. We dispersed the probe beam with a grating spectrometer (iHR320, Horiba Jobin Yvon) onto a liquid N2 cooled ▭ MCT pixel array detector with a spectral resolution of ~2 cm−1 per pixel. The liquid samples were mounted between two CaF2 windows with a 50μm Teflon spacer. The sample cell was continuously rotated as well as vertically translated during the experiments to minimize photo damage of the samples. Transient spectra are averaged for ~5 hours for each sample. The transient IR spectra at different delay times were fit to a sum of two pseudo-Voigt functions to determine the nitrile stretching frequencies and width (FWHM) of the peaks in the ground state bleach and electronic excited state. The concentration of free and enzyme bound C183 was ~ 6mM for UV-pump IR-probe experiments.
Results
Free C183 in aqueous solution at pH 5 is predominantly in its protonated form and exhibits an absorbance maximum at 355 nm (Fig. 3A). The deprotonated form at pH 9.0 absorbs maximally at 408 nm (Fig. 3A). The UV absorption maximum of C183 at pH 5 in the presence of KSI shifts from 355 nm to 408 nm indicating that KSI binding converts C183 to its anionic form (Fig. 3A), as expected for a ligand with a solution pKa of ~ 6.5 46,48,49. The fluorescence emission of C183 (λmax = 450 nm) is heavily quenched when it is bound to KSI (Fig. 3A), and its fluorescence lifetime also decreases significantly 46. In parallel to these electronic absorption features, the IR frequency of the C183-nitrile also shifts upon binding to KSI (Fig. 3B), as expected for the formation of the anion. At pH 5, C183 remains predominantly in its protonated form in solution and the nitrile absorbs at 2238.2 cm−1 (Fig. 3B), while at pH 9.0, where C183 remains predominantly in its deprotonated form in solution, the nitrile absorbs maximally at 2226.3 cm−1 nm (Fig. 3B). More precisely, the IR absorption frequency of the nitrile of C183 at pH 5 in the presence of KSI shifts from 2238.2 cm−1 to 2227.2 cm−1 (Fig. 3B). The stretching frequency of C183-nitrile when it is bound at the active site of KSI is similar to the stretching frequency of C183-nitrile in pH 9 buffer. The inset in Fig. 3B shows that the stretching frequency of the nitrile shifts to the blue upon H-bonding. The nitrile absorbs at 2228.9 cm−1 when C183 is dissolved in DMSO (an aprotic solvent), but shifts to 2238.2 cm−1 when C183 is dissolved in pH 5 buffer (a protic solvent of similar dielectric to DMSO) (Fig. 3B: inset). The blue-shifting of nitrile absorption frequency upon H-bonding to water has been extensively discussed in many previous studies 36,50–52. Thus, we can conclude both from the electronic absorption and from the nitrile stretch frequency that C183 binds at the active site in the anionic form, and further, from the nitrile stretch frequency, that the nitrile, which is buried within the substrate binding pocket, is interacting with water. We will now present a detailed discussion of how the time-dependent CN-stretch absorption spectrum induced by UV excitation of C183 reports upon the distinct manner with which water in solution and water associated with the protein solvate a functionally-relevant movement of charge.
Figure 3.

C183 binding to KSI. (A) UV-Vis absorption spectrum of C183 (shown on left hand side Y-axis in blue) when it is free in solution at pH 5 (dashed blue line) and pH 9.0 (dotted blue line). The continuous blue line shows the absorption spectra of C183 at pH 5 in presence of KSI. Fluorescence emission spectrum of C183 (shown on right hand side Y-axis in dark red) when it is free in solution at pH 7.2 (dotted dark red line) and when bound to KSI at pH 7.2 (continuous dark red line). (B) Steady state FTIR spectrum of C183 (shown in 2190–2300 cm−1 range where nitrile absorption occurs) when it is free in solution at pH 5 (dashed blue line) and pH 9.0 (dotted blue line). The continuous blue line shows the IR spectra of C183 at pH 5 in presence of KSI. The inset in panel (B) compares the IR spectrum of C183 in pH 5 buffer (dashed blue line) and DMSO (dashed-dotted black line).
As shown in Fig. 4, we used sub-picosecond duration 400 nm pulses to excite C183, and broadband mid-IR pulses to probe the transient spectra of the nitrile stretch of C183 (see Material and Methods). The transient IR spectra at different delay times were extracted (Fig. 5) and fit with a sum of two pseudo-Voigt functions to determine the nitrile stretching frequencies and width (FWHM) of the peaks in the ground state bleach and electronic excited state.
Figure 4.

Temporally and spectrally resolved transient UV pump – IR probe data for C183-nitrile. (A) C183 in pH 9 buffer; (B) C183 bound to KSI at pH 7.2. The difference IR absorbance spectra of the nitrile in the electronic excited state (dark blue) and electronic ground state bleach (reddish yellow) is plotted at various delay time between UV-pump and IR-probe pulse. The plots show the dynamics for time delays greater than 500 fs.
Figure 5.

Changes in the transient difference IR absorption spectra of the nitrile attached to C183 at different times after photo-excitation of C183 at 400 nm. (A, B) free C183 in pH 9.0 buffer; and (C, D) C183 bound to KSI at pH 7.2. The inset in panels (A) and (C) is an expanded view of the evolution of the IR-absorption frequency of –CN in the electronic excited state at different times after the photo-excitation of C183. In panels (A) and (C), empty circles represent the data and continuous solid lines are the fit to a sum of two pseudo-Voigt functions. Panels (B) and (D) show peak shift dynamics of C183-nitrile. In panels (B) and (D) the blue triangles and the dark red circles respectively show the evolution of the peak of absorption frequency of –CN in electronic ground state bleach and electronic excited state. The solid blue line through the data is a fit to a straight line equation and the solid dark red line through the data is a fit to a single exponential equation with a time constant of 12 ps. The insets in panel (B) and (D) show the change in width (FWHM) of transient IR spectra when C183 is free in solution (B) and bound to KSI (D). In both insets, the blue triangles and the dark red circles respectively show the change in width of transient IR spectra of C183–nitrile in electronic ground state bleach and electronic excited state. The solid blue and red lines through the data in insets are fits to a straight line equation.
C183 dissolved in pH 9.0 buffer shows a nitrile stretching frequency of 2226.3 cm−1 in the electronic ground state bleach (Fig. 4A; Fig. 5A and B). Upon photo-excitation to the first electronic excited state the nitrile stretching frequency initially shifts to 2171.3 cm−1. This peak shifts to the blue from 2171.3 cm−1 to 2182.3 cm−1 with delay times after photo-excitation, while the absorption frequency of the nitrile in the electronic ground state remains constant at 2226.3 cm−1 (Fig. 4A; Fig. 5A and B). The time-dependent blue shift in absorption frequency of the nitrile in the electronic excited state of C183 fits well to a single exponential with a time constant of 12 ± 2 ps (Fig. 5B). This time dependent shift in the CN-stretch frequency could originate from two physically distinct phenomena: an initial red-shift of the absorption frequency due to a combination band or hot band shift 27,53 that relaxed with a rate determined by the rate of energy relaxation from C183 to the solvation environment or a vibrational analog 24,26,27,31,33 of the dynamic fluorescence Stokes shift 2,21 where the rate of relaxation reflects the rate of dielectric relaxation of the solvent in response to the electronic excitation of C183. We attribute the observed time-dependent spectral shift of the CN-stretch frequency to a dynamic Stokes shift. The reasons for this assignment will be addressed in the Discussion.
When C183 is bound in the active site of KSI, the nitrile stretching frequency at 2227.2 cm−1 in the ground state bleach shifts to 2168.5 cm−1 upon photo-excitation to the first electronic excited state (Fig. 4B; Fig. 5C and D). In this case, however, no time-dependent blue shift of the nitrile stretching frequency is observed (Fig. 4B; Fig. 5C and D), and only a slight red shift of unknown origin with time from 2168.5 cm−1 to 2165.4 cm−1 is observed. The absorption frequency of the nitrile in the ground state bleach remains constant at 2227.2 cm−1 (Fig. 4B; Fig. 5C and D). The transient IR intensity decays with a time constant of ~ 80 ps, in both ground state bleach and electronic excited state, which is consistent with the fluorescence lifetime decay timescale of KSI-bound C183 46.
Discussion
The hydration environment in the substrate pocket of KSI has been a subject of many theoretical and experimental studies. Solvent penetration in the substrate pocket of the apo enzyme has been studied using fluorescence quenching experiments 54. The tryptophan residue at position 120 (Trp120) in the sequence of KSI makes a H-bond with the side chain carbonyl of Asp40 which plays the role of general base during catalysis. KSI variants having different active site mutations show differing degrees of quenching of fluorescence of Trp120 by acrylamide present in the solvent, implying different degrees of solvent penetration in the substrate pocket 54. Water molecules have also been found to be present in the substrate pocket of ligand-bound KSI. 19F NMR experiments on 2-fluoro-4-nitrophenolate bound to several KSI variants have indicated that the degree of solvation in the substrate pocket depends upon the nature of the mutation in the active site 55. The presence of a water molecule within hydrogen bonding distance of the enzyme-bound ligand has been identified in a recent study using a combination of 13C NMR and FTIR spectroscopy 52, which was proposed to be at a position similar to that identified previously in a high resolution crystal structure of ligand-bound KSI (PDB:3FZW) (Fig.6A).
Figure 6.

Water in the substrate pocket of KSI. Ordered water molecules (red sphere) in the KSI substrate pocket as seen in PDB files (A) 3FZW; and (B) 1OH0. KSI bound equilenin is shown with thick lines.
X-ray crystal structures of proteins generally do not show structural water molecules 8,56. Nevertheless, a water molecule near the oxyanion hole which was earlier identified in molecular dynamics simulations of the isomerization reaction catalyzed by KSI 44,57, was observed in high-resolution crystal structures of KSI bound to equilenin (PDB:3FZW) 58 (Fig. 6A). Crystallographic data for an Y16S mutant of KSI bound to equilenin (PDB: 3IPT) 55 also suggest the presence of many disordered water molecules in the substrate pocket of KSI, some of which are distal to the oxyanion hole. A recent theoretical study 11 and additional high resolution crystal structure (PDB: 1OH0) (Fig. 6B) have identified several water occupation sites in the substrate pocket that are distal to the oxyanion hole. Water molecules are found near the A and B rings of enzyme-bound equilenin some of which are in contact with the bulk solvent 11. The number and position of these water molecules depend upon the nature of the bound ligand and the mutation in the active site 11. It is expected that more water molecules will occupy the substrate pocket when C183 is bound to KSI due to its smaller size compared to equilenin (Fig. 1). Most of the above studies examined and elucidated the structural properties of active site water molecules. The dynamical properties of these water molecules and how the dynamics couple to catalysis remains poorly understood, and characterizing this coupling is the main objective of the present paper.
In the electronic ground state of C183, the nitrile of C183 senses the equilibrium configuration of water molecules surrounding it. Photo-excitation of C183 into the electronic excited state redistributes the electronic charge density of the molecule, with the negative charge moving away from the deprotonated hydroxyl atom towards the nitrile moiety (Fig. 2B). The redistributed charge density on C183 produces a non-equilibrium configuration of water molecules around C183-nitrile. These events generate a prompt red shift in the nitrile vibrational frequency (Fig. 5). The time scales for water reorientation in response to an electronic excitation of a solute molecule depends upon the structural flexibility of the solvation environment 21. When C183 is free in solution, the reorientation of the water molecules surrounding the nitrile in the electronic excited state leads to stronger H-bonds with C183-nitrile, which results in a dynamic blue shift in the vibrational absorption frequency of C183-nitrile with a time-constant of 12 ± 2 ps (Fig. 5A and B). It is important to note that a vibrational Stokes shift can be either towards the blue or red 26,27, unlike fluorescence Stokes shifts which are always to the red 21. By contrast, the absence of a dynamic spectral shift when C183 is bound at the active site of KSI implies that the water molecules in the region surrounding the C183-nitrile near the active site of KSI are highly rigid and do not reorganize (within the ~300 ps time-window probed in our experiments) in response to the changes in electrostatic environment which mimic its catalytic reaction cycle.
The 12 ± 2 ps time scale for the CN-stretch blue shift cannot be easily reconciled with the expected rates of dielectric solvation or vibrational relaxation in aqueous solution. Molecular dynamics simulations and fluorescence Stokes shift studies of non-equilibrium solvation dynamics indicate that the dielectric solvation dynamics of water in response to an electrostatic perturbation induced by electronic excitation of a solute occur predominantly with two time scales of roughly 50 fs and 1 ps 2,46,59. Molecular dynamics simulations and pump-probe spectroscopy studies of low frequency vibrational relaxation from a non-equilibrium solute to the solvent occurs on the one to a few ps time scale 60–62. The 12 ± 2 ps time constant observed in our measurement exceeds the expected time constant for either bulk dielectric solvation or vibrational relaxation.
The absence of the spectral shift for C183 bound to KSI (Fig. 5C and D), the equivalent electronic absorption spectrum for C183 bound to KSI and free in aqueous solution (Fig. 3A), and the roughly same rate of vibrational relaxation observed in aqueous solution and solvated proteins 60,63, leads us to attribute the slow spectral shift seen in aqueous solution to the short range solvation dynamics associated with reorganization of the hydrogen bonding between water and the nitrile moiety of C183. Free and enzyme-bound C183 have the same UV absorption spectra (Fig. 3A), and electronic excitation with the same wavelength of light should deposit the same amount of excess energy into the respective electronic excited states. Hence, vibrational cooling, leading to a dynamic blue shift, should also occur when C183 is bound at the active site of KSI, as it is an intrinsic property of the molecule. However, no time-dependent blue shift in absorption frequency is observed for enzyme-bound C183 (Fig. 4B; Figs. 5C and D). Secondly, vibrationally hot molecules typically show broadened vibrational spectra which narrow as the molecule cools 27,64,65. We extracted the width (FWHM) of the transient IR spectra by fitting them to pseudo-Voigt functions (Figs. 5B and D: inset). The width of the IR spectrum does not change appreciably either in the ground state bleach or electronic excited state (Fig. 5B and D: inset), both for free and KSI-bound C183, on the timescale of 12 ps which is the time constant of the vibrational dynamic Stokes shift (Fig. 5B). These observations make vibrational cooling an unlikely explanation for the time-dependent spectral shift of the CN-stretch frequency in the electronic excited state of free C183.
The attribution of time-dependent shift in the CN-stretch frequency to local changes in hydrogen bond structure matches the pronounced solvatochromic sensitivity of cyano complexes to hydrogen bond solvation 66,67. The sensitivity of a vibrational transition frequency to short-ranged hydrogen bond dynamics has been proposed to explain a wide range of time resolved measurements of hydrogen bond dynamics in water and aqueous ionic solutions 68,69. The greater sensitivity of the CN-stretch frequency to short-ranged hydrogen bond reorganization than the electronic transition frequency parallels the spatial extent of the excitation. The redistribution of charge induced by electronic excitation in C183 occurs on a length scale large compared to the size of a water molecule, whereas the charge redistribution induced by vibration excitation of the CN-stretch occurs on a length scale comparable to the size of a water molecule.
It is insightful to compare the results of the present study to a previous measurement of solvation dynamics on C183 using time-resolved fluorescence 46. Fluorescence dynamic Stokes shift experiments on KSI bound C183 showed a fast (1–2 ps) but very small solvation response which was ascribed to the water solvation of the solvent exposed electron accepting group 46. This result is consistent with the very small time-dependent shift of the nitrile stretching frequency observed in this study (Fig. 5D). For free C183, the time constant of the dielectric relaxation of the surrounding water molecules was measured to be ~ 1–2 ps 46, similar to previous measures of aqueous solvation dynamics, but in contrast to the ~ 12 ps time-constant observed in the current study (Fig. 5C). These finding support our conclusion that fluorescent probes of solvation respond to the dynamics of many solvent molecules in the first and subsequent solvation shells and hence give an average timescale of solvation 26, while molecular vibrations provide site-specific and local probes of solvation 24,26,27. In separate and somewhat related work on small molecule solvatochromism, we have found that in nitrile-containing molecules with no net molecular dipole, where no net solvation is expected, the nitrile vibrations nonetheless sense the polarity of the solvent molecules in their immediate vicinity 70. Hence, the above difference in the dielectric relaxation timescale reflects the global versus local differences in water organization and rigidity around C183.
Closing Remarks
Despite the importance of water molecules in biology 9,71–73, the functional role of water molecules near the active sites of enzymes is not well understood 15,16,56. A recent study measured the dynamics of water molecules near the active site of a metalloprotease during its catalytic cycle using terahertz spectroscopy 15. It was observed that water molecules at the active site become rigid before the formation of the functional Michaelis complex. In the present study, we have studied the dynamics of water molecules near the active site of KSI during a light-activated mimic of its catalysis, using time-resolved vibrational spectroscopy. We observe that water molecules in the active site do not reorganize dynamically in response to a catalytically relevant electrostatic perturbation. In a previous study, we observed that the protein dipoles in the active site of KSI remain static during the light activated mimic of the catalytic reaction 46,47. We propose that the rigid water molecules at the KSI active site help in the maintenance of the organized electrostatic environment required for catalysis. The approach employed in this study, to use molecular vibrations as a site-specific probe of solvation, would be very useful for other biomolecular systems and is expected to reveal novel information about differences in local solvation environments around macromolecular solutes.
Supplementary Material
Acknowledgments
Plasmids containing the genes of mutant forms of KSI were graciously provided by the Herschlag laboratory at Stanford University. We gratefully acknowledge support from the NIH (grant GM27738). We thank Nick Levinson for fruitful discussions and for his critical comments on the manuscript. M.J. and K.G. acknowledge support of the time resolved measurements through the PULSE Institute at SLAC National Accelerator Laboratory by the U.S. Department of Energy, Office of Basic Energy Sciences.
Footnotes
Supporting Information Available: This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Maroncelli M, Macinnis J, Fleming GR. Science. 1989;243:1674–1681. doi: 10.1126/science.243.4899.1674. [DOI] [PubMed] [Google Scholar]
- 2.Jimenez R, Fleming GR, Kumar PV, Maroncelli M. Nature. 1994;369:471–473. [Google Scholar]
- 3.Nandi N, Bhattacharyya K, Bagchi B. Chem Rev. 2000;100:2013–2046. doi: 10.1021/cr980127v. [DOI] [PubMed] [Google Scholar]
- 4.Zhao L, Pal SK, Xia T, Zewail AH. Angew Chem, Int Ed. 2004;43:60–63. doi: 10.1002/anie.200352961. [DOI] [PubMed] [Google Scholar]
- 5.Pal SK, Zewail AH. Chem Rev. 2004;104:2099–2124. doi: 10.1021/cr020689l. [DOI] [PubMed] [Google Scholar]
- 6.Heugen U, Schwaab G, Bründermann E, Heyden M, Yu X, Leitner DM, Havenith M. Proc Natl Acad Sci USA. 2006;103:12301–12306. doi: 10.1073/pnas.0604897103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber BH, Storm MC, Boyer PD. Arch Biochem Biophys. 1974;163:1–6. doi: 10.1016/0003-9861(74)90447-0. [DOI] [PubMed] [Google Scholar]
- 8.Levitt M, Park BH. Structure. 1993;1:223–226. doi: 10.1016/0969-2126(93)90011-5. [DOI] [PubMed] [Google Scholar]
- 9.Takano K, Funahashi J, Yamagata Y, Fujii S, Yutani K. J Mol Biol. 1997;274:132–142. doi: 10.1006/jmbi.1997.1365. [DOI] [PubMed] [Google Scholar]
- 10.Ramasubbu N, Sundar K, Ragunath C, Rafi MM. Arch Biochem Biophys. 2004;421:115–124. doi: 10.1016/j.abb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Hanoian P, Hammes-Schiffer S. Biochemistry. 2011;50:6689–6700. doi: 10.1021/bi200703y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klibanov AM. Nature. 2001;409:241–246. doi: 10.1038/35051719. [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Dordick JS, Garde S. Biophys J. 2004;87:812–821. doi: 10.1529/biophysj.104.041269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenimore PW, Frauenfelder H, McMahon BH, Young RD. Proc Natl Acad Sci USA. 2004;101:14408–14413. doi: 10.1073/pnas.0405573101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grossman M, Born B, Heyden M, Tworowski D, Fields GB, Sagi I, Havenith M. Nat Struct Mol Biol. 2011;18:1102–1108. doi: 10.1038/nsmb.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guha S, Sahu K, Roy D, Mondal SK, Roy S, Bhattacharyya K. Biochemistry. 2005;44:8940–8947. doi: 10.1021/bi0473915. [DOI] [PubMed] [Google Scholar]
- 17.Halle B, Denisov VP. Methods Enzymol. 2002;338:178–201. doi: 10.1016/s0076-6879(02)38220-x. [DOI] [PubMed] [Google Scholar]
- 18.Gottfried O. Progr NMR Spectrosc. 1997;31:259–285. [Google Scholar]
- 19.Halle B. Philos Trans R Soc Lond B Biol Sci. 2004;359:1207–1223. doi: 10.1098/rstb.2004.1499. discussion 1223–1204, 1323–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, Wang L, Kao Y-T, Qiu W, Yang Y, Okobiah O, Zhong D. Proc Natl Acad Sci USA. 2007;104:18461–18466. doi: 10.1073/pnas.0707647104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakowicz JR. Principles of fluorescence spectroscopy. 3. Springer; New York: 2006. [Google Scholar]
- 22.Cohen BE, McAnaney TB, Park ES, Jan YN, Boxer SG, Jan LY. Science. 2002;296:1700–1703. doi: 10.1126/science.1069346. [DOI] [PubMed] [Google Scholar]
- 23.Chang CW, Guo L, Kao YT, Li J, Tan C, Li T, Saxena C, Liu Z, Wang L, Sancar A, Zhong D. Proc Natl Acad Sci USA. 2010;107:2914–2919. doi: 10.1073/pnas.1000001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chudoba C, Nibbering ETJ, Elsaesser T. Phys Rev Lett. 1998;81:3010–3013. [Google Scholar]
- 25.Chudoba C, Nibbering ETJ, Elsaesser T. Journal of Physical Chemistry A. 1999;103:5625–5628. [Google Scholar]
- 26.Nibbering ETJ, Elsaesser T. Appl Phys B: Lasers and Optics. 2000;71:439–441. [Google Scholar]
- 27.Asbury JB, Wang Y, Lian T. Bull Chem Soc Jpn. 2002;75:973–983. [Google Scholar]
- 28.Rezus YLA, Madsen D, Bakker HJ. J Chem Phys. 2004;121:10599–10604. doi: 10.1063/1.1809589. [DOI] [PubMed] [Google Scholar]
- 29.Nibbering ET, Fidder H, Pines E. Annu Rev Phys Chem. 2005;56:337–367. doi: 10.1146/annurev.physchem.56.092503.141314. [DOI] [PubMed] [Google Scholar]
- 30.Zhao G-J, Han K-L. Acc Chem Res. 2011;45:404–413. doi: 10.1021/ar200135h. [DOI] [PubMed] [Google Scholar]
- 31.Prémont-Schwarz M, Xiao D, Batista VS, Nibbering ETJ. Journal of Physical Chemistry A. 2011;115:10511–10516. doi: 10.1021/jp207642k. [DOI] [PubMed] [Google Scholar]
- 32.Xiao D, Premont-Schwarz M, Nibbering ETJ, Batista VS. Journal of Physical Chemistry A. 2012;116:2775–2790. doi: 10.1021/jp208426v. [DOI] [PubMed] [Google Scholar]
- 33.Van Tassle AJ, Prantil MA, Fleming GR. J Phys Chem B. 2006;110:18989–18995. doi: 10.1021/jp0603738. [DOI] [PubMed] [Google Scholar]
- 34.Ghosh HN, Verma S, Nibbering ET. Journal of Physical Chemistry A. 2011;115:664–670. doi: 10.1021/jp108090b. [DOI] [PubMed] [Google Scholar]
- 35.Lindquist BA, Furse KE, Corcelli SA. Phys Chem Chem Phys. 2009;11:8119–8132. doi: 10.1039/b908588b. [DOI] [PubMed] [Google Scholar]
- 36.Getahun Z, Huang CY, Wang T, De Leon B, DeGrado WF, Gai F. J Am Chem Soc. 2003;125:405–411. doi: 10.1021/ja0285262. [DOI] [PubMed] [Google Scholar]
- 37.Fafarman AT, Webb LJ, Chuang JI, Boxer SG. J Am Chem Soc. 2006;128:13356–13357. doi: 10.1021/ja0650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultz KC, Supekova L, Ryu Y, Xie J, Perera R, Schultz PG. J Am Chem Soc. 2006;128:13984–13985. doi: 10.1021/ja0636690. [DOI] [PubMed] [Google Scholar]
- 39.Waegele MM, Tucker MJ, Gai F. Chem Phys Lett. 2009;478:249–253. doi: 10.1016/j.cplett.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jo H, Culik RM, Korendovych IV, Degrado WF, Gai F. Biochemistry. 2010;49:10354–10356. doi: 10.1021/bi101711a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fafarman AT, Boxer SG. J Phys Chem B. 2010;114:13536–13544. doi: 10.1021/jp106406p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hawkinson DC, Eames TC, Pollack RM. Biochemistry. 1991;30:10849–10858. doi: 10.1021/bi00109a007. [DOI] [PubMed] [Google Scholar]
- 43.Ha N-C, Choi G, Choi KY, Oh B-H. Curr Opin Struct Biol. 2001;11:674–678. doi: 10.1016/s0959-440x(01)00268-8. [DOI] [PubMed] [Google Scholar]
- 44.Feierberg I, Aqvist J. Biochemistry. 2002;41:15728–15735. doi: 10.1021/bi026873i. [DOI] [PubMed] [Google Scholar]
- 45.Ralph MP. Bioorg Chem. 2004;32:341–353. doi: 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Childs W, Boxer SG. J Am Chem Soc. 2010;132:6474–6480. doi: 10.1021/ja1007849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jha SK, Ji M, Gaffney KJ, Boxer SG. Proc Natl Acad Sci USA. 2011;108:16612–16617. doi: 10.1073/pnas.1113874108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kraut DA, Sigala PA, Pybus B, Liu CW, Ringe D, Petsko GA, Herschlag D. PLoS Biol. 2006;4:e99. doi: 10.1371/journal.pbio.0040099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Childs W, Boxer SG. Biochemistry. 2010;49:2725–2731. doi: 10.1021/bi100074s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Purcell KF, Drago RS. J Am Chem Soc. 1966;88:919–924. [Google Scholar]
- 51.Lindquist BA, Corcelli SA. J Phys Chem B. 2008;112:6301–6303. doi: 10.1021/jp802039e. [DOI] [PubMed] [Google Scholar]
- 52.Fafarman AT, Sigala PA, Herschlag D, Boxer SG. J Am Chem Soc. 2010;132:12811–12813. doi: 10.1021/ja104573b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shelby RM, Harris CB, Cornelius PA. The Journal of Chemical Physics. 1979;70:34–41. [Google Scholar]
- 54.Choi G, Ha NC, Kim MS, Hong BH, Oh BH, Choi KY. Biochemistry. 2001;40:6828–6835. doi: 10.1021/bi002767+. [DOI] [PubMed] [Google Scholar]
- 55.Kraut DA, Sigala PA, Fenn TD, Herschlag D. Proc Natl Acad Sci USA. 2010;107:1960–1965. doi: 10.1073/pnas.0911168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kornblatt JA, Kornblatt MJ. Int Rev Cytol. 2002;215:49–73. doi: 10.1016/s0074-7696(02)15005-4. [DOI] [PubMed] [Google Scholar]
- 57.Mazumder D, Kahn K, Bruice TC. J Am Chem Soc. 2003;125:7553–7561. doi: 10.1021/ja030138s. [DOI] [PubMed] [Google Scholar]
- 58.Sigala PA, Caaveiro JM, Ringe D, Petsko GA, Herschlag D. Biochemistry. 2009;48:6932–6939. doi: 10.1021/bi900713j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maroncelli M, Fleming GR. Journal of Chemical Physics. 1988;89:5044–5069. [Google Scholar]
- 60.Johnson AE, Levinger NE, Barbara PF. Journal of Physical Chemistry. 1992;96:7841–7844. [Google Scholar]
- 61.Benjamin I, Whitnell RM. Chemical Physics Letters. 1993;204:45–52. [Google Scholar]
- 62.Kliner DAV, Alfano JC, Barbara PF. Journal of Chemical Physics. 1993;98 :5375–5389. [Google Scholar]
- 63.Deak J, Chin HL, Lewis CM, Miller RJD. Journal of Physical Chemistry B. 1998;102:6621–6634. [Google Scholar]
- 64.Mohammed OF, Banerji N, Lang B, Nibbering ETJ, Vauthey E. Journal of Physical Chemistry A. 2006;110:13676–13680. doi: 10.1021/jp066079x. [DOI] [PubMed] [Google Scholar]
- 65.Eom HS, Jeoung SC, Kim D, Ha J-H, Kim Y-R. Journal of Physical Chemistry A. 1997;101:3661–3669. [Google Scholar]
- 66.Winkler JR, Creutz C, Sutin N. Journal of the American Chemical Society. 1987;109:3470–3471. [Google Scholar]
- 67.Chen PY, Meyer TJ. Chemical Reviews. 1998;98:1439–1477. doi: 10.1021/cr941180w. [DOI] [PubMed] [Google Scholar]
- 68.Moller KB, Rey R, Hynes JT. Journal of Physical Chemistry A. 2004;108:1275–1289. [Google Scholar]
- 69.Eaves JD, Tokmakoff A, Geissler PL. Journal of Physical Chemistry A. 2005;109:9424–9436. doi: 10.1021/jp051364m. [DOI] [PubMed] [Google Scholar]
- 70.Levinson NM, Fried SD, Boxer SG. J Phys Chem B. 2012 doi: 10.1021/jp301054e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levy Y, Onuchic JN. Annu Rev Biophys Biomol Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 72.Chaplin M. Nat Rev Mol Cell Biol. 2006;7:861–866. doi: 10.1038/nrm2021. [DOI] [PubMed] [Google Scholar]
- 73.Jha SK, Udgaonkar JB. Proc Natl Acad Sci USA. 2009;106:12289–12294. doi: 10.1073/pnas.0905744106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.