

Abstract
The unfolded protein response is a set of cell signaling pathways recently recognized to be activated in the lens during both normal development and endoplasmic reticulum stress induced by either unfolded proteins or oxidative damage. While mutations in the gene for connexin 50 are known to cause autosomal dominant cataracts, it has not been previously reported whether mutant connexins can activate the unfolded protein response in the lens. Mice homozygous for the S50P or G22R mutation of connexin 50 have reduced amounts of connexin 50 protein at the cell membrane, with some intracellular staining consistent with retention in the endoplasmic reticulum. Connexin 50 mutants have elevated levels of BiP expression in both lens epithelial and fiber cells from E15.5 with the most robust elevation detected in newborns. While this elevation decreases in magnitude postnatally, BiP expression is still abnormally high in adults, particularly in the perinuclear endoplasmic reticulum of cell nuclei that are inappropriately retained in adult homozygous mutant lenses. Xbp1 splicing was elevated in lenses from both connexin mutants studied, while Atf4 and Atf6 levels were not majorly affected. Overall, these data suggest that UPR may be a contributing factor to the phenotype of connexin 50 mutant lenses even though the relatively modest extent of the response suggests that it is unlikely to be a major driver of the pathology.
Keywords: UPR, BiP, Cataract, Connexin 50
Introduction
The ocular lens is a transparent, avascular tissue comprised of two polarized epithelial cell types (Beebe, 2008); a monolayer of flattened epithelial cells found on anterior side with the bulk of the lens composed of lens fibers, highly elongated cells which interlock with their neighbors via elaborate lateral membrane interdigitations (Donaldson et al., 2009; Kistler and Bullivant, 1989; Kuszak, 1995). Alterations in lens cell structure/function, as well as the physical properties of its constituent proteins can lead to the opacity of the lens known as cataract (Graw, 1997). While cataract treatment has become ubiquitous in the developed world, cataract is still the leading cause of blindness worldwide (Isaacs et al., 2004). Congenital cataract is particularly problematic since it must be treated in infancy to prevent permanent amblyopia due to early visual deprivation (Bremond-Gignac et al., 2011; Levi and Li, 2009; Rahi and Cable, 2003). Congenital cataract occurs at a rate of approximately 1.6 per 10,000 live births (Francis et al., 2000), with between 8 and 25% of these with an obvious genetic etiology only affecting the lens although some studies suggest that over 50% of congenital cataracts presenting in developed countries are associated with heritable diseases. (Rahi and Dezateux, 2000). Notably, the majority of known cataract causing mutations are found in lens structural proteins and exhibit an autosomal dominant mode of inheritance. (Hejtmancik, 2008)
The mechanisms of cataract associated with autosomal dominant mutations are likely to be diverse. Heterozygous deletion of connexin 50 (Cx50), a transmembrane protein important for the gap junctional coupling of fiber cells, results in normal lenses while lenses lacking Cx50 are small, with nuclear cataracts due to loss of appropriate gap junctional coupling in the lens. (Rong et al., 2002; White et al., 1998). However, mutations in the Cx50 gene are a major cause of autosomal dominant congenital cataract with a spectrum of lens phenotypes associated with different mutations in this gene (Gao et al., 2010; Vanita et al., 2008; Wang et al., 2009; Yan et al., 2008). In some cases, the cataract phenotype is associated with misregulation of connexin gating, which presumably results in cataract due to defects in the lens circulation (Minogue et al., 2009; Tong et al., 2011). However, one of the best studied mouse Cx50 mutations, Cx50-S50P (proline replaces serine at position 50), results in a protein that is unable to establish normal electric coupling between Xenopus oocytes due in part to the inability to efficiently traffic to the plasma membrane although this mutant protein can reach the plasma membrane in the presence of Cx46 (DeRosa et al., 2007). In another mouse mutant, Cx50-G22R, (arginine replaces glycine at position 22), the mutant protein was shown to only inefficiently reach the lens fiber cell membrane, with most protein remaining intracellular (Chang et al., 2002; Xia et al., 2006a).
Recently our laboratory has demonstrated that retention of unfolded collagen IV within the secretory pathway of lens cells can result in cataracts due to the induction of the Unfolded Protein Response (UPR) (Firtina et al., 2009). UPR comprises a set of evolutionary conserved cell signaling pathways which allow cells to adjust to increased levels of proteins transiting the secretory pathway (such as during secretory epithelium differentiation), can allow for the adjustment of cell biology to compensate for the presence of increased levels of mutant protein (by upregulating endoplasmic reticulum associated degradation; ERAD) and can also induce apoptosis when the UPR fails to achieve ER homeostasis (Lai et al., 2007; Rasheva and Domingos, 2009). UPR appears to induce cataract via diverse molecular impacts on the lens including a global inhibition of protein synthesis, inhibition of fiber cell elongation, the retention of both the endoplasmic reticulum and fiber cell nuclei during differentiation and finally apoptosis (Firtina et al., 2009).
Since defects in Cx50 trafficking to the cell membrane have been reported in Cx50 S50P and G22R mutants (DeRosa et al., 2007; Xia et al., 2006b), while the phenotype of these mutants overlaps with that induced by UPR including microphthalmia, defects in fiber cell elongation and cataract (Chang et al., 2002; DeRosa et al., 2007; Xia et al., 2006c), we hypothesized that the intracellular retention of the mutant Cx50 resulted from inefficient folding leading to its failure to pass endoplasmic reticulum (ER) quality control. Thus, we investigated whether UPR is activated in these connexin mutants.
MATERIALS AND METHODS
Generation of mutant mice
All experiments using mice were approved by the University of Delaware Institutional Animal Care and Use Committee and conform to the ARVO statement for the use of Animals in Vision Research. Wildtype mice were of the strain C57Bl/6 <har> and were either obtained directly from Harlan-Sprague Dawley (Frederick, MD) or generated in house from a breeding colony founded upon such animals. Homozygous Cx50-S50P and Cx50-G22R mice on a C57Bl/6 genetic background were obtained from Dr. Thomas White (SUNY-Stonybrook). Heterozygous mutants were obtained by mating homozygous mutants to wildtype C57Bl/6 mice. Embryos used in the study were staged by designating the day that the vaginal plug was observed in the dam as E0.5. Postnatal mice were staged by designating the day of birth as P0. For gross documentation of alterations in lens structure, lenses were isolated under a dissecting microscope, placed in Media 199 and photographed with a Zeiss dissecting microscope using dark field optics.
Immunolocalization
All immunofluorescence experiments were performed as described previously (Reed et al., 2001). Embryonic heads or postnatal eyes were isolated and immediately embedded in Optimum Cutting Temperature media (OCT, Tissue Tek, Torrance California). Sixteen micron thick sections were cut with a cryostat and mounted on ColorFrost plus slides (Fisher Scientific, Hampton, New Hampshire). For staining with most antibodies, sections were fixed in ice cold 1:1 acetone:methanol for 20 minutes at −20°C, blocked in 1% BSA for 1 hour at room temperature, followed by the incubation with the appropriate dilution of primary antibody (see table 1) for an overnight incubation at 4°C. Sections were then washed two times for 10 minutes each and detected with the appropriate AlexaFluor488 and/or 568 labeled secondary antibody (Molecular Probes, Eugene Oregon) prepared in 1% BSA containing 1:2000 diluted Draq-5 as the nuclear stain (Biostatus Limited, Leicestershire, United Kingdom). Slides were then visualized using either a Zeiss LSM780 or Zeiss LSM510 confocal microscope (Carl Zeiss Inc, Gottingen, Germany). Identical conditions were used to image wildtype and mutant sections to ensure the validity of comparisons. The images were quantitated by evaluating relative pixel intensities from the respective anatomical regions using ImageJ. The data was analyzed statistically by ANOVA.
Table 1.
Antibodies used in this study
| Antibody | Source | Cat.# | IF | WB |
|---|---|---|---|---|
| Rabbit polyclonal to Cx50 (C-term) | Abgent (San Diego, CA) | AP1548b | 1:50 | - |
| Goat polyclonal to Cx50 (K20) (used for co-localization with BiP) | Santa Cruz (Santa Cruz, CA) | SC-20876 | 1:200 | - |
| Rabbit polyclonal to BiP/Grp78 | Abcam (Cambridge, MA) | ab21685 | 1:200 | 1:1000 |
| Rabbit polyclonal to Xbp1 | Santa Cruz Inc., (Santa Cruz, CA) | sc7160 | - | 1:1000 |
| Rabbit polyclonal to Atf6α | Santa Cruz Inc., (Santa Cruz, CA) | Sc-22799 | - | 1:1000 |
| Rabbit polyclonal to Atf4/Creb2 | Santa Cruz Inc., (Santa Cruz, CA) | sc200 | - | 1:1000 |
| Rabbit polyclonal to β-actin | Cytoskeleton Inc.(Denver, CO) | AAN01 | - | 1:1000 |
Western Blotting
Lenses were isolated and immediately homogenized with 0.1 ml of ice-cold lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Halt protease and phosphatase inhibitor mixture (Thermo Scientific, Rockford, IL). The insoluble material was removed by centrifugation at 12,000xg for 30 min. Final protein concentrations were determined using a Bio-Rad protein assay kit according to the manufacturer’s specifications. Forty micrograms of total protein was resolved by SDS-polyacrylamide gel and transferred onto supported PVDF membrane (Bio-Rad). The protein blots were blocked with SuperBlock T20 blocking buffer (Thermo Scientific) overnight at 4 °C and incubated with the primary antibody (see Table 1 for dilution of antibody) in the same blocking buffer for 2 hours. After incubation with secondary antibodies conjugated with horseradish peroxidase (Calbiochem) for 1 hour at room temperature, the signals were detected using an ECL detection kit (GE Healthcare, Pittsburgh, PA).
RNA Preparation and QRT-PCR
Total RNA from lenses was extracted using the SV total RNA isolation kit (Promega). RNA levels were measured using a Nanodrop system and samples were stored at −80°C. Quantitative real time rt-PCR was used to detect the levels of spliced Xpb-1 in these samples as previously described (Firtina and Duncan, 2011).
Results
Investigation of the underlying cause of cataracts resulting from Cx50 mutations has identified diverse mechanisms for these lens defects. Defects in the regulation of gap junctional coupling and dysregulated cell signaling have both been reported, however, improper trafficking of mutant connexins has been reported in several mutants (Arora et al., 2008; DeRosa et al., 2009; Minogue et al., 2009; Xia et al., 2006c). Here we investigate whether this mistrafficked protein can activate the unfolded protein response.
Mice harboring Cx50 G22R/G22R and S50P/S50P mutations both develop cataracts associated with incomplete localization of Cx50 to the fiber cell membrane
As previously reported, lenses from mice homozygous for either Cx50 G22R or S50P mutations are smaller than normal and develop nuclear cataracts (Chang et al., 2002; Xia et al., 2006c) (Figure 1). Further, while Cx50 is still expressed in mice homozygous for both of these mutations, the protein is not strongly localized to the lens fiber cell membrane, unlike the situation in the wildtype lens (Figure 2). Instead, the Cx50 immunoreactivity is diffuse in lenses from both connexin mutants. Further, some staining is concentrated around the cell nuclei.(arrows) (Figure 2D, G).
Figure 1.

Darkfield imaging demonstrates that Cx50 mutant lenses have profound opacities and are smaller than normal at two months of age. A) wild type C57Bl/6 lens B) homozygous Cx50 G22R lens C) homozygous Cx50 S50P lens. Scale bar = 1 mm
Figure 2.

Immunostaining of two month old mouse lenses with an antibody raised against the C-terminus of Cx50. Cx50 is strongly localized to the lens fiber cell membranes in WT lenses (A) while Cx50 protein is less sharply localized in homozygous Cx50 G22R (D) and S50P (G) mutants. Further, homozygous Cx50G22R (D) and Cx50S50P (G) lenses also exhibit appreciable levels of Cx50 protein surrounding the nuclear envelope (arrows). Red-Cx50; Blue-DRAQ5 (DNA); e-epithelial cells; f- fiber cells; tz-transition zone; Scale bar=77μm
BiP levels are elevated in Cx50 mutant mouse lenses
Since we have previously reported that retention of a secreted protein (collagen IV) in the endoplasmic reticulum can activate the unfolded protein response leading to cataract (Firtina et al., 2009), we then assessed whether the expression of BiP, an ER protein whose expression levels robustly upregulate upon UPR activation, is altered in Cx50 mutant lenses (Figure 3). In two month old wildtype mice, BiP expression is largely localized to the lens epithelium and fiber cells in the earliest stages of differentiation with much lower levels in maturing fiber cells (Figure 3A–C) consistent with our prior results (Firtina and Duncan, 2011). Notably, both G22R/G22R and S50P/S50P mutants exhibit delayed lens fiber cell terminal differentiation as measured by the retention of cell nuclei in deeper lens layers than normal. Further, these abnormally retained cell nuclei are associated with a region of significantly increased BiP expression (figure 3J), particularly in the area surrounding these persistent cell nuclei (Figure 3D–I). BiP levels were elevated in the lens epithelium of adult mice as well (Figure 3J)
Figure 3.

BiP, a molecular chaperone found in the ER, is more abundant around the cell nuclei of maturing lens fibers in two month old Cx50 mutant mice (arrows). In wildtype lenses (A, B, C), BiP is predominately seen in the lens epithelium, with lower levels distributed through the lens fibers. In contrast, BiP staining is more prominent in maturing lens fibers, particularly in the perinuclear ER (arrows) in both Cx50 G22R (D, E, F,) and Cx50 S50P (G, H,I) homozygous mutant lenses. J- Quantitation of BiP levels in the lens epithelium, transition zone and cortical fibers using ImageJ. P values reported represent ANOVA analysis performed on results obtained from at least three biological replicates. Red-BiP; Blue-DRAQ5 (DNA); e-epithelial cells; f- fiber cells; tz-transition zone; Scale bar=77μm
Since the region of retained nuclei/elevated BiP expression corresponds to lens fibers generated perinatally or earlier, we next evaluated the expression of BiP in newborn Cx50 mutant lenses (Figure 4). Newborn WT lenses, similar to adults, express BiP predominately in the lens epithelium and lens fibers undergoing early differentiation (Figure 4A, B) and this Cx50 does not strongly co-localized with BiP (Figure 4C, D, E), however, lenses from mice homozygous for the G22R mutation (Figure 4F, G) have elevated levels of BiP throughout the lens fibers suggesting defects in the degradation of both the cell nuclei and endoplasmic reticulum that is a normal feature of fiber cell differentiation (Bassnett, 1995). This elevated BiP was co-localized with areas of Cx50 retention as well (Figure 4, H, I, J) Lenses from mice homozygous for the S50P mutation (Figure 4K,L) have elevated BiP expression in the cortical fiber cells as well which co-localizes with Cx50 (Figure 4M,N,O), but do not exhibit a retention of BiP in the lens center although some cell nuclei are retained there (Figure 4K,L). This upregulation of BiP protein levels in both mutants was quantitated by image analysis (Figure 4P) and confirmed by western blot analysis as well (Figure 4Q).
Figure 4.

BiP levels are elevated in newborn Cx50 mutant lenses (A, B,) Wildtype newborn lenses express BiP prominently in the lens epithelium with lower amounts in the lens fibers that have not completed organelle degradation. (C,D,E)- BiP and Cx50 are not found co-localized in wildtype lens fibers at birth (F, G,) Homozygous Cx50G22R lenses have increased levels of BiP expression, particularly in the lens fiber cells. (H,I,J)- The elevated amounts of BiP found in G22R lens fibers is co-localized with Cx50 (K,L) Cx50S50P lenses also have increased levels of BiP in the lens fibers although this increase is more confined to the cortical fiber cells and this additional BiP co-localizes with Cx50 (M,N,O). (P) Quantitation of BiP levels in G22R/G22R and S50P/S50P homozygotes in the lens epithelium, transition zone and cortical fibers using ImageJ. P values reported represent ANOVA analysis performed on results obtained from at least three biological replicates. (Q) Western blotting analysis of newborn lenses confirming the increase in BiP expression in the Cx50S50P and Cx50G22R mutants. Red-BiP; Blue-DRAQ5(DNA); Green- Cx50 e- epithelial cells; f- fiber cells; tz-transition zone; Scale bar A,B,F,G,K,L=77 μm, C,D,E,H,I,J,M,N,O= 6 μm
Since these data suggested that BiP expression was elevated in lens fiber cells generated during embryonic development, the expression of BiP was then assessed in Cx50 mutant embryos (Figure 5). At embryonic day 13.5 (E13.5), shortly after the initial formation of the lens, BiP expression is uniformly distributed throughout the lens fiber cells (Figure 5A,B), and this expression pattern was similar in both homozygous G22R (Figure 5E, F) and S50P (Figure 5I, J) and wildtype mice at this age. However, by E15.5, BiP levels are more noticeably elevated in both homozygous G22R (Figure 5G, H) and S50P (Figure 5K, L) mutants. Quantitation of these images shows this upregulation as well (Figure 5M,N).
Figure 5.

BiP upregulation begins in Cx50 mutant lenses between E13.5 and E15.5. (A, B) WT E13.5 lenses exhibit a uniform BiP distribution which is also seen in E13.5 G22R/G22R (E,F) and E13.5 S50P/S50P (I, J) connexin mutants. In WT lenses, BiP levels begin to be relatively higher in the transition zone of E15.5 lenses (C, D), BiP levels are noticeably upregulated by E15.5 in both G22R/G22R (G, H) and S50P/S50P (K,L) lenses. (M,N) Quantitation of BiP levels in G22R/G22R and S50P/S50P homozygotes in the lens epithelium, transition zone and cortical fibers at E13.5 (M) and E15.5 (N) using ImageJ. P values reported represent ANOVA analysis performed on results obtained from at least three biological replicates. Red-BiP; Blue-DRAQ5 (DNA); e- epithelial cells; f- fiber cells; tz-transition zone; Panels A,B,E,F,I,J Scale bar 67 μm; Panels C,D,G,H,K,L Scale bar=71 μm.
Homozygous Cx50 S50P and G22R mutant lenses express elevated level of spliced Xbp1 mRNA
During UPR activation, the transmembrane sensor Ire1 is activated, and catalyzes the non-canonical splicing of Xbp1 mRNA, resulting in a transcript capable of producing a transcription factor which activates the transcription of genes important for UPR (Aragon et al., 2009; Calfon et al., 2002; Lee et al., 2002). As previously reported, the WT lens expresses low, but detectable levels of spliced Xbp1 mRNA (Firtina and Duncan, 2011). In both Cx50-G22R and S50P homozygous lenses, the levels of spliced Xbp1 are similar to WT at birth but the levels of spliced Xbp1 mRNA are significantly elevated at two months postnatally in G22R homozygotes (Figure 6).
Figure 6.

Quantitative rt-PCR analysis of Xbp-1 splicing in homozygous G22R and S50P mutant lenses at birth and two months postnatal. P values are obtained from unpaired T-test.
UPR does not appear to explain the severe lens phenotype of mice heterozygous for the S50P mutation in Connexin 50
It has been previously reported that lenses from mice heterozygous for the S50P mutation in Cx50 have severe defects in fiber cell elongation during embryogenesis (Xia et al., 2006c) superficially similar to that we observed in lenses with high levels of UPR activation (Firtina et al., 2009). However, neither heterozygous Cx50-G22R nor Cx50-S50P mutant lenses exhibit extreme upregulation of BiP expression consistent with an extreme UPR response although modest elevations in BiP protein levels were observed (Figure 7). However, at birth, G22R heterozygotes sharply upregulate Xbp-1 splicing while a more modest upregulation of Xbp-1 splicing is seen in heterozygotes at 2 months of age (Figure 8). Similarly, western blot analysis of newborn lenses detected a slight elevation in the levels of Xbp1 protein translated from the spliced form (Figure 9). In contrast, no consistent differences were detected in either Atf4 or Atf6 protein levels in lenses from heterozygous or homozygous Cx50 G22R or S50P mice.
Figure 7.
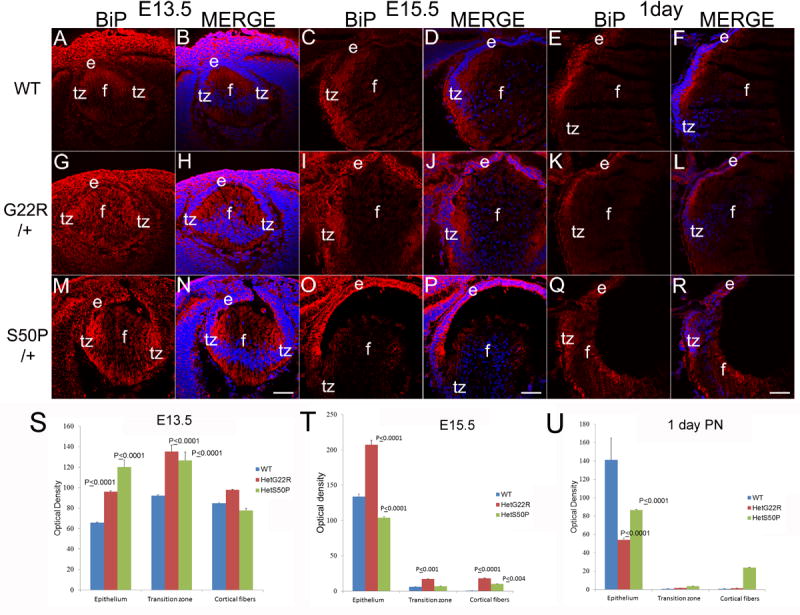
BiP levels are upregulated prenatally in heterozygous connexin mutant lenses (A,B) WT E13.5 lenses exhibit a uniform BiP distribution which is more intense in E13.5 G22R/+ (G,H) and E13.5 S50P/+ (M, N) connexin mutants. In WT lenses, BiP levels begin to be relatively higher in the transition zone of E15.5 lenses (C, D), which is similar to that seen in both G22R/+ (I, J) and S50P/+ (O,P) lenses although BiP levels may be elevated in the lens epithelium of both heterozygous mutants. By birth, BiP levels/distribution is similar between WT (E,F), G22R/+ (K,L) and S50P/+ lenses (Q,R). (S,T,U) Quantitation of BiP levels in G22R and S50P heterozygotes in the lens epithelium, transition zone and cortical fibers at E13.5 (S), E15.5 (T) and birth (U) using ImageJ. P values reported represent ANOVA analysis performed on results obtained from at least three biological replicates. Red-BiP; Blue-DRAQ5(DNA); e- epithelial cells; f- fiber cells; tz-transition zone; Scale bar 77 μm
Figure 8.

Quantitative rt-PCR analysis of Xbp-1 splicing in heterozygous G22R and S50P mutant lenses. P values are obtained from unpaired T-test.
Figure 9.
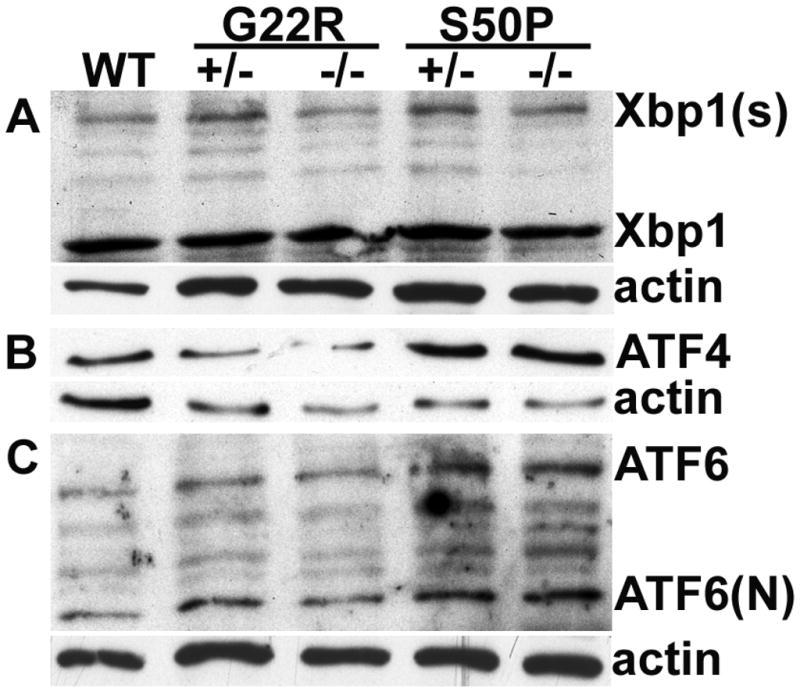
Western blotting analysis of XBP1, ATF4 and ATF6 in newborn heterozygous (+/−) and homozygous (−/−) Cx50S50P and Cx50G22R mutant lens. (A) Levels of protein derived from the spliced XBP1 message (Xbp1(s)) are apparently increased in the both heterozygous mutants although western blotting did not detect consistent differences in levels in homozygous mutants. (B) ATF4 levels are moderately higher in Cx50 S50P mutants and while no changes were seen for the Cx50 G22R mutant as compared to wildtype. (C) Atf6 protein levels are slightly higher in Cx50 S50P mutant lenses while no consistent changes were detected for Cx50 G22R mutants as compared to wildtype.
Discussion
The unfolded protein response (UPR) is a set of evolutionarily conserved cellular pathways which allows cells to respond to endoplasmic reticulum stress, first by attenuating global protein synthesis then by increasing both the area of the ER membrane and ER resident chaperone expression to relieve this stress. During normal development, this mechanism allows differentiating cells to expand the volume of their endoplasmic reticulum which is particularly important for secretory cells (Austin, 2009; Diehl et al., 2011; Iwakoshi et al., 2003). However, when the levels of terminally misfolded proteins rise in the ER beyond the ability of the cell to compensate, UPR can result in pathology due to global translational attenuation within the affected cell and in the most severe cases, apoptosis (Rasheva and Domingos, 2009; Szegezdi et al., 2006). Such pathological elevations in unfolded protein often result from point mutations in genes encoding either secreted or transmembrane proteins whose normal synthesis requires the protein folding machinery of the secretory pathway (Iwakoshi et al., 2003; Ron and Walter, 2007).
During normal lens development, we have found that the Ire1/Xbp1, Atf6, and Perk/p-eIF2α/Atf4 pathways are all activated as lens epithelial cells transition to lens fibers, presumably to mediate the expansion of ER necessary to produce the high amounts of membrane proteins necessary for lens fiber cell function (Firtina and Duncan, 2011). However, consistent with the normal function of UPR which re-establishes cellular homeostasis after the stress imposed by a burst of protein synthesis via the secretory pathway, Chop activation and apoptosis are not detected during normal lens development. In contrast, transgenic overexpression of a terminally misfolded extracellular matrix protein in lens fibers hyperactivates all three arms of the canonical UPR, leading to cataract, apparently due to a combination of attenuated protein synthesis, dysregulation of organelle degradation and defective cell elongation. While Chop activation is detected from the earliest stages of this stress response, lens fiber cells appear somewhat resistant to UPR induced apoptosis since only a very few cells were TUNEL positive even a week after the misfolded proteins began to accumulate (Firtina et al., 2009). However, the overexpression of other proteins which induce UPR in the lens elicits a more robust apoptotic response, perhaps due to the biological function of the transgenes (Reneker et al., 2011).
Between the normal physiological UPR induced during development and pathological UPR induced upon the extreme overexpression of an unfolded protein is expected to lie the response of the lens to the expression of endogenous levels of animproperly folded or terminally unfolded mutant protein. Such responses could be expected to be a significant pathogenic mechanism in the development of diverse congenital cataracts of genetic etiology since it is known that several dominant cataract mutations have more severe phenotypes than homozygous null alleles in the same gene (Graw and Loster, 2003; Hejtmancik, 2008). This is consistent with the observation that mice heterozygous for a Col4a1 null allele are morphologically normal (Poschl et al., 2004), while mice heterozygous for Col4a1 mutations have multiple organ defects including cataracts (Gould et al., 2005; Van Agtmael et al., 2005). Notably though, while we have detected chronic activation of all three arms of the UPR pathway in lenses from mice harboring one of these Col4a1 mutations, they appear to achieve homeostasis despite this chronic activation since Atf4 upregulation, Chop activation and apoptosis are not detected in these lenses (Firtina et al., 2009). This is consistent with the concept of “overall stress adaptation” wherein the homeostasis places the cell outside of the physiological range for normal function (van Anken and Braakman, 2005)
However, the possible contribution of UPR to the pathology of cataracts associated with mutations in the genes for transmembrane proteins prominently expressed in the lens had not been previously explored. Connexin 50 (Gja8, Cx50) is a major component of lens cell membranes, comprising 18% of the integral membrane proteome of weanling mouse lenses (Bassnett et al., 2009) whose physiological function is to provide the gap junctional coupling necessary for tissue function in the absence of a blood supply (Evans and Martin, 2002; Mathias et al., 2010). While complete removal of the Cx50 gene results in an autosomal recessive phenotype typified by small, cataractous lenses (Rong et al., 2002; White et al., 1998), point mutations in the Cx50 gene are commonly found in both human families and animals exhibiting autosomal dominant congenital cataracts (Vanita et al., 2006; Wang et al., 2009; Xia et al., 2006c) leading to the idea that mutant forms of Cx50 actively disrupt lens physiology. Prior investigations of the S50P and G22R Cx50 mutations have revealed that their trafficking to the plasma membrane is defective. Physiological investigations revealed that they can form heteromeric gap junctions with other connexins, although these channels have altered voltage gating which were proposed to be the cause of the dominant cataracts seen in these mutants (DeRosa et al., 2009; DeRosa et al., 2007; Xia et al., 2006b). However, one notable feature of the phenotype of Cx50 S50P/+ lenses is that they have a profound defect in lens fiber cell elongation (Xia et al., 2006c) which is superficially similar to that observed in transgenic mice overexpressing unfolded collagen IV in lens fiber cells (Firtina et al., 2009) while Cx50 G22R/+ lenses are also cataractous showing that the mutant protein is having a dominant effect (Chang et al., 2002). Thus, we sought to investigate whether pathological UPR was operative in these lenses.
Overall, the present investigation of UPR activation in lenses from mice heterozygous for these Cx50 mutations revealed that UPR activation as measured by BiP upregulation is only very modest in both cases studied. Further, mice heterozygous for these connexin 50 mutations only exhibited Ire1/Xbp1 pathway activation in embryonic and newborn lenses while the levels of spliced Xbp1 were similar between wildtype and heterozygous mutants in adulthood. Similarly, while mice homozygous for these Cx50 mutants do express elevated amounts of BiP, the only UPR pathway consistently activated at levels higher than normal from birth until adulthood is the Ire1/Xbp1 pathway. These results suggest that the lens is able to transiently activate a single branch of the UPR to reset the cellular physiology to a point where UPR activation is no longer necessary. However, it is still possible that low levels of Perk/Atf4 and Atf6 pathway activation are present in Cx50 mutant lenses but are not easily detected due to the endogenous levels of UPR which occur during normal lens fiber cell differentiation (Firtina et al., 2009). This results in a chronic increase in ER area in the lens fibers of mutant mice and may be at least partially responsible for the delay in the organelle loss normally associated with lens fiber cell differentiation in these mutants. These data show that UPR activation may contribute to the lens phenotype of Cx50 mutants although the relatively low level of activation suggests that it is not the major driver of the phenotype.
Overall, this investigation shows that the lens can use pathologic UPR to reset cellular physiology to a new homeostasis. Since other stresses on the lens such as the expression of mutant αA-crystallin (which is a cytoplasmic protein that does not enter the secretory pathway) or oxidative insult also result in UPR pathway activation (Shinohara et al., 2006; Watson and Andley, 2010), it is possible that such modifications of cellular physiology are a general mechanism either underlying or contributing to diverse cataract etiologies.
Highlights.
Connexin 50 mutants exhibit dominant cataracts
Connexin 50 G22R and S50P mutants have less connexin 50 at cell membrane
Connexin 50 mutants have elevated BiP expression
Connexin 50 mutants exhibit elevated Xbp-1 splicing
Unfolded protein response is activated in connexin 50 G22R and S50P lenses
Acknowledgments
Funding: National Eye Institute EY015279 to MKD, INBRE program grant P20 RR16472 supporting the University of Delaware Core Imaging facility; JKS was supported by both the Howard Hughes Medical Institute and a Basel V. Worgul Lens Research Fellowship from the Fight for Sight Foundation. We also thank Thomas White, SUNY-StonyBrook for providing the mice used in this study.
The abbreviations used
- ER
endoplasmic reticulum
- UPR
unfolded protein response
- RT
reverse transcription
- WT
wild type
- Atf4
activating transcription factor 4
- ATF6
activating transcription factor 6
- Xbp-1
X-box binding protein-1
- BiP/Grp78
immunoglobulin heavy chain-binding protein
- Perk
RNA-dependent protein kinase-like ER kinase
- Chop
CCAAT/enhancer-binding protein-homologous protein
- eIF2α
elongation initiation factor 2α
- IRE1
Inositol-requiring-1
- Cx50
connexin 50
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A, Minogue PJ, Liu X, Addison PK, Russel-Eggitt I, Webster AR, Hunt DM, Ebihara L, Beyer EC, Berthoud VM, Moore AT. A novel connexin50 mutation associated with congenital nuclear pulverulent cataracts. J Med Genet. 2008;45:155–160. doi: 10.1136/jmg.2007.051029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin RC. The unfolded protein response in health and disease. Antioxid Redox Signal. 2009;11:2279–2287. doi: 10.1089/ars.2009.2686. [DOI] [PubMed] [Google Scholar]
- Bassnett S. The fate of the Golgi apparatus and the endoplasmic reticulum during lens fiber cell differentiation. Invest Opthalmol Vis Sci. 1995;36:1793–1803. [PubMed] [Google Scholar]
- Bassnett S, Wilmarth PA, David LL. The membrane proteome of the mouse lens fiber cell. Mol Vis. 2009;15:2448–2463. [PMC free article] [PubMed] [Google Scholar]
- Beebe DC. Maintaining transparency: a review of the developmental physiology and pathophysiology of two avascular tissues. Semin Cell Dev Biol. 2008;19:125–133. doi: 10.1016/j.semcdb.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremond-Gignac D, Copin H, Lapillonne A, Milazzo S. Visual development in infants: physiological and pathological mechanisms. Curr Opin Ophthalmol. 2011;22(Suppl):S1–8. doi: 10.1097/01.icu.0000397180.37316.5d. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Chang B, Wang X, Hawes NL, Ojakian R, Davisson MT, Lo WK, Gong X. A Gja8 (Cx50) point mutation causes an alteration of alpha 3 connexin (Cx46) in semi-dominant cataracts of Lop10 mice. Hum Mol Genet. 2002;11:507–513. doi: 10.1093/hmg/11.5.507. [DOI] [PubMed] [Google Scholar]
- DeRosa AM, Mese G, Li L, Sellitto C, Brink PR, Gong X, White TW. The cataract causing Cx50-S50P mutant inhibits Cx43 and intercellular communication in the lens epithelium. Exp Cell Res. 2009;315:1063–1075. doi: 10.1016/j.yexcr.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRosa AM, Xia CH, Gong X, White TW. The cataract-inducing S50P mutation in Cx50 dominantly alters the channel gating of wild-type lens connexins. J Cell Sci. 2007;120:4107–4116. doi: 10.1242/jcs.012237. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Fuchs SY, Koumenis C. The cell biology of the unfolded protein response. Gastroenterology. 2011;141:38–41. 41 e31–32. doi: 10.1053/j.gastro.2011.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson PJ, Chee KS, Lim JC, Webb KF. Regulation of lens volume: implications for lens transparency. Exp Eye Res. 2009;88:144–150. doi: 10.1016/j.exer.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Evans WH, Martin PE. Gap junctions: structure and function (Review) Mol Membr Biol. 2002;19:121–136. doi: 10.1080/09687680210139839. [DOI] [PubMed] [Google Scholar]
- Firtina Z, Danysh BP, Bai X, Gould DB, Kobayashi T, Duncan MK. Abnormal expression of collagen IV in lens activates unfolded protein response resulting in cataract. J Biol Chem. 2009;284:35872–35884. doi: 10.1074/jbc.M109.060384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firtina Z, Duncan MK. Unfolded Protein Response (UPR) is activated during normal lens development. Gene Expr Patterns. 2011;11:135–143. doi: 10.1016/j.gep.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PJ, Berry V, Bhattacharya SS, Moore AT. The genetics of childhood cataract. J Med Genet. 2000;37:481–488. doi: 10.1136/jmg.37.7.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Cheng J, Lu C, Li X, Li F, Liu C, Zhang M, Zhu S, Ma X. A novel mutation in the connexin 50 gene (GJA8) associated with autosomal dominant congenital nuclear cataract in a Chinese family. Curr Eye Res. 2010;35:597–604. doi: 10.3109/02713681003725831. [DOI] [PubMed] [Google Scholar]
- Gould DB, Phalan FC, Breedveld GJ, van Mil SE, Smith RS, Schimenti JC, Aguglia U, van der Knaap MS, Heutink P, John SW. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167–1171. doi: 10.1126/science.1109418. [DOI] [PubMed] [Google Scholar]
- Graw J. The crystallins: genes, proteins and diseases. Biol Chem. 1997;378:1331–1348. [PubMed] [Google Scholar]
- Graw J, Loster J. Developmental genetics in ophthalmology. Ophthalmic Genet. 2003;24:1–33. doi: 10.1076/opge.24.1.1.13888. [DOI] [PubMed] [Google Scholar]
- Hejtmancik JF. Congenital cataracts and their molecular genetics. Seminars in Cell and Developmental Biology. 2008;19:134–149. doi: 10.1016/j.semcdb.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs R, Ram J, Apple D. Cataract blindness in the developing world: is there a solution? J Agromedicine. 2004;9:207–220. [PubMed] [Google Scholar]
- Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol. 2003;4:321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- Kistler J, Bullivant S. Structural and molecular biology of the eye lens membranes. Crit Rev Biochem Mol Biol. 1989;24:151–181. doi: 10.3109/10409238909086397. [DOI] [PubMed] [Google Scholar]
- Kuszak JR. The ultrastructure of epithelial and fiber cells in the crystalline lens. Int Rev Cytol. 1995;163:305–350. doi: 10.1016/s0074-7696(08)62213-5. [DOI] [PubMed] [Google Scholar]
- Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda) 2007;22:193–201. doi: 10.1152/physiol.00050.2006. [DOI] [PubMed] [Google Scholar]
- Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002;16:452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi DM, Li RW. Perceptual learning as a potential treatment for amblyopia: a mini-review. Vision Res. 2009;49:2535–2549. doi: 10.1016/j.visres.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RT, White TW, Gong X. Lens gap junctions in growth, differentiation, and homeostasis. Physiol Rev. 2010;90:179–206. doi: 10.1152/physrev.00034.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minogue PJ, Tong JJ, Arora A, Russell-Eggitt I, Hunt DM, Moore AT, Ebihara L, Beyer EC, Berthoud VM. A mutant connexin50 with enhanced hemichannel function leads to cell death. Investigative Ophthalmology and Visual Science. 2009;50:5837–5845. doi: 10.1167/iovs.09-3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- Rahi JS, Cable N. Severe visual impairment and blindness in children in the UK. Lancet. 2003;362:1359–1365. doi: 10.1016/S0140-6736(03)14631-4. [DOI] [PubMed] [Google Scholar]
- Rahi JS, Dezateux C. Congenital and infantile cataract in the United Kingdom: underlying or associated factors. British Congenital Cataract Interest Group. Invest Ophthalmol Vis Sci. 2000;41:2108–2114. [PubMed] [Google Scholar]
- Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- Reed NA, Oh DJ, Czymmek KJ, Duncan MK. An immunohistochemical method for the detection of proteins in the vertebrate lens. J Immunol Methods. 2001;253:243–252. doi: 10.1016/s0022-1759(01)00374-x. [DOI] [PubMed] [Google Scholar]
- Reneker LW, Chen H, Overbeek PA. Activation of unfolded protein response in transgenic mouse lenses. Investigative Ophthalmology and Visual Science. 2011;52:2100–2108. doi: 10.1167/iovs.10-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rong P, Wang X, Niesman I, Wu Y, Benedetti LE, Dunia I, Levy E, Gong X. Disruption of Gja8 (alpha8 connexin) in mice leads to microphthalmia associated with retardation of lens growth and lens fiber maturation. Development. 2002;129:167–174. doi: 10.1242/dev.129.1.167. [DOI] [PubMed] [Google Scholar]
- Shinohara T, Ikesugi K, Mulhern ML. Cataracts: role of the unfolded protein response. Med Hypotheses. 2006;66:365–370. doi: 10.1016/j.mehy.2005.08.044. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JJ, Minogue PJ, Guo W, Chen TL, Beyer EC, Berthoud VM, Ebihara L. Different consequences of cataract-associated mutations at adjacent positions in the first extracellular boundary of connexin50. Am J Physiol Cell Physiol. 2011;300:C1055–1064. doi: 10.1152/ajpcell.00384.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Agtmael T, Schlotzer-Schrehardt U, McKie L, Brownstein DG, Lee AW, Cross SH, Sado Y, Mullins JJ, Poschl E, Jackson IJ. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet. 2005;14:3161–3168. doi: 10.1093/hmg/ddi348. [DOI] [PubMed] [Google Scholar]
- van Anken E, Braakman I. Endoplasmic reticulum stress and the making of a professional secretory cell. Crit Rev Biochem Mol Biol. 2005;40:269–283. doi: 10.1080/10409230500315352. [DOI] [PubMed] [Google Scholar]
- Vanita V, Hennies HC, Singh D, Nurnberg P, Sperling K, Singh JR. A novel mutation in GJA8 associated with autosomal dominant congenital cataract in a family of Indian origin. Mol Vis. 2006;12:1217–1222. [PubMed] [Google Scholar]
- Vanita V, Singh JR, Singh D, Varon R, Sperling K. A novel mutation in GJA8 associated with jellyfish-like cataract in a family of Indian origin. Mol Vis. 2008;14:323–326. [PMC free article] [PubMed] [Google Scholar]
- Wang K, Wang B, Wang J, Zhou S, Yun B, Suo P, Cheng J, Ma X, Zhu S. A novel GJA8 mutation (p.I31T) causing autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2009;15:2813–2820. [PMC free article] [PubMed] [Google Scholar]
- Watson GW, Andley UP. Activation of the unfolded protein response by a cataract-associated alphaA-crystallin mutation. Biochem Biophys Res Commun. 2010;401:192–196. doi: 10.1016/j.bbrc.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TW, Goodenough DA, Paul DL. Targeted ablation of connexin50 in mice results in microphthalmia and zonular pulverulent cataracts. J Cell Biol. 1998;143:815–825. doi: 10.1083/jcb.143.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia CH, Cheng C, Huang Q, Cheung D, Li L, Dunia I, Benedetti LE, Horwitz J, Gong X. Absence of alpha3 (Cx46) and alpha8 (Cx50) connexins leads to cataracts by affecting lens inner fiber cells. Exp Eye Res. 2006a;83:688–696. doi: 10.1016/j.exer.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Xia CH, Cheung D, DeRosa AM, Chang B, Lo WK, White TW, Gong X. Knock-in of alpha3 connexin prevents severe cataracts caused by an alpha8 point mutation. J Cell Sci. 2006b;119:2138–2144. doi: 10.1242/jcs.02940. [DOI] [PubMed] [Google Scholar]
- Xia CH, Liu H, Cheung D, Cheng C, Wang E, Du X, Beutler B, Lo WK, Gong X. Diverse gap junctions modulate distinct mechanisms for fiber cell formation during lens development and cataractogenesis. Development. 2006c;133:2033–2040. doi: 10.1242/dev.02361. [DOI] [PubMed] [Google Scholar]
- Yan M, Xiong C, Ye SQ, Chen Y, Ke M, Zheng F, Zhou X. A novel connexin 50 (GJA8) mutation in a Chinese family with a dominant congenital pulverulent nuclear cataract. Mol Vis. 2008;14:418–424. [PMC free article] [PubMed] [Google Scholar]