

Abstract
We previously developed Bisulfite Padlock Probes (BSPPs) for the specific and parallel digital quantification of DNA methylation1. Here we report the second-generation of BSPPs with a design algorithm to generate more efficient padlock probes, a library-free protocol that dramatically reduces the time and cost of sample preparation and is compatible with automation, and an efficient bioinformatics pipeline that accurately obtains both methylation levels and genotypes from bisulfite sequencing data.
To interrogate the methylation of the most informative loci across many samples quickly and cost-effectively we developed the second generation BSPP for improved flexibility and multiplexing capability. These improvements have contributed to recent findings in mouse and human pluripotent stem cells2-5.
First, target selection and probe design is crucial for BSPP. To aid in the design of efficient padlock probes for bisulfite analysis, we developed a program called ppDesigner. It accepts as input the genome of any organism, a list of arbitrary targets desired by the user, and a set of user-desired probe constraints matching requirements of the experimental protocol. It in silico bisulfite-converts the genome on the fly (that is, it changes all cytosine to thymine) and outputs a set of padlock probes to cover the chosen targets while avoiding CpGs on the capturing arms which could be methylated and not converted to be recognized as thymine. ppDesigner uses a back-propagation neural network to predict probe efficiency (Supplementary Fig. 1). We had previously trained this network using data from probes for exomic targets6 based on seven properties. Using bisulfite capture data, we have refined the network with two additional factors. ppDesigner can explain ~50% of the variance in capturing efficiency for genomic DNA and ~20% of the variance in capturing efficiency for bisulfite converted DNA; additional variation could be due to factors such as variability in oligonucleotide synthesis and sample DNA quality. ppDesigner is extremely flexible, and has been used to design a variety of genomic and bisulfite probes for Homo sapiens2, 3, Mus musculus4, and Drosophila melanogaster7.
Key requirements for methylation analysis on large sample sizes include low cost, simple workflow, and automation compatibility. As DNA sequencing cost has rapidly decreased, sample processing has become a bottleneck in terms of cost and throughput. A complicated workflow increases variability between samples, and reduces power in large-scale studies. To address these issues, we extended a “library-free” protocol8 to multiplexed BSPP capture (Fig. 1). This method eliminates five steps from Illumina’s library construction protocol, such that multiplexed libraries can be generated from DNA in only four steps (Supplementary Table 1). Using multiplexed primers with 6 bp barcodes, we have routinely generated libraries for 96 samples in 96-well plates and sequenced all at once in a single Illumina HiSeq flowcell. Additional primers have been designed to process 384 samples per batch. As sample-specific barcodes were added, barcoded libraries can be pooled for size-selection, which is the most time consuming, contamination-prone, and error-prone step if performed individually. The protocol is compatible with multi-channel pipettes or liquid handling devices. It dramatically reduced experimental cost and time, and improved reproducibility and read mapping rates (Supplementary Tables 1 and 2). For large sample sizes, the library preparation cost (including probes) is comparable to that of the Restricted Representation and Whole Genome Bisulfite Sequencing (RRBS, WGBS) protocols, while the sequencing cost is much lower than that of WGBS due to targeting of CpG sites of interest. RRBS is more cost-effective than BSPP, but there is little flexibility in selecting specific sites or regions.
Figure 1.
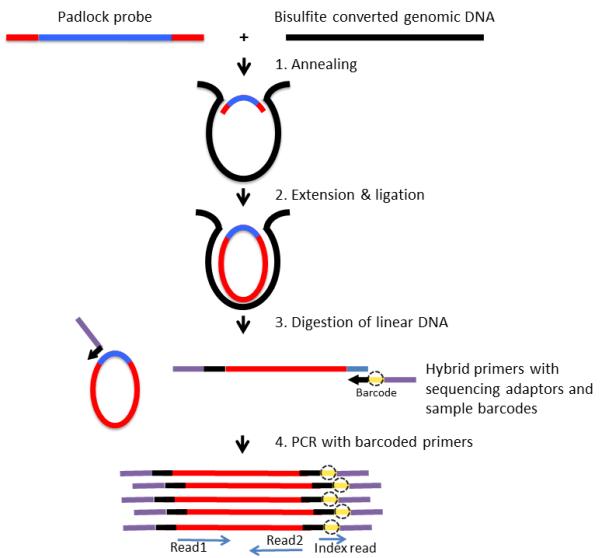
Schematic of library-free BSPP protocol. Each padlock probe has a common linker sequence flanked by two target-specific capturing arms (red) that anneal to bisulfite converted genomic DNA (black). The 3′ end is extended and ligated with the 5′ end to form circularized DNA. After removal of linear DNA, all circularized captured targets are PCR-amplified with barcoded primers and directly sequenced with an Illumina sequencing platform (GA II(x) or HiSeq). Amplicon size is 363 bp, which includes captured target (180 bp), capturing arms (55 bp), and amplification primers and adapters (128 bp). The inserts can be read through with paired-end 120 bp sequencing reads.
Another bottleneck in bisulfite sequencing is a lack of computational tools to efficiently analyze sequencing data generated from hundreds of samples. To overcome this issue, we developed an analysis pipeline for read mapping and methylation quantification called bisReadMapper (Supplementary Fig. 2). In previous padlock probe studies, reads were mapped only against target regions due to the computational requirements of sequence alignment1. In contrast, bisReadMapper maps to the full genome sequence, allowing processing of both targeted and whole genome bisulfite data. bisReadMapper also determines the origin strand of the read based on base composition and maps reads as if they were fully bisulfite-converted to a fully bisulfite-converted genome sequence, allowing mapping of both bi- and uni-directional bisulfite libraries in an unbiased manner. Another feature is the capability to call single nucleotide polymorphisms from bisulfite sequencing data; this feature not only allows for analysis of allele-specific methylation9, but also allows accurate sample tracking in large-scale experiments. Finally, bisReadMapper can call methylation levels at both CpG and non-CpG sites.
To demonstrate the effectiveness of our assay, we generated a new genome-scale probe set based on our previous results and new information about differential methylation1,10-12. Our new design was targeted to evaluate the methylation level at a set of genomic locations known to contain differentially methylated regions (DMRs) or sites (DMSs)10-13, CTCF binding sites, and DNase I hypersensitive regions. In addition, all microRNA genes and all promoters for human NCBI Reference Sequence (RefSeq) genes were targeted. Using ppDesigner, we successfully designed ~330,000 padlock probes that covered 140,749 non-overlapping regions with a total size of 34 megabases. We performed capturing experiments and end-sequencing, and found that these probes were slightly more specific (~96% on-target) and uniform than previous probes1 (Supplementary Fig. 3). These probes were further normalized using subsetting and suppressor oligonucleotides as described previously1 to improve uniformity. Roughly 500,000 CpG sites were characterizable with ~4 gigabases of sequencing reads, and additional sites became callable with deeper sequencing (Supplementary Fig. 4-5).
We used this probe set to analyze H1 embryonic stem cells (H1 ESCs), PGP1 fibroblasts (PGP1F), and two technical replicates of PGP1 fibroblast-derived induced pluripotent stem cells (PGP1-iPSC). For each sample, we sequenced on average ~3.66 gigabases and measured the methylation level for an average of 480,904 CpG sites. In order to assess whether this data could identify potential epigenetic regulation of transcription, we utilized GREAT14 to predict the cis-regulatory potential of regions around captured CpG sites. In total, the padlock probes captured CpG sites in regions predicted to regulate 98% of RefSeq genes (Supplementary Fig. 6).
The data generated by BSPP accurately represented the methylation status of the target regions. Methylation levels for the two technical replicates of PGP1-iPSC were consistent both within a single batch and between separate batches (Pearson’s correlation coefficient R = 0.97 – 0.98, Supplementary Fig. 7a,b). Additionally, when methylation levels were compared between technical replicates, no CpG site was found to be significantly different by a Fisher Exact Test with Benjamini-Hochberg multiple testing correction (FDR = 0.01, n = 439,090). In comparison, large fractions of sites were found differentially methylated due to either the process of nuclear reprogramming (27.9% DMS between PGP1-iPSC and PGP1F) or the difference in cell type (31.3% DMS between PGP1F and H1) with the same criteria (FDR = 0.01, n = 444,111 and 359,290, respectively). Our BSPP results on H1 ESCs are highly consistent with the published whole genome bisulfite sequencing data12 (Pearson’s correlation coefficient R = 0.95, Supplementary Fig. 8).
Our assay has very low technical variability. We have performed the assay on over 150 samples in 96-well plates; the yield for each was similar (Supplementary Fig. 9). Approximately 10% of CpG sites are targeted separately on each strand, allowing low-quality data sets with poor correlation between these built-in technical replicates to be identified (Supplementary Fig. 7c,d,e). As our BSPP assay measures absolute methylation levels, no normalization is necessary as long as the internal replicates are consistent. Therefore, a large number of datasets, even generated from different laboratories, can be directly compared without batch effects, which is important for case-control studies on large samples or meta-analyses. Additionally, the SNP-calling feature of bisReadMapper allowed us to characterize roughly 20,000 SNPs for each sample at an accuracy of 96% or better. This allowed us to unambiguously track samples, which is crucial for projects involving large sample sizes.
Our library-free BSPP method is flexible for different study designs. While our genome-scale probe set allows global profiling on thousands of samples, a focused assay is often necessary to follow up on tens to hundreds of candidate regions identified in genome-scale scanning. Such an assay needs to be customizable to different genomic targets, scalable to a very large sample size (1,000-100,000), and inexpensive. To further demonstrate the flexibility, we designed a second set of 3,918 probes to evaluate the methylation state 1 kbp upstream and downstream of 120 genomic regions previously known and confirmed by BSPP to carry aberrant methylation in induced pluripotent stem cells15. We acquired the oligonucleotides from a second vendor (LC Sciences). Even with shorter capturing sequences (40 bp total for capturing arms rather than 50 bp on average, Supplementary Figure 10) and a 100-fold smaller target size, an average of 56% of mappable bases were on-target, equivalent to an enrichment factor of ~6,500. With the data from three cell lines (H1 ESCs, PGP1F, and PGP1-iPSCs) we were able to identify regions of aberrant methylation in iPSCs (Supplementary Fig. 11), and demonstrated that aberrant methylation continues further upstream and downstream than observed previously. This analysis demonstrates that a focused probe set can be used to validate specific regions of interest identified in global scanning using either our genome-wide probe set or other methods.
This method can be implemented to aid in the identifying the effects of DNA methylation in any organism by using the computational tools made available on the supporting website for this paper (http://genome-tech.ucsd.edu/public/Gen2_BSPP/).
ONLINE METHODS
Probe design and read mapping algorithms, as well as probe sequences and additional information are available at http://genome-tech.ucsd.edu/public/Gen2_BSPP/. Schematic for the padlock probes is illustrated in Supplementary Fig. 10.
Bisulfite padlock probe production (Oligonucleotides from Agilent)
Libraries of oligonucleotides (~150 nt) were synthesized by ink-jet printing on programmable microarrays (Agilent Technologies) and released to form a combined library of 330,000 oligonucleotides. The oligonucleotides were amplified by PCR in 96 reactions (100 μl each) with 0.02 nM template oligonucleotide, 400 nM each of pAP1V61U primer and AP2V6 primer (Supplementary Table 3), and 50 μl of KAPA SYBG fast Universal 2× qPCR Master Mix (Kapabiosystems) at 95 °C for 30 s, 15-16 cycles of 95 °C for 3 s; 55 °C for 30 s; and 60 °C for 20 s, and 60 °C for 2 min. The amplified amplicons were purified by ethanol precipitation and re-purified with Qiaquick PCR purification columns (Qiagen). Approximately 20 μg of the purified amplicons were digested with 50 units of Lambda Exonuclease (5 U/ μl; New England Biolabs (NEB)) at 37 °C for 1 h in lambda exonuclease reaction buffer. The resulting single-strand amplicons were purified with Qiaquick PCR purification column. Approximately 5-8 μg of single strand amplicons were subsequently digested with 5 units USER (1 U/μl, NEB) at 37 °C for 1 h. The digested DNAs were annealed to 5.88 μM RE-DpnII-V6 guide oligo (Supplementary Table 3) and denatured at 94 °C for 2 min decreased the temperature to 37 °C and incubated at 37 °C for 3 min. The mixture was digested with 50 units DpnII (10U/μl, NEB) in NEBuffer DpnII at 37 °C for 2 h. Then the mixture was further digested with 5 units USER at 37 °C for 2 h followed by enzyme inactivation at 75 °C for 20 min. The USER/DpnII digested DNAs were purified with Qiaquick PCR purification column. The single-strand 102 nucleotide probes were purified with 6% denaturing PAGE (6% TB-urea 2D gel; Invitrogen).
Bisulfite padlock probe production (Oligonucleotides from LC Sciences)
The oligonucleotides (100 nt) were synthesized using a programmable microfluidic microarray platform (LC Sciences) and released to form a mix of 3,918 oligoucleotides. The oligonucleotides were amplified by two-step PCR in a 200 μl reaction with 1nM template oligonucleotides, 400 nM each of eMIP_CA1_F primer and eMIP_CA1_R primer (Supplementary Table 3), and 100 μl of KAPA SYBR fast Universal qPCR Master Mix at 95 °C for 30 s, 5 cycles of 95 °C for 5 s; 52 °C for 1 min; and 72 °C for 30 s, 10-12 cycles of 95 °C for 5 s; 60 °C for 30 s; and 72 °C for 30sec, and 72 °C for 2 min. The resultant amplicons were purified with Qiaquick PCR purification columns and re-amplified by PCR in 32 reactions (100 μl each) with 0.02 nM first round amplicons, 400 nM each of eMIP_CA1_F primer and eMIP_CA1_R primer, and 50 μl of KAPA SYBR fast Universal qPCR Master Mix at 95 °C for 30 s, 13-15 cycles of 95 °C for 5 s; 60 °C for 30 sec; and 72 °C for 30 s, and 72 °C for 2 min. The resultant amplicons were purified by ethanol precipitation and re-purified with Qiaquick PCR purification columns as described above. Approximately 4 μg of the purified amplicons were digested with 100 units of Nt.AlwI (100 U/μl, NEB) at 37 °C for 1 h in NEBuffer 2. The enzyme was heat inactivated at 80 °C for 20 min. The digested amplicons were then incubated with 100 units of Nb.BrsDI (10 U/μl, NEB) at 65 °C for 1 h. The nicked DNA was purified by Qiaquick PCR purification column. The probe molecules (with size of approximately 70 bases) were purified by 6% denaturing PAGE (6% TB-urea 2D gel).
Sample preparation and capture
Genomic DNA was extracted using the AllPrep DNA/RNA Mini kit (Qiagen) and bisulfite converted with the EZ-96 DNA methylation Gold kit (Zymoresearch) in 96-well plate. Normalized amount of padlock probes, 200 ng of bisulfite converted gDNA, and 4.2 nM oligo suppressor were mixed in 25 μl 1× Ampligase Buffer (Epicentre) in 96-well plate, denatured at 95 °C for 10 min, gradually lowered the temperature at 0.02 °C/s to 55 °C in a thermocycler, and hybridized at 55 °C for 20 h. 2.5 μl of SLN mix (100 μM dNTP, 2 U/μl AmpliTaq Stoffel Fragment (ABI) and 0.5 U/μl Ampligase (Epicentre) in 1 × Ampligase buffer) was added to the reaction for gap-filling reaction. For circularization, the reactions were incubated at 55 °C for 20 h, followed by enzyme inactivation at 94 °C for 2 min. To digest linear DNA after circularization, 2 μl of exonuclease mix (10 U/μl exonuclease I and 100 U/μl exonuclease III, USB) was added to the reactions, and the reactions were incubated at 37 °C for 2 h then inactivated at 94 °C for 2 min.
Capture circles amplification (Library-free BSPP protocol, Agilent Oligonucleotides)
10 μl circularized DNA was amplified and barcoded in 100 μl reactions with 400 nM each of AmpF6.3Sol primer (Supplementary Table 3) and AmpR6.3 indexing primer (Supplementary Table 3), 0.4 × SYBR Green I (Invitrogen), and 50 μl Phusion High-Fidelity 2 × Master Mix (NEB) at 98 °C for 30 s, 5 cycles of 98 °C for 10 s; 58 °C for 20 s; and 72 °C for 20 s, 9-12 cycles of 98 °C for 10 s; and 72 °C for 20 s, and 72 °C for 3 min.
Capture circles amplification (Library-free BSPP protocol, LC Sciences Oligonucleotides)
10 μl circularized DNA was amplified in a 100 μl reaction with 200 nM each of CP-2-FA primer and CP-2-RA primer (Supplementary Table 3) and 50 μl KAPA SYBR fast Universal qPCR Master Mix at 98 °C for 30 s, 5 cycles of 98 °C for 10 s; 52 °C for 30 s; and 72 °C for 30 s, 15 cycles of 98 °C for 10 s; 60 °C for 30 s; and 72 °C for 30 s, and 72 °C for 3 min. The resultant amplicons with the corresponding expected size of approximately 260 bp were purified with 6% PAGE (6% 5-well gel, Invitrogen) and resuspended in 12 μl of TE buffer. 30% of the gel-purified amplicons were re-amplified and barcoded in a 100 μl reaction with 200 nM each of two different sets of primers to enable SE sequencing for both ends of the amplicons (CP-2-FA.IndSol primer and CP-2-RA.Sol primer or Switch.CP-2-FA and Switch.CP-2-RA.IndSol) and 50 μl KAPA SYBR fast Universal qPCR Master Mix at 98 °C for 30 s, 4 cycles of 98 °C for 10 s; 54 °C for 30 s; and 72 °C for 30 s, and 72 °C for 3 min.
Primer barcode design for multiplexing
An in-house perl script was written to randomly generate 6 nt long sequences. A sequence was kept if it does not have more than two matching positions with another accepted barcode and if it has between two to four guanine or cytosine. The script reiterates until the desired number of barcodes have been obtained. A total of 384 primers were designed (Supplementary Table 4).
Bisulfite read mapping and data analysis
Bisulfite converted data was processed as previously described. Reference genome is computationally converted by changing all C’s to T’s on Watson and Crick strands separately. FASTQ reads are encoded by 1) predicting the mapping orientation, 2) converting all predicted forward mapping reads by changing all C’s to T’s and converting all predicted reverse complementary mapping reads by changing all G’s to A’s, the original reads are maintained. The bisulfite reads are then mapped to the converted reference separately using SOAP2Align (http://soap.genomics.org.cn/soapaligner.html) with the parameters r = 0, v = 2 (one mismatch per 40bp sequenced), Paired-End: m = 0, × = 400. Alignment files are then combined, and one alignment per read was selected. Original C calls were placed back into the alignment information. Alignments are then converted to pileup format using SamTools (http://samtools.sourceforge.net/). Raw SNPs and methylation frequency files were computed from pileup counts. Methylation frequencies and SNPs were called using a method described previously1.
Correlation of methylation levels between two samples
To check if methylation levels were similar between two samples, the Pearson’s correlation was calculated on all CpG sites characterizable in both. First, a list of CpG sites with read depth of at least 10 in both samples was generated. The methylation frequencies at these sites were obtained from bisReadMapper output, and input into the statistical package R. Finally, Pearson’s correlation for the two samples was computed using the cor() function.
Analysis of differential methylation
From the bisReadMapper output, the raw read counts showing methylation and lack of methylation were assembled for each line. Using these counts, a Fisher-Exact Test with Benjamini-Hochberg Multiple Testing Correction (FDR = 0.01) was carried out on each CpG site with minimum 10x depth coverage. This resulted in a set of differentially methylated sites (DMSs) between the two lines; at each of these sites, the methylation levels were statistically significantly different with at least 0.1 methylation level difference. Technical replicates did not show any differential methylation, while different cell types showed a large degree (~33%).
Supplementary Material
ACKNOWLEDGEMENTS
We thank A. Feinberg and J. Stamatoyannopoulos for providing informative genomic targets and E. LeProust (Agilent Technology) for long-oligonucleotide synthesis. This work is funded by grants from National Institute of Health (R01 DA025779; R01 GM097253) to K.Z. A.G. is sponsored by a California Institute of Regenerative Medicine (CIRM) pre-doctoral fellowship.
Footnotes
AUTHOR CONTRIBUTIONS
K.Z. oversaw the project. D.D. and N.P. performed experiments and bioinformatic analyses. A.G. developed a probe design algorithm. R. S. and A.G. designed probes. H.F. performed sequencing. D.D., N.P., A.G. and K.Z. wrote the manuscript.
COMPETING FINANCIAL INTERESTS
Kun Zhang is a paid consultant for Pathogenica Inc. Robert Shoemaker is a current employee of Illumina Inc.
REFERENCES
- 1.Deng J, et al. Nat Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu GH, et al. Nature. 2011;472:221–225. doi: 10.1038/nature09879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu GH, et al. Cell Stem Cell. 2011;8:688–694. doi: 10.1016/j.stem.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Y, et al. Mol Cell. 2011;42:451–464. doi: 10.1016/j.molcel.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hansen KD, et al. Nature Genet. 2011 [Google Scholar]
- 6.Gore A, et al. Nature. 2011;471:63–67. doi: 10.1038/nature09805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, et al. Genome Res. 2010;20:981–988. doi: 10.1101/gr.102921.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Nat Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shoemaker R, Deng J, Wang W, Zhang K. Genome Res. 2010 doi: 10.1101/gr.104695.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irizarry RA, et al. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doi A, et al. Nat Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lister R, et al. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Figueroa ME, et al. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLean CY, et al. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lister R, et al. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.