

Abstract
Mutations in the X-linked gene, methyl-CpG binding protein 2 (Mecp2), underlie a wide range of neuropsychiatric disorders, most commonly, Rett Syndrome (RTT), a severe autism spectrum disorder that affects approximately one in 10,000 female live births. Because mutations in the Mecp2 gene occur in the germ cells with onset of neurological symptoms occurring in early childhood, the role of MeCP2 has been ascribed to brain maturation at a specific developmental window. Here, we show similar kinetics of onset and progression of RTT-like symptoms in mice, including lethality, if MeCP2 is removed postnatally during the developmental stage that coincides with RTT onset, or adult stage. For the first time, we show that brains that lose MeCP2 at these two different stages are actively shrinking, resulting in higher than normal neuronal cell density. Furthermore, we show that mature dendritic arbors of pyramidal neurons are severely retracted and dendritic spine density is dramatically reduced. In addition, hippocampal astrocytes have significantly less complex ramified processes. These changes accompany a striking reduction in the levels of several synaptic proteins, including CaMKII α/β, AMPA, and NMDA receptors, and the synaptic vesicle proteins Vglut and Synapsin, which represent critical modifiers of synaptic function and dendritic arbor structure. Importantly, the mRNA levels of these synaptic proteins remains unchanged, suggesting that MeCP2 likely regulates these synaptic proteins post-transcriptionally, directly or indirectly. Our data suggest a crucial role for MeCP2 in post-transcriptional regulation of critical synaptic proteins involved in maintaining mature neuronal networks during late stages of postnatal brain development.
Introduction
Mutations in the X-linked gene methyl-CpG-binding protein 2 (Mecp2) underlie most cases of Rett Syndrome (RTT), a postnatal progressive neurodevelopmental disorder that manifests in girls during early childhood. Several mouse models with germline mutations in Mecp2 have been generated that recapitulate RTT (Chen et al., 2001, Guy et al., 2001, Shahbazian et al., 2002). Other studies have indicated that symptomatic MeCP2-null male mice can be rescued by reactivation of global MeCP2 expression (Giacometti et al., 2007, Guy et al., 2007), suggesting that the neuronal damage can be reversed. Recently, we and others have shown that, in addition to neurons, MeCP2-dysfunction in glia also contributes to RTT (Ballas et al., 2009, Maezawa et al., 2009, Maezawa and Jin, 2010), specifically to disease progression (Lioy et al., 2011, Derecki et al., 2012).
In addition to the genetic studies, the impact of MeCP2 dysfunction on brain anatomy as well as neuronal structure and function has been further supported by several studies indicating that RTT patients and mice show increased neuronal cell density (Kishi and Macklis, 2004), reduced brain size (Chen et al., 2001), reduced dendritic arborization (Armstrong et al., 1995, Kishi and Macklis, 2004, Ballas et al., 2009), and spine density (Belichenko et al., 1994).
Although mutations in Mecp2 arise in the germline, the onset of overt neurological symptoms occurs at early postnatal stages (typically 4–6 weeks of age in male mice) (Chen et al., 2001, Guy et al., 2001). This has led to the idea, supported most recently by studies of the critical period in the visual system (Noutel et al., 2011), that MeCP2 function is required for brain maturation during a specific developmental window. Yet, why MeCP2 would be critical at this specific postnatal stage and whether there is a similar or different functional requirement for MeCP2 past this stage is poorly understood. A recent study has shown that depletion of MeCP2 in adult male mice results in manifestation of RTT-like symptoms followed by lethality (McGraw et al., 2011). However, the underlying mechanisms of symptom appearance at the adult stage were not examined. The present study addresses these questions.
Specifically, we show that inducible postnatal loss of MeCP2 in mice at two different stages—late juvenile stage, which represents the time of onset of RTT, or adult stage—results in equal manifestation of RTT-like symptoms, with immediate onset and parallel kinetics of symptom progression and lethality. Our analysis of the brain of symptomatic male mice revealed that postnatal loss of MeCP2 in otherwise healthy mice, whether at late juvenile or adult stage, results in severe abnormalities, which include global shrinkage of the brain, increased neuronal cell density, severe retraction of dendritic arbors, reduction in dendritic spine density, as well as significant reduction in complexity of astrocytic processes. Importantly, we show that the levels of several synaptic proteins, but not the levels of the corresponding mRNAs, are reduced dramatically, suggesting that MeCP2 likely regulates these proteins post-transcriptionally, either directly or indirectly.
Materials and Methods
Animals.
All animal studies were approved by the Institutional Animal Care and Use Committees at Stony Brook University and were in line with the guidelines established by the National Institutes of Health.
CreER (B6.Cg-Tg(CAG-cre/Esr1)5Amc/J) and Mecp2loxB/y (Guy et al., 2001) (B6;129P2-Mecp2tm1Bird/J) transgenic mice were obtained from the Jackson Laboratory. CreER (B6.Cg-Tg(CAG-cre/Esr1)5Amc/J) mice were maintained in a pure C57BL/6 background. The Mecp2loxB/y mice were backcrossed to C57BL/6 for eight generations. The Mecp2loxJ/y (Ballas et al., 2001) (B6;129S4-Mecp2tm1Jae/Mmcd) transgenic mice were obtained from MMRRC and were crossed as mix background. To generate the double transgenic Mecp2loxJ/y /CreER males, heterozygous Mecp2loxJ/+/CreER females, and all their control littermates (WT, CreER, and Mecp2loxJ/y or Mecp2loxJ/+), we crossed female mice heterozygous for the Mecp2-lox allele (Mecp2loxJ/+) with male mice heterozygous for the CreER transgene. A similar strategy was used to generate the Mecp2loxB/y/CreER males and all the necessary control male mice. The flox and Cre sequences were identified by PCR on tail biopsies with the following sets of primers: For Mecp2loxJ allele; forward 5′-CAC CAC AGA AGT ACT ATG ATC-3′, reverse 5′-CTA GGT AAG AGC TCT TGT TGA-3′. For Mecp2loxB allele; forward 5′-TGG TAA AGA CCC ATG TGA CCC AAG-3′, reverse 5′-GGC TTG CCA CAT GAC AAG AC-3′. For the CreER allele; forward 5′-CCG TAC ACC AAA ATT TGC C-3′, reverse 5′-ATC GCG AAC ATC TTC AGG-3′.
Tamoxifen treatment.
Tamoxifen (Tam; Sigma) was prepared in the vehicle solution consisting of 10% ethanol/ 90% sunflower oil (Sigma) by sonication, and aliquots were stored at −20°C. Male or female mice (5 or 10 week old) were injected intraperitoneally with 100 mg/kg body weight of tamoxifen or vehicle (Veh), daily for seven consecutive days. Excision of the Mecp2lox cassette was verified by genotyping of tail biopsies using the following primers: forward 5′-ATG CTG ACA AGC TTT CTT CTA-3′, reverse 5′-CAC CAC AGA AGT ACT ATG ATC-3′.
Phenotypic scoring and behavioral assays.
Male and female mice were scored weekly or biweekly, respectively. Mice were removed from their home cage and placed onto a bench, at the same time during the day when possible. The scoring was performed essentially as previously described (Guy et al., 2007), with several modifications. Each of five symptoms was scored as 0 (symptom absent), 1 (symptom present), or 2 (symptom severe): Mobility: 0 = active without extended freezing period. 1 = reduced movement and extended freezing period when first placed on the bench. 2 = no spontaneous movement when placed on the bench. Gait: 0 = as wild-type. 1 = hind legs are spread wider than wild-type or display a “waddling” gait when walking or running. 2 = more severe abnormalities: tremor when feet are lifted, walks backward or “bunny hops” by lifting both rear feet at once. Hindlimb clasping: When holding by the tail, mouse was scored 0 = legs splayed outwards. 1 = one leg is pulled into the body. 2 = both legs are pulled touching each other or touching the body. Tremor: Mouse observed while standing on the flat palm of the hand. 0 = no tremor. 1 = intermittent mild tremor. 2 = continuous tremor or intermittent violent tremor. General condition: 0 = clean shiny coat, clear eyes, normal stance. 1 = eyes dull, coat dull/ungroomed. 2 = eyes crusted or narrowed, piloerection, hunched posture or fur loss and self-injury. Mice were also weighed at each scoring session.
Behavioral assays.
For all behavioral studies, the observer was blinded to genotype.
Digiscan activity monitor.
The mouse was placed in the center of an empty rat cage (44 × 21 cm) situated inside an Opto-Varimex-Minor animal activity meter (Columbus Instruments). Fifteen × 15 infrared beams running in the x/y coordinates and 2 cm above the base of the machine recorded beam breaks associated with ambulatory activity (consecutive beam breaks) and total activity (all beam interruptions) during the 20 min observation period.
Rotarod.
Mice were tested on a Medical Associates (model ENV-575M) Rotarod and ran by the “rotarod” program on a Dell Optiplex with a Windows 98 operating platform. The apparatus was a mechanized 30-cm-long rod divided into five 6 cm sections by four plastic white discs. The speed of rotation increased from 4 rpm to 40 rpm through the course of 5 min session. The latency in seconds to drop off the rotarod was the main dependent measure of motor competence. Each mouse was tested for three consecutive trials with a 10 min rest between each session.
Wire hang test.
The mice were placed on a 44 cm long stainless steel wire suspended 54 cm off the ground by two wooden posts so that only their front paws contact the wire. The latency to fall onto a foam pad below the apparatus was recorded. The maximum hang time was 60 s before removal. Each mouse was tested on three consecutive trials without resting.
Dowel assay.
The tested mouse was placed in the middle of a wooden dowel (diameter, 1 cm; length, 16 cm, 30 cm above the ground) and was allowed to move freely. The latency to fall was measured for a maximum time of 2 min.
RNA isolation and quantitative real-time PCR analysis.
Total RNA was prepared from tissue using TRIZOL Reagent (Invitrogen) and treated with RNase-free DNase (Ambion, DNA-free kit). For reverse transcription, AMV-RT (New England BioLabs) was used and quantitative real-time RT-PCR was performed in an ABI StepOnePlus real-time PCR system using SYBR-green PCR master mix (Applied Biosystems). The relative abundance of the specific mRNAs was normalized to β-actin mRNA. The specific primers used were as follows: Mecp2, forward 5′-AGG CAG GCA AAG CAG AAA CAT CAG-3′ and reverse 5′-TCA TAC TTT CCA GCA GAT CGG CCA-3′; β-actin, forward 5′-GGC TGT ATT CCC CTC CAT CG-3′ and reverse 5′-CCA GTT GGT AAC AAT GCC ATG T-3′; for Synaptotagmin 1, forward 5′-GAA CTG CAT AAA ATC CCA TTG CC-3′ and reverse 5′-CAG CAG GTC ACG ACT AGA AGG-3′; Vglut1, forward 5′-CCG GCA GGA GGA GTT TCG GAA G-3′ and reverse 5′-GTG CAG TCC ACC ACA GGC GG-3′; PSD-93, forward 5′-CTT GTG CAT GTG TCG GAA AAG-3′ and reverse 5′-TCA GAG GAG AGA TGT GAG ACT G-3′; CaMKIIβ, forward 5′-CGT TTC ACC GAC GAG TAC CAG-3′ and reverse 5′-GCG TAC AAT GTT GGA ATG CTT C-3′; CaMKIIα, forward 5′-CGC ATC TGC CGC TTG TT-3′ and reverse 5′-TCC TCG GAG ATG CTG TCA TG-3′; NMDAR2A, forward 5′-AGC CCC CTT CGT CAT CGT AGA-3′ and reverse 5′-ACC CCT TGC AGC ACT TCT TCA C-3′; GABABR2, forward 5′-ATG CAA TAA AGT ATG GGC CGA A-3′ and reverse 5′-CGC GAT GAT AGA GGT GAC AGA-3′; for GluR2, forward 5′-TTC TCC TGT TTT ATG GGG ACT GA-3′ and reverse 5′-CCC TAC CCG AAA TGC ACT GTA-3′; GluR3, forward 5′-GTG CAG TTA TAC AAC ACC AAC CA-3′ and reverse 5′-GAG CAG AAA GCA TTA GTC ACA GA-3′.
Western blot analysis.
Protein extracts were prepared from different parts of the brain using the modified Dignam method (Ballas et al., 2009). Tissues were homogenized in a Dounce homogenizer with the loose pestle, directly in high-salt lysis buffer. For whole-brain extract, tissues were homogenized in a Dounce homogenizer with the loose pestle in lysis buffer containing 5 mm sodium phosphate, pH 7.2, 320 mm sucrose, 1 mm sodium fluoride, and 1 mm sodium orthovanadate and protease inhibitors (Roche). The lysates were centrifuged at 4°C for 15 min at 700 × g. Supernatants were collected and Western blotting was performed as described previously (Ballas et al., 2009). The primary antibodies used are as follows: chicken anti-MeCP2 antibody (a generous gift from J.M. LaSalle, University of California, Davis, Davis, CA) (1:10,000) or a mouse anti-MeCP2 (Sigma) (1:2000), mouse anti-CaMKIIα (Millipore) (1:2000), rabbit anti-CaMKIIβ (Abcam) (1:5000), rabbit anti-GluR2/3 (Millipore) (1:500), mouse anti-GABABR2 (Neuromab) (1:2000), mouse anti-Vglut1 (Neuromab) (1:250), rabbit anti-NMDAR2A (Abcam) (1:200), mouse anti-PSD-93 (Neuromab) (1:500), rabbit anti-Synaptotagmin 1 (Synaptic Systems) (1:1000), rabbit anti-synapsin 1 (Abcam) (1:1000), mouse anti-PSD-95 (Neuromab) (1:1000), and rabbit anti-β actin (Abcam) (1:2000) was used for loading control. The secondary antibodies used were IRDye800 or IRDye700 conjugated (LI-COR Biosciences) (1:10,000). Membranes were scanned using the Odyssey-Infrared Imaging System (LI-COR Biosciences), and the intensities of the bands of interest were determined from the captured images using the Odyssey imaging software.
Immunohistochemistry.
Mice were fixed by transcardial perfusion with PBS-buffered 4% paraformaldehyde as described previously (Ballas et al., 2009). Briefly, brains were removed and postfixed overnight with PBS-buffered 4% paraformaldehyde at 4°C. The fixed tissue was cryoprotected with 30% sucrose in PBS buffer and frozen in optimal cutting medium (Neg50, Richard-Allan Scientific). Coronal sections were obtained by sectioning the tissue on a HM505E Microm Cryostat (Cryostat Industries) at 40 μm and preserved at −20°C in PBS-buffered 50% glycerol until analyzed. The sections were immunostained using rabbit anti-MeCP2 antibody (a generous gift from M. Greenberg, Harvard Medical School, Boston, MA) (1:500), chicken anti-GFAP (Millipore Bioscience Research Reagents) (1:500), or mouse anti-NeuN (Millipore Bioscience Research Reagents) (1:150) followed by incubation with the appropriate secondary antibodies conjugated to cyanine (Jackson ImmunoResearch). MeCP2 immunostaining was enhanced using biotinstreptavidin-conjugated secondary antibodies. Images were collected on a Zeiss confocal laser-scanning LSM 510 microscope.
Nissl staining.
Brains were harvested and sectioned as described above. Consecutive coronal sections (40 μm) were mounted on gelatin-coated slides, rehydrated, stained with cresyl violet dye (Sigma), and mounted on coverslips with DPX. Images were taken with a Zeiss Axioskop microscope with SPOT software (Diagnostic Instrument). To measure the thickness of the pyramidal layer of the CA1 region of the hippocampus and the density of cells in layer V of the cortex, images of several sections from each animal (n = 3) were analyzed using the ImageJ software. Five different measurements were performed along the pyramidal layer of the CA1 region for each image. The number of cells present in each image of the Layer V were counted and normalized per square micrometer.
Golgi staining.
Golgi staining was performed using a modified Golgi method (EZ Golgi method, Weill Cornell Medical College, New York, NY) and performed according to the manufacturer's instructions. Three brains of Veh- or Tam-injected MeCP2loxJ/y/CreER mice were incubated in the impregnation solution and sectioned at a thickness of 150 μm. Sixteen to seventeen pyramidal neurons in the CA1 region of the hippocampus and 15 astrocytes located between the myelinated fibers and the compacted neuronal cell body layers of the CA1 region were traced and reconstructed using a camera lucida device blinded to genotype and treatment. Reconstructed neurons and astrocytes were analyzed using Neurolucida explorer software. For Sholl analysis (Sholl, 1953), the concentric circles were 10 μm. Dendritic spines were counted along 100 μm lengths.
Brain-weight assessment.
Mice were anesthetized at the appropriate age with isoflurane, and the brains were harvested and immediately weighted. Whole brains were excised by a single cut at the caudal edge of the cerebellum between the brain and the spinal cord.
Results
Inducible loss of MeCP2 in late juvenile or adult mice results in severe RTT-like phenotype with similar kinetics of symptom initiation and progression
To induce MeCP2 loss at specific postnatal stages, we crossed a transgenic mouse line carrying a Tam-inducible Cre recombinase (CreER) transgene driven by the ubiquitous CMV/β-actin promoter with a mouse line carrying floxed Mecp2 allele (Mecp2loxJ) (Chen et al., 2001), which generate mutation in Mecp2 upon excision of the loxP cassette (Fig. 1A, top). Male progeny harboring the CreER and the Mecp2lox alleles (Mecp2loxJ/y/CreER) were injected with Tam for seven consecutive days starting at two different time points: (1) a late juvenile stage (5 week old, referred to as 5W-Tam injected), a time at which the brain is still growing and developing and that parallels the time period of onset of RTT symptoms in male mice with germline mutation in Mecp2 (Chen et al., 2001, Guy et al., 2001); or (2) an adult stage (10 week old, referred to as 10W-Tam injected), where the brain has matured in term of reaching its full size and dendritic complexity (Fig. 1A, bottom). We determined the efficiency of the floxed Mecp2 excision and found that MeCP2 was undetectable by immunostaining in most neurons and astrocytes in brains of 5W-Tam-injected Mecp2loxJ/y/CreER mice (Fig. 1B,C). Quantitative Western blot analysis showed 80–90% reduction in the MeCP2 protein content in all areas of the brain including cortex, hippocampus, cerebellum, and brainstem regions (Fig. 1D). Importantly, Mecp2-lox excision in the 10W-Tam-injected Mecp2loxJ/y/CreER mice was as efficient as in the 5W-Tam-injected Mecp2loxJ/y/CreER mice under the same conditions (Fig. 1E,F), allowing a direct and unbiased comparison of the effect of MeCP2 loss at the late juvenile and adult stages.
Figure 1.
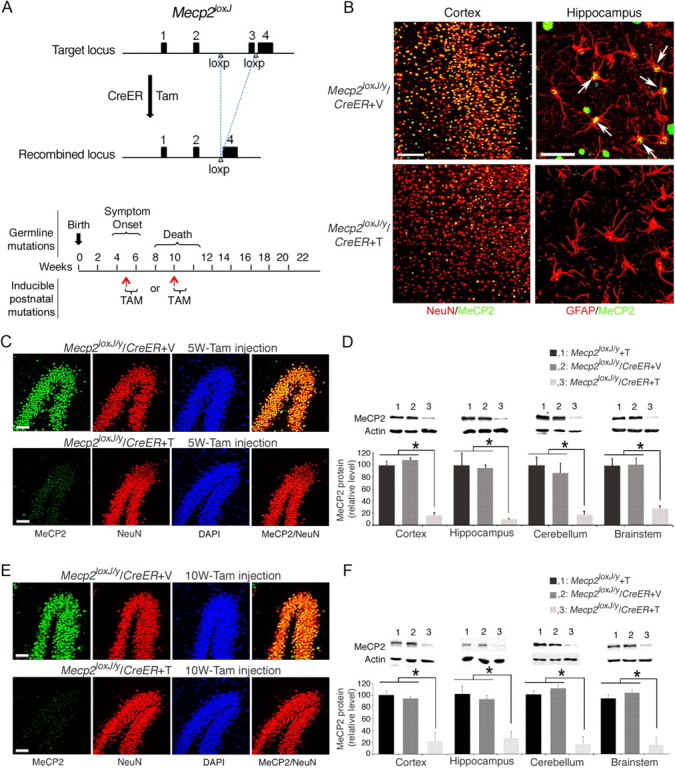
MeCP2 protein is efficiently depleted in brains of Mecp2loxJ/y/CreER mice injected with Tamoxifen at 5 or 10 weeks of age. A, Top, Schematic diagram showing the Tam-induced excision of the floxed Mecp2 cassette using the Jaenisch (J) Mecp2-lox mouse line and the CreER mouse line. Bottom, Schematic diagram showing the time period of onset and progression of RTT symptoms in male mice with germline mutations in Mecp2 and the postnatal stages at which Mecp2 was inactivated upon Tam injections. Red arrows indicate the age of the mice when first injected with tamoxifen. B, Representative images of immunostaining of brain sections of 5W-Tam- or Veh-injected Mecp2loxJ/y/CreER mice immunolabeled for MeCP2 (green) and NeuN (red), 19 weeks after Tam (T) or Veh (V) injection. Note that MeCP2 (green) is absent in most neurons (NeuN+, red) and astrocytes (GFAP+, red) in the cortex and hippocampus of the Tam-injected mice, respectively. Scale bars: left panels, 100 μm; right panels, 20 μm. C, E, Representative images of the dentate gyrus area of 5W- (C) or 10W- (E) Tam- or Veh-injected Mecp2loxJ/y/CreER mice immunolabeled for MeCP2 (green), and NeuN (red). DAPI (blue) represents nuclear staining. Scale bar, 50 μm. D, F, Quantitative Western blot showing the levels of MeCP2 in the indicated brain areas of 5W- (D) or 10W- (F) Tam or Veh-injected Mecp2loxJ/y/CreER mice and Tam-injected Mecp2loxJ/y mice. Actin served as loading control. All mice were analyzed 18 weeks after injection. One-way ANOVA followed by appropriate post hoc for multiple-comparisons test was used to determine differences between groups. Error bars are mean ± SEM *p < 0.05. n = 3 mice per genotype.
We first analyzed whether MeCP2 loss-of-function at a late juvenile stage, in otherwise normal healthy brain, could cause overt RTT-like phenotypes characteristic of the corresponding germline Mecp2-null mouse model (Mecp2J−/y) (Chen et al., 2001) and the kinetics of the appearance of symptoms. We used the observational phenotypic scoring system (0–10) described previously (Guy et al., 2007) for five typical phenotypes manifested in germline MeCP2-null mice, which we termed as classic RTT: mobility, gait, hindlimb clasping, tremors, and general condition. Weekly scoring of the Tam-injected Mecp2loxJ/y/CreER progeny showed dramatic progression of aggregate symptom scores including all five categories, beginning as early as 1 week after the last Tam injection and reaching a mean score of 8 at 18 weeks after injection followed by lethality, with 50% survival at 16 weeks after Tam injection (Fig. 2A–C). Veh-injected Mecp2loxJ/y/CreER mice and all other Tam-injected control mice remained equally healthy (Fig. 2A–C). Similar results were obtained when a different floxed Mecp2 mouse line (Guy et al., 2001) was used to generate Tam-inducible Mecp2loxB/CreER mice (Fig. 2D–F), suggesting that the severe RTT phenotype observed was related directly to the functional loss of MeCP2 and not dependent on the specific mouse line or type of Mecp2 mutation generated upon Cre excision. Furthermore, the phenotypic deficits were similar to those observed in mice with germline mutation in Mecp2 (classic RTT). These data show that RTT-like phenotypes can be initiated and efficiently progressed as soon as Mecp2 is mutated at 5 weeks of age, a postnatal stage, which coincides with the onset of classic RTT in male mice.
Figure 2.
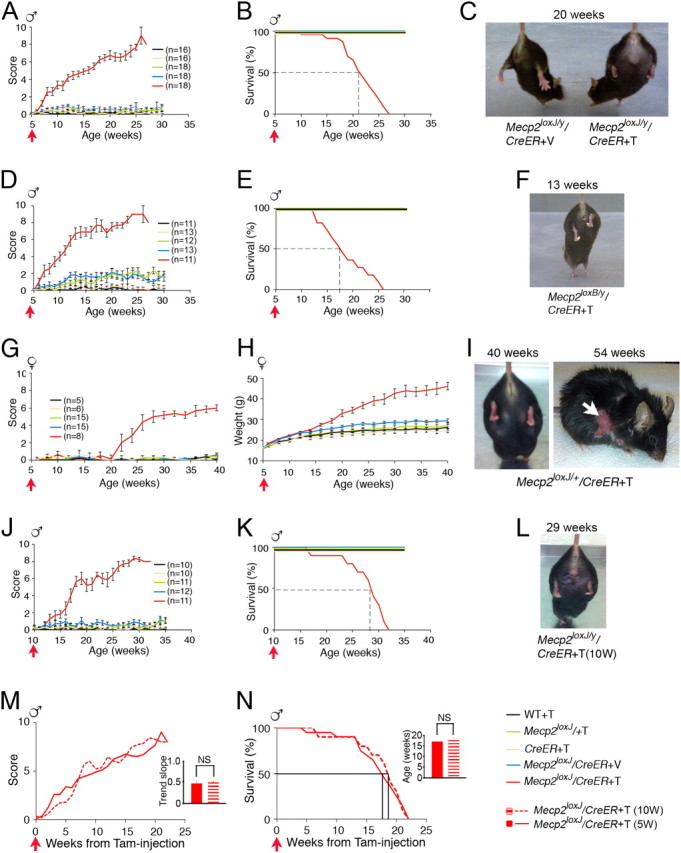
The loss of MeCP2 at late juvenile or adult stages induces RTT-like phenotypes in male and female mice. A, B, Phenotypic scores and Kaplan–Meier survival time course, respectively, of 5W-Tam-injected Mecp2loxJ/y/CreER and the indicated control mice. The number of mice in B is similar to the indicated in A. D, E, Phenotypic scores and Kaplan–Meier survival time course, respectively, of 5W-Tam-injected Mecp2loxB/y/CreER and the indicated control mice. The number of mice in E is similar to the indicated in D. G, H, Phenotypic scores and weight gain time course, respectively, of 5W-Tam-injected Mecp2loxJ/+/CreER female mice and the indicated control mice. The number of mice in H is similar to G. J, K, Phenotypic scores and Kaplan–Meier survival time course graphs, respectively, of 10W-Tam-injected Mecp2loxJ/y/CreER and the indicated control mice. The number of mice in K is similar to the indicated in J. M, Overlay of the aggregate symptom scores from the time of injection of the 5W-Tam-injected Mecp2loxJ/y/CreER and the 10W-Tam-injected Mecp2loxJ/y/CreER mice. Inset shows no significant differences in the mean of the trend slopes between the 5W- and 10W-Tam-injected mice (NS, p > 0.05). Error bars are mean ± SEM. t test was used to compare within the 5W- and the 10W-Tam-injected groups. N, Overlay of the survival curves of the 5W- and 10W-Tam-injected Mecp2loxJ/y/CreER mice. A LogRank survival test showed no significant difference between the survival time of the 5W- and the 10W-Tam-injected Mecp2loxJ/y/CreER mice. Inset shows no significant differences in the mean age of death between the 5W- and 10W-Tam-injected mice (NS, p > 0.05). Error bars are mean ± SEM. t test was used to compare within the 5W- and the 10W-Tam-injected groups. C, F, I, L, Images illustrating the hindlimb clasping phenotype of the indicated Tam-injected MeCP2loxJ/CreER or Tam-injected MeCP2loxB/CreER mice. Note that the Tam-injected MeCP2loxJ/y/CreER mice (male and female) are overweight and that the female is injured due to overgrooming (white arrow). All the mice in the images were injected with Tam at 5 weeks of age except in L, where the mouse was injected at 10 weeks of age (10 W). Red arrows show the time of Tam or Veh injection.
Because human RTT affects mostly girls, we sought to determine whether the loss of MeCP2 in heterozygous female mice at a late juvenile stage could also initiate RTT-like phenotypes. The 5W-Tam-injected Mecp2loxJ/+/CreER female mice showed efficient excision with up to 50% reduction in Mecp2 mRNA (data not shown). Biweekly scoring of the Tam-injected Mecp2loxJ/+/CreER mice showed that, unlike in the male mice, the onset of symptoms appeared 16 weeks after Tam injection, reaching an average score of 5–6 at 22 weeks postinjection followed by stabilization of the symptoms (Fig. 2G). All other control mice remained healthy throughout the period analyzed (Fig. 2G). None of the Tam-injected Mecp2loxJ/+/CreER mice died, but 4 of 8 mice had to be killed due to a severe self-injurious overgrooming (Fig. 2I, right image), a repetitive autistic-like behavior. Furthermore, female mice became extremely obese with up to 100% increase in body weight (Fig. 2H; I, left image). The delayed onset and the slow kinetics of the appearance of symptoms followed by their stabilization, as well as the type of symptoms, are closely correlated with the classic form of RTT in female mice (Mecp2−/+) (Chen et al., 2001).
Next, we examined the effect of MeCP2 loss at the adult stage, when the brain is mature and has attained its full size. For this, Mecp2loxJ/y/CreER mice were injected with Tam at 10 weeks of age, and analyzed for overt RTT-like symptoms. Our data show, that like the juvenile mice, adult mice also developed RTT-like symptoms beginning as early as 1 week after the last Tam injection and reaching an average score of 8, 16 weeks from injection (Fig. 2J,L). Furthermore, like the juvenile mice, the Tam-injected adult mice all died by 21 weeks after injection with 50% survival at 16–17 weeks after Tam injection (Fig. 2K). The inducible loss of MeCP2 at late juvenile and adult stages (Fig. 1) allowed us to directly compare the effect of MeCP2 loss at the two different developmental stages. Surprisingly, comparison of the kinetics of symptom progression and the survival rate from the time of Tam injection showed no significant differences between juvenile and adult mice (Fig. 2M,N), suggesting similar and absolute requirement for MeCP2 at the critical stage of brain maturation (late juvenile) and after, when the brain is already mature (adult).
We further compared the Tam-injected juvenile and adult male mice for behavioral deficits at 13 weeks after injection, a stage when they were not severely symptomatic but reached a median score of 6 (Fig. 2M). We found that the total activity in a digiscan activity monitor of the 5W- and 10W-Tam-injected Mecp2loxJ/y/CreER mice was reduced by 40–50% relative to all other control mice (Fig. 3A,C). In a wire hang test, the 5W- and the 10W-Tam-injected Mecp2loxJ/y/CreER mice showed altered forelimb strength with shorter latency to fall (Fig. 3D,F), and their motor coordination evaluated on an accelerating rotarod was impaired, as evident by their short latency to fall (Fig. 3G,I). We also evaluated the 5W-Tam-injected heterozygous female mice at 27 weeks after Tam injection, a stage when their symptoms stabilized and reached a mean score of 6, and found poor performance in all tests (Fig. 3, compare B,E,H,K, A,D,G,J). In fact, the female mice performed worse than the male mice, likely due to their severe obesity. These behavioral deficits are comparable to the known behavioral deficits in classic RTT (Ricceri et al., 2008).
Figure 3.

Mice that lost MeCP2 at a late juvenile or adult stage show similar behavioral deficits. Male and female mice were subjected for the indicated behavioral tests at 13 weeks or 27 weeks after injection, respectively. A–K, Total activity test in digiscan activity monitor (A–C), wire-hang test (D–F), rotarod test (G–I), and dowel test (J, K). The colors of the graphs and bar graphs correspond to the indicated genotypes in the right bottom corner. A, D, G, J, 5W-Tam-injected Mecp2loxJ/y/CreER and the indicated control mice. B, E, H, K, 5W-Tam-injected Mecp2loxJ/+/CreER and the indicated control female mice. C, F, I, 10W-Tam-injected Mecp2loxJ/y/CreER and the indicated control male mice. The number of mice in D and G is similar to that indicated in A; in E and H it is similar to B; in F and I it is similar to C. Note that one outlier 10W-Tam-injected Mecp2loxJ/y/CreER mouse whose score was more than two SDs from the group mean was removed from the rotarod analysis in I. One-way ANOVA followed by appropriate post hoc for multiple-comparisons test was used to determine differences between groups. Error bars are mean ± SEM. *p < 0.05 represents significant differences between the different Tam-injected Mecp2loxJ/CreER mouse groups and all control littermate groups.
Inducible loss of MeCP2 at late juvenile or adult stage causes global brain shrinkage and compaction of neuronal cell bodies
To understand the underlying cause of RTT-like phenotype and behavioral deficits in mice that lost MeCP2 at a late juvenile or adult stage, we analyzed in depth the anatomy of the brain of the symptomatic mice and the morphology of their neurons and glia. Because in our mouse models the brain was similar in size to the wild-type brain at the time of Tam injection, we first asked whether the loss of MeCP2 would negatively affect the brain size. Unexpectedly, we found that the loss of MeCP2 upon Tam treatment of Mecp2loxJ/y/CreER mice, whether at a late juvenile or adult stage, both resulted in significantly reduced brain size when compared with Veh-injected Mecp2loxJ/y/CreER or Tam-injected Mecp2loxJ/y age-matched littermates (Fig. 4A,B). Time course analysis of normal brains from wild-type mice showed that the brain reaches its full size only at ∼10 weeks of age (Fig. 4C). Thus, Tam injection at 5 or 10 weeks of age represents two different developmental stages in terms of global brain maturation. 5W-Tam-injected Mecp2loxJ/y/CreER mice, like all other control male mice, had similar brain weights by the end of the seventh consecutive injection (Fig. 4E, 6 weeks of age). However, at 24 weeks of age (18 weeks after injection), while the size of the brains of all control mice were similar and significantly larger than the brain size of the corresponding mice at 6 weeks of age (Fig. 4C,E), the brains of the Tam-injected Mecp2loxJ/y/CreER mice were significantly smaller (by 10%) than the brains of all control mice at 24 weeks of age (Fig. 4C,E). In fact, at 24 weeks of age, the brains of the symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice were even smaller (by 5%) than the normal wild-type brains at 5 weeks of age, the starting point of Tam injections, and smaller than all other control mice right after the last Tam injection (6 weeks of age) (Fig. 4C,E). These results suggest that the reduced brain size is likely caused, at least in part, by shrinkage of the developing brain. This phenomenon is even more noticeable in brains of symptomatic Mecp2loxJ/y/CreER mice treated with Tam at 10 weeks of age, a time by which the normal brain reaches its full size. Indeed, the brains of all control mice as well as the Tam-injected Mecp2loxJ/y/CreER mice were similar in size right after injection of Veh or Tam (11 weeks of age) and similar to the brain size of the control mice at 28–32 weeks of age (Fig. 4D,F). However, at 28–32 weeks of age, the brain size of symptomatic 10W-Tam-injected Mecp2loxJ/y/CreER mice was reduced by 10% relative to the brain size of the different control mice or to the brain size of the same Mecp2loxJ/y/CreER mice right after injection (Fig. 4D,F). To verify that the reduced brain size of symptomatic 5W- or 10W-Tam-injected Mecp2loxJ/y/CreER mice is brain specific and not due to simply reduced body size, we analyzed the body weight from the time of injection to their severe symptomatic period. This analysis revealed that in fact the 5W- or 10W-Tam-injected Mecp2loxJ/y mice were somewhat overweight compared with all other control littermate mice (Fig. 4H,I). The body weight gain was previously reported with the same mutation in male mice with classic RTT (Chen et al., 2001). Remarkably, similar to the males, the brains of the 5W-Tam-injected Mecp2loxJ/+/CreER heterozygous female mice, which were extremely obese at their symptomatic stage (Fig. 2G–I), were also reduced in size by 10% (Fig. 4G). This suggests that even though MeCP2 loss is mosaic and occurs only in 50% of the cells at most, the effect on the anatomy of the female brain is global. Together, these data strongly suggest that postnatal loss of MeCP2, whether at late juvenile or adult stage, elicited shrinkage of the mature healthy brain in hemizygous males as well as in heterozygous females.
Figure 4.

The loss of MeCP2 at late juvenile or adult stage leads to shrinkage of the brain. A, B, Representative images showing that symptomatic Mecp2loxJ/y/CreER mice (loxJ/y/Cre+T), injected with Tam at 5 (A) or 10 (B) weeks of age, have smaller brains than their control littermates, Mecp2loxJ/y+T (loxJ/y+T) and Mecp2loxJ/y/CreER+V (loxJ/y/Cre+V) mice. Brains are from mice at 16 weeks after injection. C, D, Age-dependent changes in brain weights of WT, 5W- (C) or 10W- (D) Tam- or Veh-injected Mecp2loxJ/y/CreER and Mecp2loxJ/y mice. Fragmented lines distinguish between the brains that are smaller (below) or bigger (above) than the Mecp2loxJ/y/CreER brains right after the last Tam injection (6W or 11W). E, Brains of the 5W-Tam-injected Mecp2loxJ/y/CreER mice are smaller than their control littermates at 24 weeks of age (18 weeks after injection), and smaller than the WT brains at 5 weeks of age and the 5W-Tam-injected Mecp2loxJ/y/CreER at 6 weeks of age (after the last Tam injection). F, Brains of the 10W-Tam-injected Mecp2loxJ/y/CreER mice are smaller than the brains of the 10W-Veh-injected Mecp2loxJ/y/CreER mice at 29–32 weeks of age (18–21 weeks after injection) and smaller than the WT brains at 10 weeks of age and the 10W-Tam-injected Mecp2loxJ/y/CreER brains at 11 weeks of age (after the last Tam injection). G, Brains of the 5W-Tam-injected Mecp2loxJ/+/CreER female mice are smaller than the brains of their control littermates at 39–50 and 79–99 weeks of age. Two-way ANOVA analysis followed by appropriate post hoc for multiple-comparison tests were performed to determine differences between Veh/Tam groups and between the different age groups. Error bars are mean ± SEM. *p < 0.05. n = 4–6 mice per genotype. H, I, Time course analysis of body weight gain of 5W- (H) or 10W- (I) Tam-injected Mecp2loxJ/y/CreER mice and the indicated control male mice. n indicates the number of mice used. Red arrows indicate the time of Tam or Veh injection.
Analysis of the histology of the brains of the 5W- and 10W-Tam-injected Mecp2loxJ/y/CreER mice showed global shrinkage of the brain with no other detectable malformations (Fig. 5A,B). Detailed analysis of the hippocampus showed overall shrinkage of all areas, including CA1, CA2, CA3, and the dentate gyrus, of both 5W- and 10W-Tam-injected Mecp2loxJ/y/CreER mice in their symptomatic stage compared with the hippocampus of Veh-injected Mecp2loxJ/y/CreER littermate mice (Fig. 5C,D). Furthermore, the thickness of the pyramidal layer of CA1 area in the symptomatic brains was significantly reduced (up to 30%), and neuronal cell bodies appeared more compact (Fig. 5E–H). The cortical area of symptomatic 5W-and 10W-Tam-injected Mecp2loxJ/y/CreER mice was affected similarly with 10–15% higher cell density than in the cortex (layer V) of Veh-injected Mecp2loxJ/y/CreER mice (Fig. 5I–L).
Figure 5.
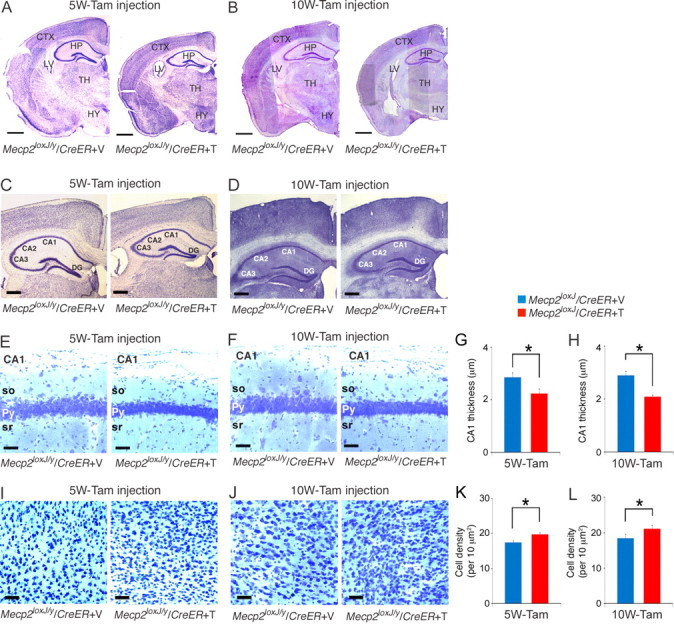
The brain anatomy is severely affected upon the loss of MeCP2 at a late juvenile or adult stage. A, B, Images of Nissl staining showing smaller brain structures for the 5W- (A) and 10W- (B) Tam-injected mice at 24 and 30 weeks of age, respectively, when compared with their control Veh-injected littermates. CTX, Cortex; LV, lateral ventricle; HP, hippocampus; TH, thalamus; HY, hypothalamus. Scale bars, 800 μm. C, D, Representative images of Nissl staining showing smaller hippocampal structure for the 5W- (C) and 10W- (D) Tam-injected mice. DG, Dentate gyrus. Scale bars, 400 μm. E, F, The cell bodies of the pyramidal (Py) neurons in the CA1 area of the hippocampus of the 5W- (E) and 10W- (F) Tam-injected Mecp2loxJ/y/CreER mice are more compact. so, Stratum oriens; py, pyramidal layer; sr, radiatum. Scale bars, 20 μm. G, H, The thickness of the pyramidal neuronal cell bodies in the CA1 area of the 5W- (G) and 10W- (H) Tam-injected Mecp2loxJ/y/CreER mice is significantly reduced. I, J, Representative images of Nissl staining showing a higher density of neuronal cell bodies in layer V of the motor cortex of the 5W- (I) or 10W- (J) Tam-injected mice when compared with 5W- or 10W-Veh-injected Mecp2loxJ/y/CreER mice. Scale bars, 40 μm. K, L, The density of the neurons in layer V of the motor cortex of 5W-(K) and the 10W- (L) Tam-injected Mecp2loxJ/y/CreER mice is significantly higher than the Veh-injected control mice. t test was used to compare within Veh and Tam groups. Error bars are mean ± SEM. *p < 0.05. n = 3 mice per genotype.
The loss of MeCP2 at late juvenile or adult stage induces retraction of dendritic arbor structures of mature neurons and reduces the complexity of astrocytic processes
Next, we analyzed the morphology of the pyramidal neurons in the CA1 area of the hippocampus in mice that lost MeCP2 at a late juvenile or adult stage. For this, we evaluated the hippocampal dendritic complexity of symptomatic Tam-injected Mecp2loxJ/y/CreER mice, 18 weeks after injection, using Golgi impregnation. Interestingly, comparison of the CA1 pyramidal neurons of the 5W-Veh- and Tam-injected Mecp2loxJ/y/CreER mice revealed that the Tam-injected Mecp2loxJ/y/CreER mice developed severe dendritic anomalies (Fig. 6A). While the number of primary dendrites was unchanged, Sholl and branch analyses showed that the pyramidal neurons of Tam-injected Mecp2loxJ/y/CreER mice exhibited fewer and shorter basal and apical dendritic branches when compared with Veh-injected Mecp2loxJ/y/CreER littermates at 24 weeks of age (18 weeks after injection) (Fig. 6A–G). Such abnormalities in dendritic arbor structure were previously found in human RTT and RTT mouse models with germline mutations in MeCP2 (Armstrong et al., 1995, Kishi and Macklis, 2004, Ballas et al., 2009). Potentially, these differences in dendritic arbors could be due to developmental stagnation, but they could also be caused by retraction of dendritic arbors. The ability to induce postnatal loss of MeCP2 in our mouse model allowed us to distinguish between these two possibilities. For this, we analyzed the dendritic complexity of 5W-Tam-injected Mecp2loxJ/y/CreER mice immediately after the last Tam injection (6 weeks of age). At this time, MeCP2 was still present, but was reduced by 30–40% of its normal level (data not shown), and the mice appeared healthy (Fig. 2A). Comparison of the dendritic complexity of the Tam-injected Mecp2loxJ/y/CreER mice at 6 weeks of age and of the Veh-injected Mecp2loxJ/y/CreER mice at 24 weeks of age indicated that the dendritic complexities of the pyramidal neurons are largely similar to each other except for the basal dendritic branches closer to the soma, which seemed less complex in the 6-week-old Tam-injected Mecp2loxJ/y/CreER mice (Fig. 6B). At 24 weeks of age, however, the Tam-injected Mecp2loxJ/y/CreER mice showed significant reduction in the number and length of both apical and basal dendritic branches even when compared with Tam-injected Mecp2loxJ/y/CreER mice right after injection (6 weeks of age) (Fig. 6B–G). These results suggest that the loss of MeCP2 in the developing 5-week-old mice mediated retraction of the already existing dendritic arbors. Interestingly, the observed reduced dendritic complexity was even more robust than previously reported for classic RTT, suggesting a central role for MeCP2 in maintaining the dendritic complexity of the maturing and mature neurons. Importantly, analysis of the dendritic spines showed robust reduction (up to 40%) in spine density of pyramidal neurons of the 5W-Tam-injected Mecp2loxJ/y/CreER mice at their symptomatic stage when compared with Veh-injected Mecp2loxJ/y/CreER or Tam-injected wild-type age-matched littermates (Fig. 6H,I). Furthermore, the associated dendritic branches showed morphological abnormalities, such as swelling and thickening (Fig. 6H).
Figure 6.
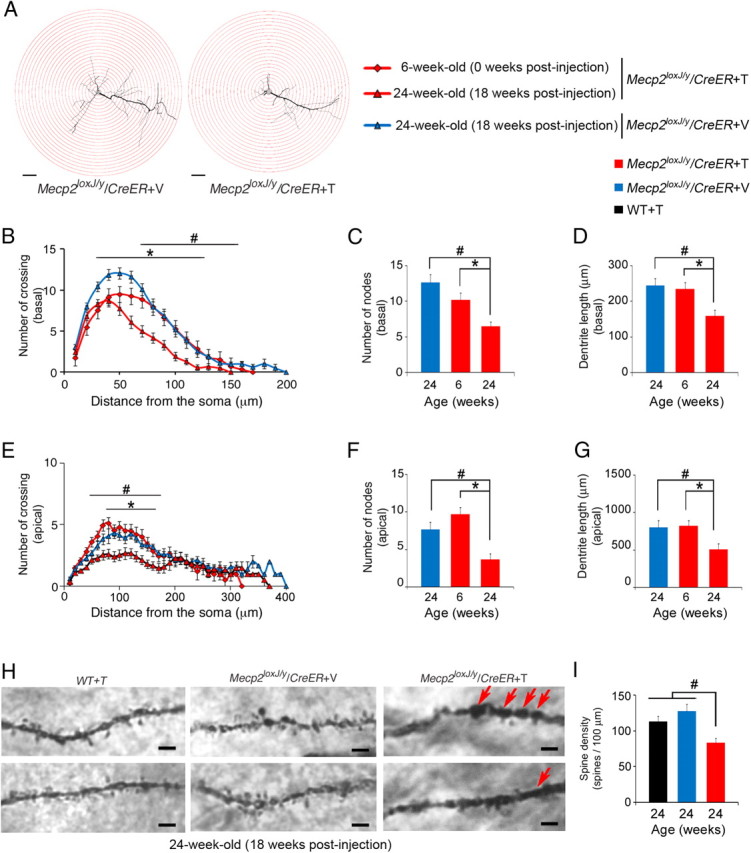
Dendritic complexity of hippocampal pyramidal neurons is severely affected upon the loss of MeCP2 at late juvenile stage. A, Representative Camera lucida tracing of CA1 pyramidal neurons in the hippocampus of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER and the 5W-Veh-injected Mecp2loxJ/y/CreER control littermate mice. Concentric circles for Sholl analysis in 10 μm radius increments are superimposed in red. Scale bars, 40 μm. B, E, Sholl analysis of the 5W-Tam-injected mice showing that the basal (B) and apical (E) dendrites of CA1 pyramidal neurons at 18 weeks after the last Tam injection (24 weeks of age) are significantly less complex than those of the 5W-Veh-injected Mecp2loxJ/y/CreER age-matched littermate mice and even less complex than the dendrites of Tam-injected mice right after the last injection (6 weeks of age). Two-way repeated-measures ANOVA followed by appropriate post hoc for multiple-comparisons test was used to determine differences within Veh and Tam groups at 18 weeks after injection or within 0 and 18 weeks after the last Tam injection of 5W-Tam-injected Mecp2loxJ/y/CreER mice. C, D, F, G, CA1 pyramidal neurons of 5W-Tam-injected Mecp2loxJ/y/CreER mice exhibit fewer and shorter basal (C, D) and apical (F, G) dendritic branches at 18 weeks after Tam injection. Mann and Whitney test was used to compare within Veh and Tam groups at 18 weeks after injection or within the 5W-Tam-injected Mecp2loxJ/y/CreER mice at 0 and 18 weeks after injection. H, Representative images of Camera lucida tracing of CA1 pyramidal neurons in the hippocampus showing structural abnormalities in spines and the associated secondary dendrites of the 5W-Tam-injected Mecp2loxJ/y/CreER mice when compared with Veh-injected Mecp2loxJ/y/CreER or Tam-injected wild-type (WT) mice, 18 weeks after injection. Arrows indicate swelling or thickening of the associated dendritic branches. Scale bars, 2 μm. I, CA1 pyramidal neurons of 5W-Tam-injected Mecp2loxJ/y/CreER mice demonstrate significant reduction in dendritic spine density. Spine density per 100 μm length was measured on secondary dendrites. One-way ANOVA followed by appropriate post hoc for multiple-comparison tests was used to determine differences between groups. Error bars are mean ± SEM. *p < 0.05 significant differences in dendritic complexity of the 5W-Tam-injected Mecp2loxJ/y/CreER at 0 and 18 weeks after the last injection. #p < 0.05 significant differences between 5W-Tam and Veh-injected Mecp2loxJ/y/CreER mice at 24 weeks of age (18 weeks after injection). n = 3 mice per genotype and age. n = 5–6 neurons per animal.
Notably, analysis of the pyramidal neurons of adult 10W-Tam-injected Mecp2loxJ/y/CreER mice, 18 weeks after injection (29 weeks of age), showed similar aberrations in basal and apical dendritic complexity when compared with 10W-Veh-injected Mecp2loxJ/y/CreER age-matched littermates (Fig. 7A–F). Similarly, comparison of the 10W-Tam-injected Mecp2loxJ/y/CreER mice, immediately after injection (11 weeks of age), when MeCP2 levels are reduced by 30–40% (data not shown), and at their symptomatic stage (29 weeks of age), shows dramatic reduction in both basal and apical dendritic complexity (Fig. 7A–F), further supporting the idea that dendritic arbors were retracted from a more mature state in the absence of MeCP2. Similar to the 5W-Tam-injected mice, the 10W-Tam-injected mice showed also significant reduction in spine density (Fig. 7H). Furthermore, the associated dendritic branches showed morphological abnormalities, such as swelling and thickening, similar to the 5W-Tam-injected mice (Fig. 7G). Together, these data indicate that inducible loss of MeCP2, whether at late juvenile or adult stage, mediates dendritic abnormalities, reflected in significant reduction in dendritic arborization as well as in spine density.
Figure 7.
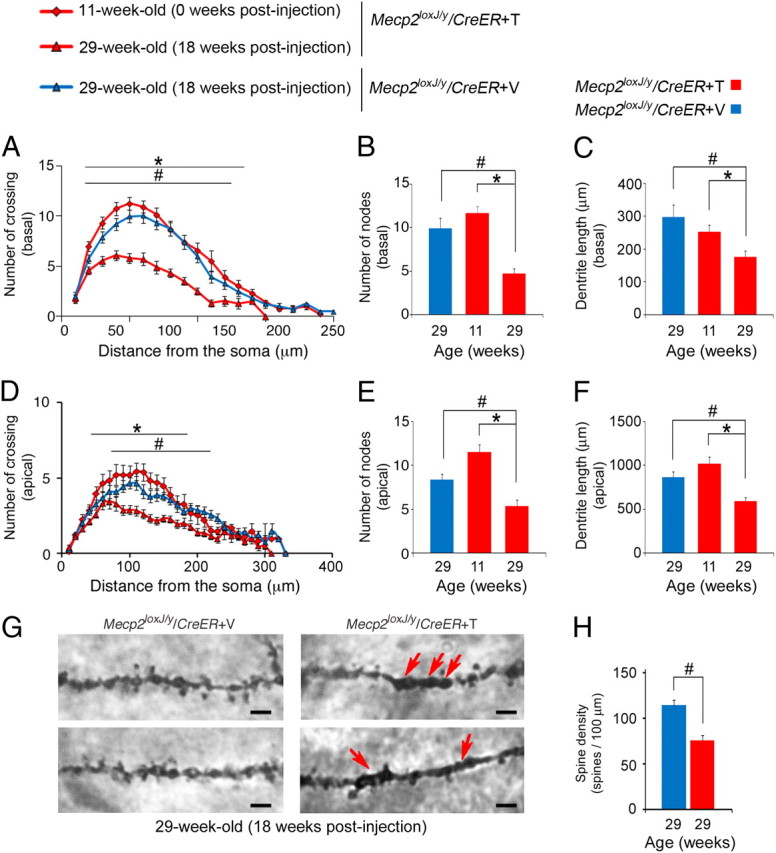
The loss of MeCP2 at adult stage induces reduction in dendritic complexity and spine density. A, D, Sholl analysis of the 10W-Tam-injected mice showing that the basal (A) and apical (D) dendrites of CA1 pyramidal neurons at 29 weeks of age (18 weeks after Tam injection) are significantly less complex than those of 11 weeks of age (right after the last Tam injection) and less complex than the dendrites of the 10W-Veh-injected Mecp2loxJ/y/CreER age-matched littermates. Two-way repeated-measures ANOVA followed by appropriate post hoc for multiple-comparisons test was used to determine differences within Veh and Tam groups at 18 weeks after injection or within 0 and 18 weeks after the last Tam injection of 10W-Tam-injected Mecp2loxJ/y/CreER mice. B,C, E, F, The CA1 pyramidal neurons of 10W-Tam-injected Mecp2loxJ/y/CreER mice exhibit fewer and shorter basal (B, C) and apical (E, F) dendritic branches at 29 weeks of age (18 weeks after injection). Mann and Whitney test was used to compare within Veh and Tam groups at 18 weeks after injection or within the 10W-Tam-injected Mecp2loxJ/y/CreER mice at 0 and 18 weeks after injection. G, Representative images of Camera lucida tracing of CA1 pyramidal neurons in the hippocampus showing structural abnormalities in spines and the associated secondary dendrites of the 10W-Tam-injected Mecp2loxJ/y CreER mice when compared with Veh-injected Mecp2loxJ/y/CreER mice, 18 weeks after injection. Arrows indicate swelling or thickening of the associated dendritic branches. Scale bars, 2 μm. H, CA1 pyramidal neurons of 10W-Tam-injected Mecp2loxJ/y/CreER mice demonstrate significant reduction in dendritic spine density. Spine density per 100 μm length was measured on secondary dendrites. t test was used to compare within Veh and Tam groups. Error bars are mean ± SEM. *p < 0.05 significant differences in dendritic complexity of the 10W-Tam-injected Mecp2loxJ/y/CreER at 0 and 18 weeks after the last injection. #p < 0.05 significant differences between 10W-Tam and Veh-injected Mecp2loxJ/y/CreER mice at 29 weeks of age (18 weeks after injection). n = 3 mice per genotype and age. n = 5–6 neurons per animal.
Our previous studies showed that not only neurons, but also astrocytes contribute to manifestation of RTT neuropathology (Ballas et al., 2009) (Lioy et al., 2011). Therefore, we asked whether the morphology of astrocytes is altered upon depletion of MeCP2. To this end, we analyzed the processes of hippocampal astrocytes in the CA1 area in brains that had been subject to Golgi impregnation. We found that astrocytes of the symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice (18 weeks after injection) had abnormal morphology compared with astrocytes of the Veh-injected Mecp2loxJ/y/CreER age-matched littermates (Fig. 8A), supporting our previous finding of glial involvement in RTT. While the number of primary processes radiating from the cell body of astrocytes of symptomatic mice was not altered, they possessed significantly less ramified processes (30% reduction) as assessed by Sholl analysis (Fig. 8B) and fewer nodes (50% reduction) (Fig. 8C). Moreover, the average length of processes projected was significantly shorter (28% reduction) in astrocytes of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice when compared with astrocytes of Veh-injected Mecp2loxJ/y/CreER age-matched littermates (Fig. 8D). These data indicate that postnatal loss of MeCP2 affects the morphology of neurons as well as glia.
Figure 8.

Processes projected from hippocampal astrocyte cell bodies are severely affected upon the loss of MeCP2 at late juvenile stage. A, Representative Camera lucida tracing of astrocytes in CA1 area of the hippocampus of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER and the 5W-Veh-injected Mecp2loxJ/y/CreER control littermate mice. Concentric circles for Sholl analysis in 10 μm radius increments are superimposed in red. Scale bars, 20 μm. B, Sholl analysis of the 5W-Tam-injected mice showing that the processes of hippocampal astrocytes at 18 weeks after the last Tam injection (24 weeks of age) are significantly less complex than those of the 5W-Veh-injected Mecp2loxJ/y/CreER age-matched littermate mice. Two-way repeated-measures ANOVA followed by appropriate post hoc for multiple-comparisons test was used to determine differences within Veh and Tam groups at 18 weeks after injection. C, D, Hippocampal astrocytes of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice exhibit fewer (C) and shorter (D) branches at 18 weeks after Tam injection. t test was used to compare between Veh and Tam groups. #p < 0.05 significant differences between 5W-Tam and Veh-injected Mecp2loxJ/y/CreER mice at 24 weeks of age (18 weeks after injection). Error bars are mean ± SEM. n = 3 mice per genotype. n = 5 astrocytes per animal.
Postnatal loss of MeCP2 affects expression of both excitatory and inhibitory synaptic proteins
Finally, we asked whether the reduced dendritic complexity and spine density also accompanied alterations in the amounts of synaptic proteins. To this end, we analyzed the levels of several known synaptic proteins with key roles in synaptic structure and plasticity. These experiments revealed dramatic reduction, up to 50%, in the levels of several excitatory synaptic proteins, such as CaMKIIα, CaMKIIβ, and NMDAR2A, as well as the two synaptic vesicle proteins, Vglut1 and Synapsin1, and up to 35% in the level of GluR2/3, in symptomatic Tam-injected Mecp2loxJ/y/CreER mice when compared with Tam-injected Mecp2loxJ/y or Veh-injected Mecp2loxJ/y/CreER littermate mice (Fig. 9A). Furthermore, the level of GABABR2, an inhibitory synaptic protein, was also reduced by up to 40% (Fig. 9A). This reduction in synaptic protein levels was not global, but rather specific, as the levels of the presynaptic protein, Synaptotagmin 1 (Syt1), and the postsynaptic scaffolding proteins, PSD93 and PSD95, were not altered (Fig. 9A). We analyzed more systematically the alteration in CaMKIIα, one of the critical synaptic proteins implicated not only in synaptic plasticity but also in stabilization of dendritic arbor structure. Our results show that the level of CaMKIIα protein was dramatically reduced in all areas of the brains of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice, including cortex and hippocampus (50% reduction), cerebellum, and brainstem (75–80% reduction) (Fig. 9B). Furthermore, the 10W-Tam-injected Mecp2loxJ/y/CreER mice exhibited similar reduction (up to 60%) in CaMKIIα in all brain areas (Fig. 9C). Importantly, using another mouse model with different mutation in Mecp2 (Tam-injected Mecp2loxB/y/CreER; Fig. 2D–F), we found similar dramatic decreases in the level of CaMKIIα in all brain areas (Fig. 9D). These data strongly suggest that the impaired expression of CaMKIIα and likely other synaptic proteins is directly related to the functional loss of MeCP2. Importantly, when we analyzed the mRNA levels of the genes whose protein levels were altered, we found no significant changes (Fig. 9E), suggesting that MeCP2 likely regulates these synaptic proteins by posttranscriptional mechanism, directly or indirectly.
Figure 9.

The loss of MeCP2 at late juvenile and adult stage results in specific reduction of synaptic protein expression in symptomatic Tam-injected Mecp2loxJ/y/CreER mice. A, Western blot showing significant reduction in the levels of MeCP2, and the following synaptic proteins: CaMKIIα, CaMKIIβ, GluR2/3, GABABR2, Vglut1, NMDAR2A, and Synapsin 1 in the whole brain of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice when compared with their levels in the brains of the 5W-Tam-injected Mecp2loxJ/y or the 5W-Veh-injected Mecp2loxJ/y/CreER mice. B–D, Western blot showing significant reduction in the levels of CaMKIIα protein in the indicated areas of the brain of symptomatic Tam-injected Mecp2loxJ/y/CreER (3) male mice injected at 5 (B) or 10 (C) weeks of age, as well as of symptomatic Mecp2loxB/y/CreER (3) mice injected with Tam at 5 weeks of age (D). Tam-injected Mecp2loxJ/y or Mecp2loxB/y (1) and Veh-injected Mecp2loxJ/y/CreER or Mecp2loxB/y/CreER (2) are control littermates. Actin served as loading control. One-way ANOVA followed by appropriate post hoc for multiple-comparison tests were used to determine differences between groups. E, Quantitative RT-PCR analysis showing significant reduction in the level of Mecp2 mRNA in whole brain extracts of symptomatic 5W-Tam-injected Mecp2loxJ/y/CreER mice but no significant differences in the levels of the indicated mRNAs encoding the specific synaptic proteins when compared with the mRNA levels in brains of Veh-injected Mecp2loxJ/y/CreER control littermates. t test was used to compare within Veh and Tam groups. Error bars are mean ± SEM. *p < 0.05. n = 3 mice per genotype.
Discussion
Our results indicate that RTT-like symptoms, which are caused normally by germline mutations in Mecp2, can be recapitulated if MeCP2 is lost during the postnatal stage that coincides with the onset period of classic RTT (late juvenile stage). The immediate onset of symptoms in males, as soon as Mecp2 is mutated, and the severity of symptoms, including death, indicate that MeCP2 function is critical at this juncture. Interestingly, although MeCP2 loss in heterozygous female mice was induced by tamoxifen at a similar postnatal stage as in males, the onset of symptoms was delayed until they were 20 weeks old, similar to the onset of symptoms in female mice with germline mutations in Mecp2. Furthermore, the type of symptoms and the kinetics of symptom progression and stabilization were similar to heterozygous female mice with germline mutation in Mecp2. These data further support a critical role for MeCP2 at the specific postnatal time window where RTT symptoms normally appear in males and females, and further indicate that the absence of MeCP2 at earlier stages is not a prerequisite for manifestation of a severe RTT-like phenotype at later stages. Remarkably, a direct comparison of the loss of MeCP2 at late juvenile and adult stages showed parallel onset and progression of RTT symptoms, including the rate of lethality, and suggested an equal and critical requirement for MeCP2 function at these two different developmental stages.
Different mechanisms could potentially cause similar RTT-like symptoms at late juvenile and adult stages or upon the loss of MeCP2 in germ cells and postnatal stages. However, our detailed analysis of the brains that lost MeCP2 at postnatal stages suggests that although the state of brain maturation is different at late juvenile and adult stages, the loss of MeCP2 mediated similar brain abnormalities (Fig. 10). The brain abnormalities caused by the loss of MeCP2 at these postnatal stages are comparable in nature to the abnormalities caused by germline mutations in MeCP2; however, they are clearly more robust and unequivocal. These results are surprising because manifestation of similar brain phenotypes upon loss of MeCP2 in adult brain requires regression from a more mature state of the brain, as opposed to developmental stagnation, which is thought to occur in RTT patients and mouse models with germline mutation in Mecp2. Indeed, our analysis shows that while the reduced brain size upon the loss of MeCP2 at a late juvenile stage was due partly to developmental stagnation and partly to shrinkage; at the adult stage, a time at which the brain already reached its full size, the reduced brain size was mediated solely by shrinkage of the brain (Fig. 10). The increase in neuronal cell packing, which is reciprocal to the reduced brain size, suggests that shrinkage of the brain is likely not due to neuronal degeneration. Interestingly, the loss of MeCP2 in heterozygous female mice elicited shrinkage of their brain similar to hemizygous male mice, suggesting that even mosaic depletion of MeCP2 in 50% of the cells in the brain is sufficient to affect the brain globally. Our detailed analysis of the pyramidal neurons in the hippocampus of symptomatic mice that lost MeCP2 either at late juvenile or adult stage clearly shows that dendritic complexity was significantly reduced, mainly due to retraction of dendritic arbors from a more advanced state of neuronal maturation. Our analysis further shows that dendritic spine density is dramatically reduced, suggesting that the loss of MeCP2 in the maturing or mature brain mediates alterations in synaptic structural plasticity. It is likely that in human RTT patients and RTT mouse models, in addition to developmental stagnation, shrinking of the maturing brain and retraction of dendritic arbor structures also take place. This scenario could explain the regression in RTT girls that manifests in loss of the already acquired developmental milestones, such as speech and motor coordination, which may not be simply a consequence of halting developmental processes, but rather, a result of actual regression of fundamental anatomical and functional neuronal features in the brain. Previously, it was shown that RTT symptoms can be reversed upon global reactivation of MeCP2 (Guy et al., 2007) and attenuated by reactivation of MeCP2 in glia (Lioy et al., 2011) (Fig. 10). Our present studies, which demonstrate that RTT-like brain abnormalities could be elicited similarly in normal adult brain, provide further support for the idea that the function of MeCP2 in maintaining mature neuronal networks may be dynamic and that RTT has the potential to be fully reversed.
Figure 10.

Schematic model for the function of MeCP2 during critical stage of postnatal brain development and in the mature brain. A, During postnatal development, the size and weight of normal brain are increased while neurons mature as evident by the increase in dendritic complexity and synaptogenesis. The addition of synaptic proteins during late maturation increases synaptic strength, which in turn increases dendritic branching and stability of dendritic arbor structure. This process involves neuron:astrocyte interactions. B, In classic RTT caused by germline mutations in Mecp2, the brain is smaller than normal and neurons exhibit reduced dendritic arborization and synaptic protein expression. C, D, Inactivation of Mecp2 in otherwise normal mice during their late juvenile period of development (C) or at adult age (D) results in brain phenotypes typical for classic RTT. However, while the reduced brain size in late juvenile mice is caused partly by developmental stagnation and partly by shrinkage of the normally developed brain, in adult mice it is caused solely by shrinkage. In both cases, synaptic protein expression is reduced, and dendritic arbors of the already matured neurons are retracted. Similar to neurons, the morphology of astrocytes is also altered exhibiting fewer and shorter processes which together likely affect neuron:astrocyte interactions and therefore disruption of the mature neuronal networks. The loss of MeCP2, whether due to germline mutations or induced at postnatal stages, during late developmental stage (late juvenile stage) or adult stage, results in similar brain abnormalities, which lead to RTT phenotypes. E, In the reverse situation, global reexpression of MeCP2 in symptomatic RTT mice with germline mutation in Mecp2 results in reversal of RTT symptoms and neuronal function (Guy et al., 2007), and postnatal reexpression of MeCP2 in astrocytes inhibits the progression of RTT phenotypes and restores dendritic arbor structure and the level of at least the synaptic protein Vglut1 (Lioy et al., 2011).
Importantly, our structural analysis of the hippocampal astrocytes shows that, like neurons, they acquired altered morphology upon the loss of MeCP2 (Fig. 10). Given that astrocytes not only provide metabolic support to neurons, but are in fact an integral part of synapses, with their processes ensheathing synapses (Bolton and Eroglu, 2009), it is not surprising that such alteration in complexity of astrocytic processes could negatively affect the structure/function of the entire neuronal networks in the RTT brain.
Interestingly, our analysis of synaptic protein expression showed that, of the array of proteins analyzed, few were unaltered, more were robustly downregulated, and none were upregulated. The selective rather than global downregulation of synaptic protein expression may indicate that MeCP2 regulates specific synaptic proteins, directly or indirectly, to upregulate their expression in the normal maturing or mature brain (Fig. 10). Importantly, none of the genes whose protein expression was altered showed significant changes in their mRNA expression, suggesting that either MeCP2 directly regulates these genes post-transcriptionally, or indirectly by as yet unidentified primary target genes. These data could also explain why only subtle changes were mostly observed in mRNA levels in brains of conventional RTT mouse models (Tudor et al., 2002, Chahrour et al., 2008). Recent studies show aberrant expression of microRNA genes in a mouse model of RTT (Urdinguio et al., 2010, Wu et al., 2010). It is possible that specific microRNAs bind to the mRNAs of these specific synaptic proteins and inhibit their translation. Among the synaptic proteins that were downregulated, we identified presynaptic and postsynaptic proteins, both inhibitory and excitatory, supporting previous studies demonstrating that specific loss of MeCP2 in excitatory or inhibitory neurons could initiate RTT-like phenotypes (Chen et al., 2001, Chao et al., 2010). In normal brain, dendritic arbor growth is highly dynamic, and accumulating evidence suggests that synaptic strength and branch stability are concurrent (Cline and Haas, 2008). Interestingly, several synaptic proteins, which were dramatically reduced upon postnatal loss of MeCP2, such as CaMKIIα, CaMKIIβ, AMPA receptors (GluR2/3 subunits), and NMDAR2A are involved in excitatory synapse maturation as well as in increasing and stabilizing dendritic arbor structures (Fink et al., 2003, Haas et al., 2006). It is likely that the reduction in the levels of these synaptic proteins upon loss of MeCP2 mediates the retraction of dendritic arbors. However, we cannot exclude the possibility that the loss of dendritic arbors and synaptic contacts may trigger the degradation of proteins localized within these compartments. The effect of MeCP2 loss on the level of specific synaptic proteins in classic RTT mouse models remains ambiguous and partially controversial; however, several synaptic proteins that we found downregulated upon postnatal loss of MeCP2 are also downregulated in RTT mice with germline mutations in Mecp2. Examples are NMDAR2A (Asaka et al., 2006) and Vglut1 (Chao et al., 2007), which we also recently showed reduced in a RTT mouse model and rescued by reactivation of MeCP2 in astrocytes (Lioy et al., 2011). Similarly, GABAA receptor subunits are downregulated in classic RTT (Samaco et al., 2005, Medrihan et al., 2008), and our data showed that GABAB receptor subunit (GABABR2) is also downregulated as it is in postmortem brain of autistic patients (Fatemi et al., 2009). It is likely that reduced levels of both ionotropic and metabotropic GABA receptor subunits are also involved in manifestation of RTT symptoms. The reduction in the levels of synaptic proteins together with the reduction in dendritic complexity and spine density support the view that MeCP2 is required during late stages of brain maturation to establish and maintain mature neuronal circuitry throughout life (Fig. 10). Although studies using mouse models with germline mutations in Mecp2 helped to advance our knowledge about RTT development, a mouse model presented here, which allows to induce loss of MeCP2 at desired timing in otherwise healthy brain, provides a powerful tool for analyzing the direct effect of MeCP2 loss on brain structure/function. Further identification of key synaptic proteins and the mechanism by which MeCP2 regulates these proteins could provide important insights into the molecular pathways underlying the manifestation of brain abnormalities and identify new potential targets for therapeutic intervention of RTT.
Footnotes
This work was supported by grants from the National Institute of Health (N.B., G.M.), International Rett Syndrome Foundation (N.B.), and Targeted Research Opportunities, School of Medicine, Stony Brook University (N.B.). G.M. is an Investigator of the Howard Hughes Medical Institute. We thank Dr. M. Covey for providing expertise with animal procedures, and Drs. N. Bukhari and C. Trocme for their help in statistical analysis.
References
- Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54:195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21:217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, Oldfors A, Hagberg B, Dahlström A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5:1509–1513. [PubMed] [Google Scholar]
- Bolton MM, Eroglu C. Look who is weaving the neural web: glial control of synapse formation. Curr Opin Neurobiol. 2009;19:491–497. doi: 10.1016/j.conb.2009.09.007. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, Ekker M, Rubenstein JL, Noebels JL, Rosenmund C, Zoghbi HY. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–1517. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABA(B) receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink CC, Bayer KU, Myers JW, Ferrell JE, Jr, Schulman H, Meyer T. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron. 2003;39:283–297. doi: 10.1016/s0896-6273(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104:1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K, Li J, Cline HT. AMPA receptors regulate experience-dependent dendritic arbor growth in vivo. Proc Natl Acad Sci U S A. 2006;103:12127–12131. doi: 10.1073/pnas.0602670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27:306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, Chen C. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron. 2011;70:35–42. doi: 10.1016/j.neuron.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricceri L, De Filippis B, Laviola G. Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav Pharmacol. 2008;19:501–517. doi: 10.1097/FBP.0b013e32830c3645. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio RG, Fernandez AF, Lopez-Nieva P, Rossi S, Huertas D, Kulis M, Liu CG, Croce CM, Calin GA, Esteller M. Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics. 2010;5:656–663. doi: 10.4161/epi.5.7.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Tao J, Chen PJ, Shahab A, Ge W, Hart RP, Ruan X, Ruan Y, Sun YE. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18161–18166. doi: 10.1073/pnas.1005595107. [DOI] [PMC free article] [PubMed] [Google Scholar]