

Summary
The direction of causation between measures of disrupted sleep, anxiety and depression is not well understood. Under certain conditions, cross sectional analysis based on genetically informative data can provide important information about the direction of causation between variables. Two community-based samples of 7,235 Australian twins aged 18 to 87 years were mailed an extensive questionnaire that covered a wide range of personality and behavioral measures. Included were self-report measures of disrupted sleep as well as symptoms of anxiety and depression. Among all females, modeling the direction of causation did not support the hypothesis of sleep having a direct causal impact on risk of anxiety or depression. Among older females, we found evidence that both anxiety and depression interact reciprocally with disrupted sleep whereas among younger women, both anxiety and depression appear to have a causal impact on sleep. Results for males were equivocal. The nosological implications of our findings are discussed.
Key words & terms: Disrupted sleep, anxiety, depression, twins, genes, environment, direction of causation
Introduction
The relationship between disturbed sleep, anxiety and depression is probably complex. Disturbed sleep is an associated feature of lifetime DSM-IV (American Psychiatric Association, 1994) generalized anxiety disorder or major depression (Johnson et al., 2006) as well as non-clinical measures of mood and affect (Dealberto, 1992). Defined in terms of difficulties in initiating or maintaining sleep or non-restorative sleep, disrupted sleep is one of the most common symptoms of mood and anxiety syndromes, occurring in up to 90% of patients with major depression (Tsuno et al., 2005). Among the diagnostic criteria, symptoms of disrupted sleep remain among the most persistent. For example, following remission periods of 12 weeks or more post treatment patients with recent histories of major depression or generalized anxiety disorder tend to continue experiencing symptoms of disrupted sleep (Dombrovski et al., 2007).
Evidence from twin and family studies has demonstrated that familial aggregation for the symptoms of disrupted sleep (Gehrman et al., 2011), generalized anxiety disorder (Hettema et al., 2001) and major depression (Sullivan et al., 2000) is each best explained by additive genetic effects. Twin studies have also shown that covariation between measures of disrupted sleep and recently experienced symptoms of anxiety and depression is attributable to common genetic and environmental effects (Gehrman et al., 2011). Circadian clock genes are considered the most likely candidates for genetic risks (Gehrman et al., 2011) whereas life stressors, early adverse childhood experiences, shift work, medical illnesses and medication may explain aspects of the non-shared environmental risks.
Notwithstanding differences arising from lifetime versus recent diagnoses or clinical versus non-clinical measures, disrupted sleep is prevalent and associated with mood and anxiety. However, the question of causality remains. Despite the short-lived anti-depressant effect of sleep deprivation in certain samples (Walker and van der Helm, 2009) a number of longitudinal studies have shown that symptoms of disrupted sleep are predictive of major depression especially in women (Baglioni et al., 2010). This is consistent with a ‘disrupted sleep causes depression’ uni-directional hypothesis. In three reviews (Riemann and Voderholzer, 2003, Harvey, 2011, Baglioni et al., 2011) of epidemiological studies with at least one follow-up suggest that overall, insomnia at baseline significantly predicts an increased risk of depression at follow-up one to three years later. Historically, a number of criteria have been proposed for assessing evidence of causality (see Hill, 1965). In the absence of (i) double-blind random case-control experiments, (ii) genetically informative discordant twin pair methods or (iii) cross-panel designs then any association in terms of causality versus correlated liability (whereby longitudinal phenotypic associations arise because of correlated, unmeasured background genetic or environment effects) cannot be determined. Only one report has employed a genetically informative longitudinal cross panel design to test and compare causal hypotheses (Gregory et al., 2009). Based on a small, ascertained sample of 300 twin pairs measured at ages eight and ten, sleep problems at age eight best predicted depression at age ten but not conversely. Moreover, the sleep-depression association was best explained by shared additive genetic risk. Although more formal tests of causality are required, in lieu of costly genetically informative longitudinal data, alternative and innovative statistical methods can be applied instead. One novel approach is to model causation based on pairs of genetically informative relatives measured on a single occasion (Heath et al., 1993) followed, where possible, by replication.
Direction of causation modeling on cross-sectional and genetically informative data has been applied to a number of complex behavioral phenotypes (Duffy and Martin, 1994, Neale et al., 1994, Gillespie et al., 2003). Provided several assumptions are satisfied (Heath et al., 1993), differences in the patterns of cross-twin cross-trait correlations can allow one to falsify strong hypotheses about the direction of causation between two variables measured on a single occasion. The power to do this is increased when there are differences in the causes of variation in one trait versus another. Figure 1 provides an illustrative example of this approach. Let us assume that variable A is best explained by a combination of shared (C) and non-shared (E) environmental effects while variable B is best explained by additive genetic (A), dominant genetic (D) and non-shared (E) environment effects. Using Wright’s (Wright, 1934) path tracing rules one can calculate how the ‘A-to-B’ and ‘B-to-A’ hypotheses each generate very different expected monozygotic and dizygotic cross-twin cross-trait correlations (e.g. correlation between twin-1 variable A [At1] and twin-2 variable B [Bt2]) whose goodness of fit can be compared with likelihood-ratio chi-squared tests.
Figure 1.

Uni-directional causal modeling between two variables with the expected cross-twin cross-trait correlations for monozygotic (rmz) and dizygotic (rdz) twin pairs (t1 & t2) under the (i) A causes B and (ii) B causes A hypotheses.
A, C, E and D refer to additive genetic, shared environment, non-shared environment and genetic dominance respectively.
Double headed arrows illustrate the expected twin pair (cross-twin) correlations. DZ twin pairs share on average half of the DNA so the expected twin pair correlations are ½ and ¼ for additive genetic and dominance effects respectively. Expected cross-twin cross-trait correlations are derived using Wright’s (1934) path tracing rules.
Unfortunately, the statistical power for resolving these alternative hypotheses in Figure 1 is greatly reduced because non-shared environmental variance components (E) necessarily contain measurement error. Our remedy was to use multiple indicators and then model the causation between latent constructs (Heath et al., 1993) which assumes that measurement error occurs, not at the latent variable level but at the level of the indicator variables and is uncorrelated across the indicator variables.
Our aim was to fit and compare a series of competing direction of causation hypotheses using cross sectional data comprising self-report measures of disrupted sleep and symptoms of anxiety, as well as disrupted sleep and symptoms of depression. This study is based on two much larger population-based adult twin cohorts that permit us to replicate findings as well as fit separate DOC models across sex. We fitted a model that predicted that the association between disrupted sleep and symptoms of anxiety (or depression) was explained by shared genetic and environmental risk factors, a model that predicted reciprocal causation between A and B, two uni-directional causation models (A-to-B and B-to-A) and finally a model that predicted no association at all.
Method
Sample
Data come from two twin cohorts separately ascertained through the Australian National Health and Medical Research Council (NHMRC) and Australian Twin Register (ATR). This is a volunteer registry founded in 1978 with approximately 25,000 pairs of all types and all ages enrolled and in various stages of active contact. Informed consent was obtained from all participants prior to assessment. Recruitment protocols were independently reviewed and approved by the Queensland Institute of Medical Research Human Research Ethics Committee that was established in accordance with the guidelines set out by the Australian NHMRC as well as the ATR. We estimate that this represents 10–20% of living twins in Australia. Numerous analyses have shown that these twins are typical of the Australian population in many respects including the prevalence of psychiatric symptoms although the sample tends to be slightly more middle class and educated than average, particularly for males (Baker et al., 1996).
The first cohort (Cohort 1) consists of 3808 twin pairs born before 1964 and a younger cohort (Cohort 2) of 4269 twin pairs born 1964–74. An advantage of using these two separately ascertained population based twin samples is that the second cohort can be used for replication of findings from the first cohort. In order to improve our power to resolve direction of causation we jointly analyzed male and female twin data within each cohort.
First surveyed in 1980–82, data for the older Cohort 1 come from a follow-up between 1988–90 to investigate persistence and changes in drinking habits. Data for these analyses are based on a follow-up survey containing a self-report Health and Lifestyle Questionnaire (HLQ) that incorporated many of the questions sent out to the same twins eight years previously. The HLQ assessments included: age, sex, zygosity, tobacco use, alcohol consumption, personality, socio-demographic variables, psychiatric symptoms, and numerous other behavioural measures. In order to reduce postage cost and maximize response, considerable effort was made to verify the addresses of twins prior to mailing. However, after an eight-year hiatus since completing the first questionnaire, large numbers of twins were lost and extensive efforts were made to locate these twins in 1988–1990. This involved telephoning non-responding twins, their co twins or the parents who had initially enrolled them.
Data for the younger Cohort 2 come from a 1989 study using a self-report questionnaire containing many of the same assessments used in the follow up questionnaire for Cohort 1. Cohort 2 had been recruited when at school some ten years earlier as part of the earlier study, so it was not surprising that, despite extensive follow-up efforts, we were unable to re-establish contact with 1000 pairs. Twins who failed to return a completed questionnaire were followed-up by telephone up to five times, at which point they were asked to complete an abbreviated telephone interview to obtain basic demographic information only.
Measures& construct validity
Apart from assessing general demographic information, the HLQ’s sent to both cohorts included the same measures of personality, social behavior and attitudes, psychiatric symptoms, general health / illness and the occurrence of life stressors. Zygosity was determined based on twins’ responses to standard questions about similarity and the degree to which others confused them.
The data analyzed in this paper included three disrupted sleep (SD), nine anxiety and seven depression items based on factor analyses of the Delusion Symptoms States Inventory and Symptom Checklist (SCL-90)(see Gillespie et al., 2003). Summarized in Table 1, all items were phrased to conform to the DDSI/sAD format of inquiry, “Recently I have had…” rather than use the SCL-90 format, “In the past two weeks…”. For the questionnaire, a four-point ordinal response set was used (“not-at-all”, “a little”, “a lot”, “unbearably”). For each symptom the 2nd, 3rd and 4th response categories were combined in order to improve computational efficiency.
Table 1.
Disrupted sleep, depression and anxiety items based on factor analyses (Gillespie, 2003) of the Delusion Symptoms States Inventory (Bedford & Deary, 1997; Foulds & Bedford, 1975)and Symptom Checklist (SCL-90) (Derogatis, 1973).
| Disrupted Sleep |
| 1. So miserable that I have had difficulty with my sleep |
| 2. Worry has kept me awake all night |
| 3. Have woken early in the morning |
| Depression |
| 1. Been depressed without knowing why |
| 2. Gone to bed not caring if I never awake |
| 3. Low in spirits, sit for ages and do nothing |
| 4. Future seems hopeless |
| 5. Lost interest in just about everything |
| 6. So depressed thoughts of doing away with myself |
| 7. Felt worthless |
| Anxiety |
| 1. Feelings of panic for no reason |
| 2. Felt nervous or shaky inside |
| 3. Felt fearful |
| 4. Had spells of terror or panic |
| 5. Felt afraid in open spaces or in street |
| 6. Afraid to travel on buses or trains |
| 7. Avoid certain things that frighten me |
| 8. Felt uneasy in crowds |
| 9. Felt nervous when left alone |
It is important to note that although our analyses were not based on clinical diagnoses, there is strong empirical evidence which has convincingly shown that these brief symptom scales are not only optimal screening instruments but also capture most of the genetic and large proportions of the environmental variance in lifetime internalizing disorders including lifetime major depression and generalized anxiety disorder (Foley et al., 2001). Previous analyses have shown that single latent factor models can best explain variation in the anxiety and depression symptoms (Gillespie et al., 2003) which are internally reliable and have modest heritability (Gillespie et al., 2000).
Preliminary analyses show that the age and sex adjusted correlations between the three sleep items were moderate to high. The first to second eigenvalue ratio, averaged across cohort and sex, was 2.1 to 0.7 strongly indicating a best fitting single latent factor model. Cronbach alphas for the disrupted sleep factor when adjusted for the effects of sex and age were 0.65 and 0.61 for Cohort 1 and Cohort 2 respectively.
For Cohort 1, the number of complete and incomplete female monozygotic (MZ) twin pairs with sleep, depression and anxiety data were 895 and 142 respectively. The number of complete and incomplete female dizygotic (DZ) twin pairs with sleep, depression and anxiety data were 519 and 112 respectively. The numbers of complete and incomplete male MZ twin pairs with disrupted sleep, depression and anxiety data were approximately 386 and 84 respectively. For DZ males, the numbers of complete and incomplete twin pairs were approximately 220 and 76 respectively.
For Cohort 2, the number of complete and incomplete female MZ twin pairs with sleep, depression and anxiety data were 446 and 118 respectively whereas the numbers of complete and incomplete female DZ twin pairs were 302 and 138 respectively. The numbers of complete and incomplete male MZ twin pairs were 248 and 87 respectively while for DZ males the same numbers were 160 and 126 respectively.
Latent factor score estimation& ordinal data analysis
Because measurement error reduces the statistical power for resolving alternative causal hypotheses (Neale et al., 1994) our solution (shown in Figures 2a and 2b) was to model the association between latent disrupted sleep, anxiety and depression factor scores. This approach assumes that measurement error occurs at the level of indicator variables or observed symptoms and so is uncorrelated across factors (Heath et al., 1993). The disrupted sleep, anxiety and depression factors were based on the within factor item correlations, internal reliabilities and previous factor analyses of the same DSSI and SCL-90 items (Gillespie et al., 2003). After fitting single dimension factor structural models to the disrupted sleep, anxiety and depression items we calculated individual factor scores, using Maximum Likelihood estimation, in the Mx software package (Neale et al., 2006), which were then transformed onto 3-point ordinal scales to reduce skew and permit ordinal data analyses.
Figure 2.

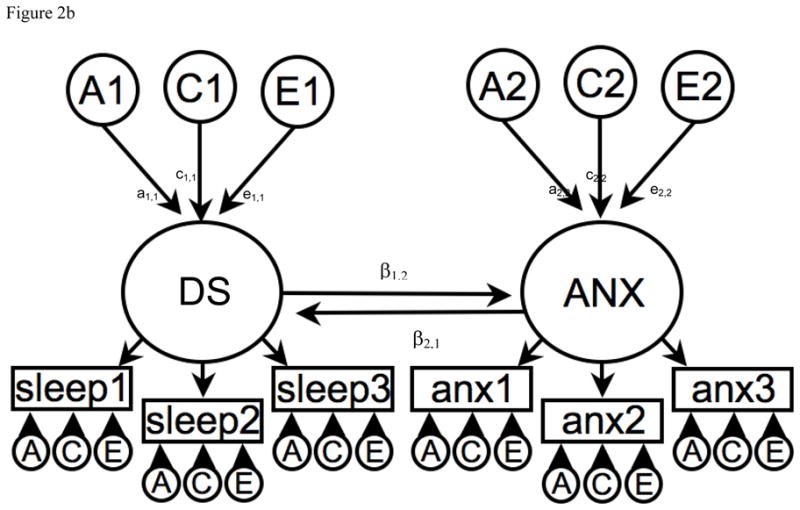
Figure 2a. Correlated liabilities model to explain the phenotypic association between Disrupted Sleep (DS) and Anxiety (ANX).
A1, C1 & E1 = latent additive genetic shared and non-shared environmental risks for Disrupted Sleep (DS).
A2, C2 & E2 = latent additive genetic shared and non-shared environmental risks for Anxiety (ANX).
DS and ANX are common factors indicated by observed phenotypic symptoms for sleep 1–3 and anx 1–3 respectively which have their own item specific latent genetic and environmental risk factors.
The non-causal association is represented by the pathway coefficients between the DS and ANX common factors i.e. via a1,1a2,1, c1,1c2,1 and e1,1e2,1.
Figure 2b. Reciprocal interaction model to explain the phenotypic association between Disrupted Sleep (DS) and Anxiety (ANX).
A1, C1 & E1 = latent additive genetic shared and non-shared environmental risks for Disrupted Sleep (DS).
A2, C2 & E2 = latent additive genetic shared and non-shared environmental risks for Anxiety (ANX).
DS and ANX are common factors indicated by observed phenotypic symptoms for sleep 1–3 and anx 1–3 respectively which have their own item specific latent genetic and environmental risk factors.
The causal association is represented between the DS and ANX common factors i.e. β1,2 from DS to ANX and β2,1 from ANX to DS.
Based on multivariate normal theory we then applied raw data methods to the recoded ordinal data. This enabled the preliminary testing of basic assumptions concerning the equality of response (threshold) distributions within twin pairs and across zygosity. This method is analogous to testing the equality of means and covariance structure when analyzing continuous data (Lange et al., 1976). For all items, except for Anxiety in Cohort 1 (p=0.04), there was no significant change in the model fit when the thresholds were equated within twin pairs and across zygosity. The marginally significant threshold difference for Anxiety was well within chance expectations.
Univariate & multivariate genetic analyses
Based on standard biometrical genetic model fitting methods (Neale and Cardon, 1992) which exploit the expected the genetic and environmental correlations for MZ and DZ twin pairs, our models assumed that the total variance in an observed variable can be decomposed into additive (A) genetic, shared environment (C) and non-shared or unique (E) environmental variance components. Because MZ twin pairs are genetically identical correlations for the A effects were 1.0 whereas for DZ twin pairs, who on average share half of their genes, the correlations for the A effects were 0.5. An important assumption of biometrical models is that shared environmental effects (C) correlate to an equal extent in MZ and DZ twin pairs. Non-shared environmental effects are by definition uncorrelated and also reflect measurement error including short-term fluctuations.
We used these methods first to estimate the contribution of genetic and environmental risks (A, C and E) in the univariate analyses of the disrupted sleep, anxiety and depression factors. In the multivariate analyses we then tested the fit of five direction of causation models using Maximum Likelihood estimation in the Mx software package (Neale et al., 2006). The models were: (i) a comparison or correlated liability model illustrated in Figure 2a; (ii) a reciprocal causation model illustrated in Figure 2b; (iii-iv) two uni-directional causation models; and (v) one non-causal no association model. The correlated liability model is identical to a bivariate Cholesky decomposition and is the null hypothesis because it predicts no causal association between the latent disrupted sleep, anxiety or depression factors. Instead, any phenotypic association is attributable to common or correlated genetic and environmental latent effects i.e. via the a11 and a21, c11 and c21, and e11 and e21 pathways. Under the reciprocal causation and uni-directional models any association between disrupted sleep and anxiety or depression arises because of direct phenotypic causality i.e. β12 and β21, between the latent sleep, anxiety or depression factors. Under the non-causal no association model, all causal pathways between disrupted sleep and anxiety or depression are removed.
Model comparisons
The reciprocal and uni-directional causation models were nested within the null correlated liability model and because we were modeling three sources of variance (A, C and E) on each phenotypic construct (SD, anxiety and depression), the goodness-of-fit for the full ACE correlated liability was compared to the sub-models using likelihood-ratio chi-squared tests. The best fitting model was chosen on the basis of parsimony i.e. non-significant changes in the chi-square and the smallest number of parameters. To this end, the Akaike Information Criterion (AIC) was calculated for each model and the model with the lowest index value was chosen as the best fitting.
Results
Univariate analyses
Standardized variance components attributable to additive genetic (A), shared environment (C) and non-shared environment (E) effects are summarized in Table 2. For all three factors, familial aggregation was entirely explained by additive genetic effects ranging from 10% to 49% with no evidence of shared environmental variance.
Table 2.
Univariate analyses: standardized variance components attributed to additive genetic (A), shared environment (C), and non-shared environmental (E) effects.
| Females | Males | |||||
|---|---|---|---|---|---|---|
| Cohort 1 | A | C | E | A | C | E |
| Disrupted Sleep | 0.33 | 0.00 | 0.67 | 0.10 | 0.00 | 0.90 |
| Anxiety | 0.40 | 0.00 | 0.60 | 0.49 | 0.00 | 0.51 |
| Depression | 0.35 | 0.00 | 0.65 | 0.35 | 0.00 | 0.65 |
| Cohort 2 | A | C | E | A | C | E |
| Disrupted Sleep | 0.20 | 0.00 | 0.80 | 0.29 | 0.00 | 0.71 |
| Anxiety | 0.37 | 0.00 | 0.63 | 0.34 | 0.00 | 0.66 |
| Depression | 0.28 | 0.00 | 0.72 | 0.37 | 0.00 | 0.63 |
Polychoric correlations between latent factors
Based on the null or correlated liability model, polychoric correlations between the recoded latent factor scores for disrupted sleep, anxiety and depression appear in Table 3. The within-twin cross-trait correlations were all high whereas the cross-twin cross-trait correlations ranged from small to moderate across sex and cohort. For Cohort 1, the male cross-twin cross-trait correlations were lower than the same female correlations whereas in Cohort 2, the cross-twin cross-trait correlations observed in males were either identical or similar to the female correlations.
Table 3.
Polychoric correlations between latent factor scores for Disrupted Sleep, Anxiety and Depression based on the correlated liability (Cholesky decomposition) null model. Males are above the diagonal shaded. The cross-twin cross-trait correlations (constrained equal within twin pairs) appear in bold.
| Sleep & Anxiety | Sleep & Depression | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 | 1. | 2. | 3. | 4. | 1. | 2. | 3. | 4. | ||
| 1. Twin 1 Sleep | 1 | 0.52 | 0.16 | 0.17 | 1. Twin 1 Sleep | 1 | 0.49 | 0.13 | 0.09 | |
| 2. Twin 1 Anxiety | 0.55 | 1 | 0.17 | 0.44 | 2. Twin 1 Depression | 0.58 | 1 | 0.09 | 0.31 | |
| 3. Twin 2 Sleep | 0.33 | 0.27 | 1 | 0.52 | 3. Twin 2 Sleep | 0.33 | 0.26 | 1 | 0.49 | |
| 4. Twin 2 Anxiety | 0.27 | 0.40 | 0.55 | 1 | 4. Twin 2 Depression | 0.26 | 0.35 | 0.58 | 1 | |
| Cohort 2 | 1. | 2. | 3. | 4. | 1. | 2. | 3. | 4. | ||
| 1. Twin 1 Sleep | 1 | 0.58 | 0.29 | 0.23 | 1. Twin 1 Sleep | 1 | 0.51 | 0.29 | 0.17 | |
| 2. Twin 1 Anxiety | 0.58 | 1 | 0.23 | 0.33 | 2. Twin 1 Depression | 0.66 | 1 | 0.17 | 0.35 | |
| 3. Twin 2 Sleep | 0.20 | 0.20 | 1 | 0.58 | 3. Twin 2 Sleep | 0.20 | 0.17 | 1 | 0.51 | |
| 4. Twin 2 Anxiety | 0.20 | 0.38 | 0.58 | 1 | 4. Twin 2 Depression | 0.17 | 0.28 | 0.66 | 1 | |
Direction of causation modeling fitting
Results for modeling the association between disrupted sleep and anxiety using data from Cohort 1 and Cohort 2 appear in Table 4. In both cohorts and for males and females alike, the model with no phenotypic causal-association when compared to the correlated liability model failed in all cases.
Table 4.
Direction of causation model fitting results for males and females in Cohort 1 and Cohort 2.
| (a) | Females | Males | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sleep & Anxiety | χ2 | df | Δχ2 | Δdf | p | AIC | χ2 | df | Δχ2 | Δdf | p | AIC | |
| Cohort 1 | Correlated liability | 11904.94 | 6153 | - | - | - | −01.06 | 4943.752 | 2661 | - | - | - | −78.25 |
| Reciprocal causation | 11904.98 | 6154 | 0.04 | 1 | 0.843 | −403.02 | 4944.969 | 2662 | 1.22 | 1 | 0.270 | −379.03 | |
| Anxiety →DS | 11908.82 | 6155 | 3.88 | 2 | 0.144 | −401.18 | 4947.896 | 2663 | 4.14 | 2 | 0.126 | −378.10 | |
| DS→Anxiety | 11916.98 | 6155 | 12.04 | 2 | 0.002 | −393.02 | 4948.211 | 2663 | 4.46 | 2 | 0.108 | −377.79 | |
| No association | 12486.82 | 6156 | 581.88 | 3 | 0.000 | 174.82 | 5122.440 | 2664 | 178.69 | 3 | 0.000 | −205.56 | |
| Sleep & Depression | |||||||||||||
| Correlated liability | 11462.78 | 6153 | - | - | - | −843.22 | 5083.97 | 2662 | - | - | - | −240.03 | |
| Reciprocal causation | 11463.98 | 6154 | 1.20 | 1 | 0.273 | −844.02 | 5083.97 | 2663 | 0.00 | 1 | 0.975 | −242.03 | |
| Depression→DS | 11468.72 | 6155 | 5.94 | 2 | 0.051 | −841.28 | 5088.21 | 2664 | 4.23 | 2 | 0.120 | −239.80 | |
| DS→Depression | 11471.21 | 6155 | 8.44 | 2 | 0.015 | −838.79 | 5084.42 | 2664 | 0.45 | 2 | 0.799 | −243.58 | |
| No association | 12108.52 | 6156 | 645.74 | 3 | 0.000 | −203.48 | 5248.94 | 2665 | 164.97 | 3 | 0.000 | −81.06 | |
| (b) | Females | Males | |||||||||||
| Sleep & Anxiety | χ2 | df | Δχ2 | Δdf | p | AIC | χ2 | df | Δχ2 | Δdf | p | AIC | |
| Cohort 2 | Correlated liability | 6898.64 | 3493 | - | - | - | −87.36 | 4029.74 | 2045 | - | - | - | −60.26 |
| Reciprocal causation | 6898.64 | 3494 | 0.000 | 1 | 1.000 | −89.36 | 4029.82 | 2046 | 0.09 | 1 | 0.771 | −62.18 | |
| Anxiety →DS | 6898.98 | 3495 | 0.333 | 2 | 0.847 | −91.03 | 4030.52 | 2047 | 0.78 | 2 | 0.677 | −63.48 | |
| DS→Anxiety | 6903.36 | 3495 | 4.720 | 2 | 0.094 | −86.64 | 4031.55 | 2047 | 1.82 | 2 | 0.404 | −62.45 | |
| No association | 7275.63 | 3496 | 376.99 | 3 | 0.000 | 283.63 | 4255.48 | 2048 | 225.74 | 3 | 0.000 | 159.48 | |
| Sleep & Depression | |||||||||||||
| Correlated liability | 6933.98 | 3493 | - | - | - | −52.02 | 4199.82 | 2047 | - | - | - | 105.82 | |
| Reciprocal causation | 6933.99 | 3494 | 0.007 | 1 | 0.933 | −54.01 | 4199.84 | 2048 | 0.02 | 1 | 0.879 | 103.84 | |
| Depression→DS | 6934.40 | 3495 | 0.412 | 2 | 0.814 | −55.61 | 4200.25 | 2049 | 0.43 | 2 | 0.806 | 102.25 | |
| DS→Depression | 6934.84 | 3495 | 0.858 | 2 | 0.651 | −55.16 | 4200.14 | 2049 | 0.32 | 2 | 0.852 | 102.14 | |
| No association | 7458.80 | 3496 | 524.81 | 3 | 0.000 | 466.80 | 4379.86 | 2050 | 180.04 | 3 | 0.000 | 279.86 | |
AIC=Akaike Information Criteria
Females
For older females in Cohort 1 the unidirectional disrupted sleep-to-anxiety and disrupted sleep-to-depression could be rejected. Although both the anxiety-to-disrupted sleep and depression-to-sleep provided a good fit to the data, the reciprocal causation models had the lowest AICs and were judged as the best fitting.
For the younger females in Cohort 2 the unidirectional disrupted sleep-to-anxiety and disrupted sleep-to-depression could again be rejected. Both reciprocal causation models provided a good fit to the data. However, the uni-directional anxiety-to-disrupted sleep and depression-to-disrupted sleep models could not be rejected either, and with the lowest AIC values these models were judged as the best fitting.
Males
For the older males in Cohort 1 we found an almost identical pattern of results for anxiety. Although both unidirectional models provided a good fit to the data, the reciprocal with the lowest AIC value was judged as the best fitting. For depression, results were different; the disrupted sleep-to-depression model was marginally better than the reciprocal.
For males in Cohort 2, both unidirectional causation models in the anxiety and depression analyses provided a good fit to the data, however based on the AIC values, it was the anxiety-to-disturbance which provided the best fits to the data whereas for depression the reciprocal causation model proved a better fit.
Follow-up analyses
Because our causal modeling relied on disrupted sleep latent factor scores we tested the possibility that the three self-reported sleep items were differentially related to our measures of anxiety or depression. For instance, one item deals with difficulty initiating sleep while another deals with early-morning waking. Clinical lore also predicts that problems with initiating sleep are correlated with anxiety while problems of early morning awakening are associated with depression, there is little empirical evidence to support this distinction (see Benca et al., 1992). In follow-up analyses based on re-coded ordinal sum scores using males from both cohorts we found that the correlations between depression and anxiety with the difficulty initiating sleep item ranged from 0.41 to 0.44. Although lower, the correlations between depression and anxiety with early-morning waking were again very similar ranging from 0.24 to 0.26. Although the eigenvalues and results from our factor analyses and measures of internal consistency suggested these items were indexing an internally reliable construct we nevertheless estimated the degree of genetic and environmental overlap between the disrupted sleep symptoms. The correlations in Table 5 suggested some environmental specificity particularly for early morning wakening whereas the additive genetic correlations indicated that these three items were mostly indexing the same genetic liability or risk. As illustrated in Figure 2b, we modeled causality between latent factors capturing common variance. The advantage of this approach is that causal parameters were unbiased by any symptom specific effects; along with measurement error, these effects were assumed to occur at the level of the individual indicator variables.
Table 5.
Phenotypic polychoric correlations between the three Disrupted Sleep items and correlations between the latent additive genetic and non-shared environmental factors underpinning variation in the same items.
| Phenotypic correlations | Additive genetic factor correlations | Non-shared env’ correlations | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1. So miserable that I have had difficulty with my sleep2 | 1.00 | 1.00 | 1.00 | ||||||
| 2. Worry has kept me awake all night11 | 0.80 | 1.00 | 0.99 | 1.00 | 0.72 | 1.00 | |||
| 3. Have woken early in the morning26 | 0.45 | 0.47 | 1.00 | 0.70 | 0.59 | 1.00 | 0.36 | 0.44 | 1.00 |
All correlations adjusted for the effects of age at interview, sex and cohort on item thresholds using the definition variable option in the Mx software package (Neale et al., 2006).
Discussion
Univariate estimates of the genetic and environmental risks in depression, anxiety and disrupted sleep are comparable to those based on the same or similar items reported elsewhere (Gehrman et al., 2011). Although risks of disrupted sleep, anxiety and depression can each be explained by a combination of latent genetic and unique environmental risk factors our modeling suggests that the observed phenotypic associations cannot be explained by shared or correlated latent genetic and environmental risks. Instead, among older females the relationship between disrupted sleep and anxiety or disrupted sleep and depression was best explained by reciprocal causation whereas among younger women the association appears causal with anxiety and depression both having a causal impact on the liability to disrupted sleep. We found no evidence to support disrupted sleep having a causal impact on the symptoms of anxiety of depression. For males, results were equivocal. There was no clear trend emerging in terms of the reciprocal versus unidirectional models. This was likely attributable to the smaller sample sizes and cross-twin cross-trait correlations for males.
Modeling anxiety, depression and disrupted sleep at the latent factor level provided an optimal means of explaining the covariance between symptoms while testing causal hypotheses. In contrast to this approach, Borsboom (Borsboom et al., 2003) has suggested that psychiatric disorders can be better explained by correlated networks of symptoms, as opposed to unobserved latent factors that causally interact. One recent study (Cramer et al., 2010) compared these two models using DSM-IV criteria for major depression in a large community sample. The authors found that a correlated network model in which the symptoms of anxiety and sleep difficulties act and interact with each other causally fit the observed data much better than modeling between common factors. It is important to note that although our conceptual framework was not exhaustive and that alternate approaches might include latent class or factor mixture modeling the symptom data. Nevertheless, our method provided a practical means of attenuating the contribution of measurement error form the causal parameters. Moreover, as part of a robust nosological approach, this latent variable methods remain more parsimonious and have been very generative in studies of psychopathology (Krueger et al., 2010).
Limitations
Our findings must be interpreted in the context of three potential limitations. The first is power. We estimated in Mx the sample sizes required to reject with 80% power the reciprocal causation model in favor of the (i) anxiety to disrupted sleep and (ii) disrupted sleep to anxiety models. In Cohort 1 the samples were 12,707 and 262 whereas for Cohort 2 the samples were 929,156 and 232 respectively indicating that there was sufficient power to reject the disrupted sleep causes anxiety model.
Second, this study relied on self-reported sleep problems. Modeling direction of causation in future will be improved with more objective measures such as actigraphy or polysomnography, or by using more commonly employed clinical thresholds of disrupted sleep based on severity, frequency and duration (Gehrman et al., 2011).
Three, our modeling was not exhaustive. In a review, Baglioni (Baglioni et al., 2010) suggests that insomnia is linked to dysregulation of emotional reactivity and argues that associations between insomnia and psychiatric disorders are regulated by deficits in emotional regulation that result in higher negative and lower positive emotions. Whether this ‘modulation’ is describing mediation or moderation in the Baron and Kenny (Baron and Kenny, 1986) sense is unclear. However, the extent to which the association between disrupted sleep and depression (or anxiety) is dependent upon variation in emotional reactivity suggests that it is less causal and more attributable to a correlated liability to emotional reactivity. This would be analogous to accumulated evidence from twin studies suggests that the association between Neuroticism, Anxiety and Depression showing a non-causal relationship best predicted by correlated or shared genetic risk factors (Kendler et al., 1986). Future studies could test the improvement of model fit when including Neuroticism (or correlated measures of emotional lability) as a mediator or moderator of the causal pathways between sleep disruption and depression or anxiety.
Conclusions
We found no evidence to suggest that disrupted sleep causes anxiety or depression in females. Among older females, the relationship between disrupted sleep and anxiety or disrupted sleep and depression appears reciprocal whereas in younger women both anxiety and depression appear to have a causal impact on sleep. Results for males were equivocal.
Acknowledgments
Funding was received from the US National Institute on Drug Abuse (R00DA023549, 1K99DA023549-01A2). This work was supported by NIH grants AA07535, AA07728, AA10249, AA11998, AA13321, AA13326 and R21-MH-079187 and Australia National Health and Medical Research Council grants 941177 and 971232. We also thank the twins for their cooperation.
Footnotes
Conflict of interests: All authors have declared no conflict of interest.
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders: DSM-IV. 4 American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- Baglioni C, Battagliese G, Feige B, et al. Insomnia as a predictor of depression: a meta-analytic evaluation of longitudinal epidemiological studies. J Affect Disord. 2011;135:10–9. doi: 10.1016/j.jad.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Baglioni C, Spiegelhalder K, Lombardo C, Riemann D. Sleep and emotions: a focus on insomnia. Sleep Med Rev. 2010;14:227–38. doi: 10.1016/j.smrv.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Baker LA, Treloar SA, Reynolds CA, Heath AC, Martin NG. Genetics of educational attainment in Australian twins: sex differences and secular changes. Behav Genet. 1996;26:89–102. doi: 10.1007/BF02359887. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: Conceptual, strategic, and statistical consideration. J Pers Soc Psychol. 1986;51:1173–82. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- Benca RM, Obermeyer WH, Thisted RA, Gillin JC. Sleep and psychiatric disorders. A meta-analysis. Arch Gen Psychiatry. 1992;49:651–68. doi: 10.1001/archpsyc.1992.01820080059010. discussion 69–70. [DOI] [PubMed] [Google Scholar]
- Borsboom D, Mellenbergh GJ, Van Heerden J. The theoretical status of latent variables. Psychol Rev. 2003;110:203–19. doi: 10.1037/0033-295X.110.2.203. [DOI] [PubMed] [Google Scholar]
- Cramer AO, Waldorp LJ, Van Der Maas HL, Borsboom D. Comorbidity: a network perspective. The Behavioral and brain sciences. 2010;33:137–50. doi: 10.1017/S0140525X09991567. discussion 50–93. [DOI] [PubMed] [Google Scholar]
- Dealberto MJ. Sleep disorders in psychiatric diseases. Epidemiological aspects. L’Encephale. 1992;18:331–40. [PubMed] [Google Scholar]
- Dombrovski AY, Mulsant BH, Houck PR, et al. Residual symptoms and recurrence during maintenance treatment of late-life depression. J Affect Disord. 2007;103:77–82. doi: 10.1016/j.jad.2007.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy DL, Martin NG. Inferring the direction of causation in cross-sectional twin data: theoretical and empirical considerations [see comments] Genet Epidemiol. 1994;11:483–502. doi: 10.1002/gepi.1370110606. [DOI] [PubMed] [Google Scholar]
- Foley DL, Neale MC, Kendler KS. Genetic and environmental risk factors for depression assessed by subject-rated symptom check list versus structured clinical interview. Psychol Med. 2001;31:1413–23. doi: 10.1017/s0033291701004755. [DOI] [PubMed] [Google Scholar]
- Gehrman PR, Byrne E, Gillespie NA, Martin NG. The genetics of insomnia. Sleep Medicine Clinics. 2011;6:191–202. [Google Scholar]
- Gillespie NA, Zhu G, Heath AC, Hickie IB, Martin NG. The genetic aetiology of somatic distress. Psychol Med. 2000;30:1051–61. doi: 10.1017/s0033291799002640. [DOI] [PubMed] [Google Scholar]
- Gillespie NA, Zhu G, Neale MC, Heath AC, Martin NG. Direction of causation modeling between measures of distress and parental bonding. Behav Genet. 2003;33:383–96. doi: 10.1023/a:1025365325016. [DOI] [PubMed] [Google Scholar]
- Gregory AM, Rijsdijk FV, Lau JY, Dahl RE, Eley TC. The direction of longitudinal associations between sleep problems and depression symptoms: a study of twins aged 8 and 10 years. Sleep. 2009;32:189–99. doi: 10.1093/sleep/32.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey AG. Sleep and circadian functioning: critical mechanisms in the mood disorders? Annu Rev Clin Psychol. 2011;7:297–319. doi: 10.1146/annurev-clinpsy-032210-104550. [DOI] [PubMed] [Google Scholar]
- Heath AC, Kessler RC, Neale MC, Hewitt JK, Eaves LJ, Kendler KS. Testing hypotheses about direction of causation using cross-sectional family data. Behav Genet. 1993;23:29–50. doi: 10.1007/BF01067552. [DOI] [PubMed] [Google Scholar]
- Hettema JM, Neale MC, Kendler KS. A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am J Psychiatry. 2001;158:1568–78. doi: 10.1176/appi.ajp.158.10.1568. [DOI] [PubMed] [Google Scholar]
- Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965;58:295–300. doi: 10.1177/003591576505800503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EO, Roth T, Breslau N. The association of insomnia with anxiety disorders and depression: exploration of the direction of risk. J Psychiatr Res. 2006;40:700–08. doi: 10.1016/j.jpsychires.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Heath A, Martin NG, Eaves LJ. Symptoms of anxiety and depression in a volunteer twin population. The etiologic role of genetic and environmental factors. Arch Gen Psychiatry. 1986;43:213–21. doi: 10.1001/archpsyc.1986.01800030023002. [DOI] [PubMed] [Google Scholar]
- Krueger RF, Deyoung CG, Markon KE. Toward scientifically useful quantitative models of psychopathology: the importance of a comparative approach. The Behavioral and brain sciences. 2010;33:163–64. doi: 10.1017/S0140525X10000646. [DOI] [PubMed] [Google Scholar]
- Lange K, Westlake J, Spence MA. Extensions to pedigree analysis. II. Recurrence risk calculation under the polygenic threshold model. Human Hereditary. 1976;26:337–48. doi: 10.1159/000152825. [DOI] [PubMed] [Google Scholar]
- Neale MC, Boker SM, Xie G, Maes HH. Mx: Statistical Modeling. 7 Box 126 MCV, Richmond, VA 23298: Department of Psychiatry; 2006. [Google Scholar]
- Neale MC, Cardon LR. Methodology for Genetic Studies of Twins and Families. 1 Kluwer Academic Publishers; Dordrecht: 1992. [Google Scholar]
- Neale MC, Walters E, Heath AC, et al. Depression and parental bonding: cause, consequence, or genetic covariance? Genet Epidemiol. 1994;11:503–22. doi: 10.1002/gepi.1370110607. [DOI] [PubMed] [Google Scholar]
- Riemann D, Voderholzer U. Primary insomnia: a risk factor to develop depression? J Affect Disord. 2003;76:255–9. doi: 10.1016/s0165-0327(02)00072-1. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic Epidemiology of Major Depression: Review and Meta-Analysis. Am J Psychiatry. 2000;157:1552–62. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- Tsuno N, Besset A, Ritchie K. Sleep and depression. J Clin Psychiatry. 2005;66:1254–69. doi: 10.4088/jcp.v66n1008. [DOI] [PubMed] [Google Scholar]
- Walker MP, Van Der Helm E. Overnight therapy? The role of sleep in emotional brain processing. Psychol Bull. 2009;135:731–48. doi: 10.1037/a0016570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S. The method of path coefficients. Annals of Mathematical Statistics. 1934;5:161–215. [Google Scholar]