

Abstract
We have initiated a broad-based program aimed at understanding the molecular basis of fluorine specificity in enzymatic systems, and in this context, we report crystallographic and biochemical studies on a fluoroacetyl-coenzyme A (CoA) specific thioesterase (FlK) from Streptomyces cattleya. Our data establish that FlK is competent to protect its host from fluoroacetate toxicity in vivo and demonstrate a 106-fold discrimination between fluoroacetyl-CoA(kcat/KM=5×107M−1 s−1) and acetyl-CoA(kcat/KM=30 M−1 s−1) based on a single fluorine substitution that originates from differences in both substrate reactivity and binding. We show that Thr 42, Glu 50, and His 76 are key catalytic residues and identify several factors that influence substrate selectivity. We propose that FlK minimizes interaction with the thioester carbonyl, leading to selection against acetyl-CoA binding that can be recovered in part by new C=O interactions in the T42S and T42C mutants. We hypothesize that the loss of these interactions is compensated by the entropic driving force for fluorinated substrate binding in a hydrophobic binding pocket created by a lid structure, containing Val 23, Leu 26, Phe 33, and Phe 36, that is not found in other structurally characterized members of this superfamily. We further suggest that water plays a critical role in fluorine specificity based on biochemical and structural studies focused on the unique Phe 36 “gate” residue, which functions to exclude water from the active site. Taken together, the findings from these studies offer molecular insights into organofluorine recognition and design of fluorine-specific enzymes.
Although the identification of halogenated natural products containing chlorine, bromine, and iodine has rapidly accelerated over the last 30 years as greater biodiversity has been explored, the incidence of fluorinated natural products remains limited to a relatively small number of structurally related compounds (Chart 1) (1–6). Fluorine has unique properties arising from the combination of its small size, high electronegativity, and high C–F bond strength that have found utility in the design and development of new pharmaceuticals (7–10). It is often used as a sterically conservative replacement for hydrogen in small molecules, allowing them to retain binding to their macromolecular targets while adjusting several molecular properties such as stability, reactivity, conformation, lipophilicity, and in vivo distribution and metabolism (7–9, 11). Furthermore, the ease of elimination of fluoride from fluorine-substituted cellular metabolites has allowed the rational design of several bioactive mechanism-based inhibitors, such as 2′,2′-difluoro-2′-deoxycytidine (Gemzar) (12–14) and 5-fluorouracil (Adrucil, Carac, Efudex, and Fluoroplex) (15, 16).
Chart 1.

Biogenic Organofluorine Natural Products Found in the Environment
Fluoroacetate is a naturally occurring organofluorine that is surprisingly poisonous considering its simple structure, with toxicity rivaling some of the most potent anticancer therapeutics (17, 18). Like Gemzar and fluorouracil, it can be metabolized by the normal host machinery before generating a mechanism-based inhibitor that shuts down the target pathway upon fluoride elimination. Specifically, fluoroacetate can be activated to form fluoroacetyl-CoA,1 which is then converted to fluorocitrate via the TCA cycle (19–22). While the aconitase-catalyzed formation of the fluoro-cis-aconitate intermediate parallels the reaction pathway of citrate, addition of water in the next step leads to stoichiometric fluoride elimination rather than formation of fluoroisocitrate (23–26). The product of this reaction is a strong noncovalent inhibitor that remains bound in the aconitase active site with an extremely slow rate of displacement by citrate (26, 27).
Although fluoroacetate production has been mainly noted in the plant kingdom (28–32), our understanding of organofluorine biosynthesis has come exclusively from studies of Streptomyces cattleya, a soil microbe that is the only known genetic host for production of both fluoroacetate and fluorothreonine (2,4,5,33,34). Similar to other antibiotic-producing hosts (35–40), S. cattleya is expected to contain pathways for detoxification in order to protect itself from its own production of fluoroacetate. A fluoroacetyl-CoA-specific thioesterase (FlK) was recently identified in S. cattleya that selectively hydrolyzes fluoroacetyl-CoA over acetyl-CoA (41). The gene encoding FlK is found clustered with the C–F bond-forming fluorinase (flA), raising the possibility that FlK-catalyzed hydrolysis of fluoroacetyl-CoA plays a role in fluoroacetate resistance in S. cattleya by preventing the entrance of fluoroacetyl-CoA into the TCA cycle (34). Given the close structural similarity between fluoroacetyl-CoA and acetyl-CoA, the discrimination between these two substrates exhibited by FlK is remarkable. The recently reported crystal structure of FlK demonstrates that this enzyme belongs to the hot dog-fold family of thioesterases (42). From these studies, a catalytic triad composed of Thr 42, His 76, and an active site water was proposed. Based on a computational model of the FlK-substrate complex, Arg 120 and Gly 69 were hypothesized to serve as the main selectivity elements by orienting the substrate for hydrolysis through fluorine-specific interactions.
Our group is interested in further studying the origin of substrate specificity in FlK to develop a better understanding of the design principles that drive fluorine-based selectivity in protein–ligand interactions. FlK provides a particularly interesting system for study because evolutionary selection against hydrolysis of the nonfluorinated substrate congener, acetyl-CoA, is likely important for maintaining normal cell growth and has tuned discrimination based on a single fluorine substitution. We quantify this fluorine-specific substrate discrimination to be surprisingly high (106-fold) and demonstate that it is derived from differences in both substrate reactivity and binding by carrying out steady-state kinetic characterization of FlK with respect to both fluoroacetyl-CoA and acetyl-CoA. Through a combination of structural and biochemical studies of FlK and a series of mutants, we have identifed several features unique to FlK that provide a molecular basis for fluorine specificity. We propose a model in which FlK minimizes interactions with the carbonyl unit common to both substrates, which contribute to substrate recognition and catalysis in typical members of the superfamily, selecting for reaction with the more activated fluoroacetyl-CoA substrate and decreasing general acyl-CoA binding. We further hypothesize that fluorine-specific interactions are recovered through the unusual lid structure of FlK, which provides a hydrophobic environment that both maximizes dipolar interactions with the fluorine atom and provides an entropic driving force for C–F bond recognition.
MATERIALS AND METHODS
Commercial Materials
Restriction enzymes, T4 DNA ligase, and Phusion polymerase were purchased from New England Biolabs (Ipswich, MA). Platinum Taq High Fidelity (HF) polymerase and One-Shot TOP10 chemically competent cells were purchased from Invitrogen (Carlsbad, CA). Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). DNA sequencing was performed by Quintara Biosciences (Berkeley, CA). DNA purification kits and Ni-NTA agarose were purchased from Qiagen (Valencia, CA). Complete Mini EDTA-free protease inhibitor was purchased from Roche Applied Science (Penzberg, Germany). Spectra Multicolor low-range protein ladder was purchased from Fermentas (Glen Burnie, MD). Amicon Ultra 3000 MWCO centrifugal concentrators and 5000 MWCO regenerated cellulose ultrafiltration membranes were purchased from Millipore (Billerica, MA). Acrylamide/bisacrylamide (40%, 19:1, and 30%, 37:1) and Gel Filtration Standard were purchased from Bio-Rad Laboratories (Hercules, CA). Luria–Bertani (LB) broth Miller and LB agar Miller were purchased from EMD Biosciences (Darmstadt, Germany). Carbenicillin, isopropyl β-D-thiogalactopyranoside (IPTG), tris(hydroxymethyl) aminomethane hydrochloride (Tris-HCl), sodium chloride, and dithiothreitol (DTT) were purchased from Fisher Scientific (Pittsburgh, PA). Streptomycin sulfate, ammonium persulfate, oxalyl chloride, N,N-dimethylformamide (Sure-Seal), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), sodium fluoroacetate, coenzyme A trilithium salt (CoA), acetyl-CoA, N, N,N′,N′-tetramethylethane-1,2-diamine (TEMED), and tricine were purchased from Sigma-Aldrich (St. Louis, MO).
Gene Synthesis
A synthetic gene encoding flK was codon-optimized for Escherichia coli using Gene Designer (DNA 2.0; Menlo Park, CA) and synthesized using PCR assembly. Gene2Oligo (43) was used to convert the gene sequence into primer sets using default optimization settings. Primers for gene assembly are listed in Supporting Information Table S1, and a primer map is shown in Supporting Information Figure S1. To assemble the synthetic gene, each primer was added at a final concentration of 1 μM to the first PCR (50 μL) containing Platinum Taq HiFi buffer (20 mM Tris-HCl, 50 mM KCl, pH 8.4), MgSO4 (1.5 mM), dNTPs (250 μM each), and 5 units of Platinum Taq HF (Invitrogen). The following thermocycler program was used for the first assembly reaction: 95 °C for 5 min; 95 °C for 30 s; 55 °C for 2 min; 72 °C for 10 s; 40 cycles of 95 °C for 15 s, 55 °C for 30 s, 72 °C for 20 s plus 3 s/cycle; these cycles were followed by a final incubation at 72 °C for 5 min. The second assembly reaction (50 μL) contained 16 μL of the unpurified first PCR with standard reagents for Platinum Taq HF. The thermocycler program for the second PCR was as follows: 95 °C for 30 s; 55 °C for 2 min; 72 °C for 10 s; 40 cycles of 95 °C for 15 s, 55 °C for 30 s, 72 °C for 80 s; these cycles were followed by a final incubation at 72 °C for 5 min. The second PCR(16 μL) was transferred again into fresh reagents and run using the same program. Following gene construction, the DNA smear at the appropriate size was gel purified and used as a template for amplification with Platinum TaqHF and rescue primers sFlK F100 and sFlKR100 (Supporting Information Table S1) under standard conditions. The PCR product was digested with NdeI and BamHI and ligated into the NdeI–BamHI sites of pET16b. The sequence of the synthetic gene was verified by sequencing.
Construction of Vectors for Protein Expression
The synthetic gene encoding FlK was PCR amplified from pET16b with Platinum TaqHF using the primers sFlKF13 and sFlK R15 (Supporting Information Table S1). The PCR product was digested with SfoI and XhoI and ligated into the SfoI–XhoI sites of pET23a vector modified to encode a His10 tag and a tobacco etch virus (TEV) protease cleavage site (Macrolab, UC Berkeley). The resulting plasmid was verified by sequencing.
Site-Directed Mutagenesis
Plasmids encoding mutant FlKs were prepared using oligonucleotide primers containing the desired mutation (Supporting Information Table S2). Each mutagenesis reaction contained forward and reverse primers (2 μM each), pET23a-His10-Tev-sFlK template (50–100 ng), dNTPs (200 μMeach), Phusion polymerase, and Phusion buffer. The reaction mixture was subjected to the following thermocycler program: 98 °C for 3 min; 16 cycles of 98 °C for 15 s, 55 °C for 15 s, 72 °C for 3 min 30 s; a final extension at 72 °C for 7 min. Reactions were digested with DpnI and transformed into One- Shot TOP10 chemically competent cells. All mutants were then verified by sequencing of the flK gene. The E50Q mutant was subcloned into vector pSV272 (Macrolab, UC Berkeley) using primers sFlK F13 and sFlK R15 and using the SfoI and XhoI restriction sites to generate a His10-MBP-Tev-sFlK-E50Q expression construct.
E. coli Viability Assay
Electrocompetent E. coli BL21-(DE3) cells were transformed with empty pET23a-His10-Tev, pET23a-His10-Tev-FlK, and pET23a-His10-Tev-FlK-H76A, and cells were grown on LB agar containing 50 μg/mL carbenicillin. Saturated cultures were diluted to OD600nm=0.3 in LB, and serial 10-fold dilutions were made in a 96-well plate. Dilutions (2 μL) were spotted onto LB agar containing either 50 μg/mL carbenicillin or 50 μg/mL carbenicillin and 20 mM sodium fluoroacetate. Plates were incubated at 37 °C for 16 h and then photographed.
Expression and Purification of FlK Variants
LB containing carbenicillin (50 μg/mL) was inoculated to OD600nm=0.05 with an overnight LB culture of freshly transformed E. coli BL21(DE3) containing the overexpression plasmid. The cultures were grown at 37 °C at 200 rpmtoOD600nm=0.8, at which point they were cooled on ice for 20 min. Isopropyl β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM, and cells were incubated at 16 °C for 12–16 h. Cells were harvested by centrifugation at 3696g at 4 °C for 15 min. The cell pellet was resuspended in 5 mL of lysis buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 20 mM imidazole, 100 μM phenylmethanesulfonyl fluoride, one Complete Mini EDTA-free tablet per 50 mL buffer) per gram of cell pellet wet weight and lysed by one pass through a French pressure cell (Thermo Fisher) at 14000 psi. In soluble material was removed by centrifugation at 15316g at 4 °C for 20 min. A 10% (w/v) solution of streptomycin sulfate was added to the supernatant to a final concentration of 0.9% (w/v), and precipitated DNA was removed by centrifugation at 15316g for 20 min. The supernatant was loaded onto aNi-NTA agarose column (1 mL resin/L of cell culture) using an ÄKTA Purifier FPLC system (GE Healthcare). The column was washed with 10 column volumes of wash buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 20 mM imidazole) and 10 column volumes of wash buffer plus 8% elution buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 250 mM imidazole). The column was eluted using a gradient from 8% to 100% elution buffer over 10 column volumes. Protein-containing fractions were pooled by their absorbance at 280 nm and concentrated to 10–15 mL using a 5000 MWCO ultrafiltration membrane. TEV protease (purified as described previously (44)) was added at a 1:50 mass ratio, and the mixture was dialyzed against TEV cleavage buffer (50 mM sodium phosphate, pH 8.0, 300 mM NaCl, 1 mM DTT) for 16 h. The dialysate was passed over a Ni-NTA column to remove the His6-tagged TEV protease and the His10 peptide, and the column was washed with 2 column volumes of wash buffer. The flow-through and wash fractions were concentrated to 2 mL and loaded onto a Superdex 75 16/60 pg (GE Healthcare) column equilibrated with size-exclusion buffer (20 mM Tris-HCl, pH 7.6, 50 mM NaCl). FlK eluted from the column as a dimer (Supporting Information Figure S2) and was concentrated to 1–10 mg/mL and stored at 4 °C. For long-term storage, 10% glycerol was added to the concentrated protein, and it was stored at −80 °C. Both His10-tagged and His10-tagged MBP fusion proteins were purified following this protocol (Supporting Information Figure S3).
Crystallization and Structure Determination
Purified FlK in size-exclusion buffer was concentrated to 17 mg/mL. Crystals of apo-FlK were obtained using the hanging drop vapor diffusion method by combining equal volumes of protein solution and reservoir solution containing 0.1 M Tris-HCl, pH 7.6, and 25% polyethylene glycol 3350. Clusters of plates grew within 2 days. Single crystals were obtained by gently agitating the clusters. The crystals were cryoprotected by soaking briefly in a solution containing 0.1 M Tris-HCl, pH 7.6, 25% polyethylene glycol 3350, and 25% (v/v) ethylene glycol and flash cooled in liquid nitrogen. Data were collected at beam line 8.2.2 at the Advanced Light Source (Lawrence Berkeley National Laboratory). Data were processed with XDS and scaled with XSCALE (45). Molecular replacement with Phaser (46) using a polyalanine model of Thermus thermophilus hypothetical protein TTHA0967 (PDB ID 2CWZ) identified two FlK monomers in the asymmetric unit arranged with typical hot dog-fold topology. Maps were improved by Phenix AutoSolve and AutoBuild (47). The resulting maps were of sufficient quality to discard the original model, and Arp/wARP (48) was used to build a near-complete chain trace. The remaining parts of the model were manually built using Coot (49). The structure was solved at 1.9 Å resolution and refined to an Rwork/Rfree of 20.1%/23.1% using Phenix Refine.
Crystals of the open conformation of FlK were obtained by soaking apo-FlK crystals in a solution of 1 mM fluoroacetyl-CoA in mother liquor for 10 min followed by brief cryoprotection by soaking in mother liquor supplemented with 25% (v/v) ethylene glycol. Crystals were then flash-cooled, and data were collected at beam line 8.3.1 (Advanced Light Source, Lawrence Berkeley National Laboratory). Molecular replacement performed with Phaser using the refined apo-FlK structure as a search model identified two monomers per asymmetric unit. The open FlK structure was manually rebuilt using Coot and refined using Phenix Refine to Rwork = 18.8% and Rfree = 22.0% at 2.0 Å resolution. Crystals of the FlK-fluoroacetate complex were obtained by soaking apo-FlK crystals in 2.5 mM fluoroacetate. The initial model of the protein was obtained as for the open conformation of FlK. Unmodeled peaks in the map were identified using Coot, and ligands corresponding to the density were modeled when possible. The structure was solved to 2.5 Å resolution and refined with Phenix Refine to Rwork/Rfree of 19.4%/24.0%
Crystals of FlK-F36A were obtained as for wild-type FlK. FlK-F36A crystals grew in a larger C2 crystal form that had a Matthews coefficient consistent with six monomers per asymmetric unit. Molecular replacement performed with Phaser using the refined apo-FlK structure as a search model unambiguously identified three dimers per asymmetric unit, in agreement with the Matthews coefficient. The structure was solved to 2.3 Å resolution, manually rebuilt using Coot, and refined to Rwork/Rfree of 22.0%/24.7%. Crystals of the FlK-F36A product complex were obtained in the same conditions as apo-FlK-F36A by cocrystallization with 1 mM fluoroacetyl-CoA and were found to contain three dimers per asymmetric unit. Maps were obtained, and the model was built as for FlK-F36A. The structure was solved to 2.0 Å resolution and refined with Phenix Refine to Rwork/Rfree of 21.8%/25.1%.
Fluoroacetyl-CoA Synthesis
Fluoroacetyl-CoA was synthesized as described previously (41) with some modification. Sodium fluoroacetate (100 mg, 1 mmol) was dried under vacuum in an oven-dried round-bottom flask equipped with a stir bar and a reflux condenser. Dry tetrahydrofuran (2 mL), dry N,N-dimethylformamide (100 μL), and oxalyl chloride (2 M in dichloromethane, 1 mmol) were added to the flask by syringe, and the reaction mixture was stirred and heated at 65 °C for 2–3 h under nitrogen atmosphere. After the flask was allowed to cool, the reaction mixture (650 μL) was added to stirred solution of coenzyme A trilithium salt (50 mg, 0.06 mmol) dissolved in 1 mL of 10% sodium bicarbonate. The mixture was stirred vigorously for 3 min under a stream of nitrogen and then immediately injected onto an Agilent Eclipse XDB-C18 column (9.4× 250 mm, 5 μm) and purified by reverse-phase HPLC (0–100% B over 30 min at 3 mL/min; A, H2O; B, acetonitrile) using an Agilent 1200 binary pump coupled to a diode-array detector. Fractions (1.5 mL) were flash frozen in liquid nitrogen and lyophilized. The lyophilized fluoroacetyl-CoA was dissolved in water and characterized using an Agilent 6130 single-quadrupole electrospray ionization mass spectrometer (ESI-MS) (m/z (MH+), 828.1; m/z (MH+calc), 828.1). The purified substrate was also assayed for contaminating coenzyme A by comparing the total coenzyme A absorbance at 260 nm (ε260 nm=13100 M−1 cm−1) with free thiol content measured by reaction with DTNB (ε412nm=13600 M−1 cm−1 for 2-nitro-5-thiobenzoate). Only fluoroacetyl-CoA of >95% purity was used for assays.
Kinetic Measurements
The thioesterase activity of FlK was measured by spectrophotometrically monitoring the increase in absorbance at 412 nm due to reaction of enzymatically generated free coenzyme A with DTNB in a Beckman Coulter DU-800 spectrophotometer as described previously (41) with some modification. FlK activity was measured immediately after purification. Assays were performed at 25 °C in a total volume of 500 μL containing 100 mM Tris-HCl, pH 7.6, 0.5 mM DTNB, the appropriate amount of fluoroacetyl-CoA or acetyl-CoA, and FlK (0.5 nM, fluoroacetyl-CoA; 10 μM, acetyl-CoA) or FlK mutant (50 nM−1 μM, fluoroacetyl-CoA; 10 μM for acetyl-CoA) in an amount sufficient to detect activity. Commercial acetyl-CoA was also tested for contaminating CoA as described above. A CoA standard curve from 0 to 50 μM was generated using the appropriate amounts of CoA in a solution containing 100 mM Tris-HCl, pH 7.6, and 0.5 mM DTNB. Kinetic parameters (kcat and KM) were determined by fitting the data using Kaleidagraph 3.51 (Synergy Software, Reading, PA) to the equation:
where vo is the initial rate and [S] is the substrate concentration.
RESULTS AND DISCUSSION
Toxicity of Fluoroacetyl-CoA and Rescue by FlK
To test whether FlK is competent to confer fluoroacetate resistance in a heterologous bacterial system, we expressed the flK gene in E. coli and measured its viability by spotting 1:10 dilutions of culture onto solid media (Figure 1). In the presence of fluoroacetate, the viability of E. coli containing an empty plasmid or a plasmid encoding an FlK mutant with diminished activity was severely reduced, while E. coli harboring the plasmid encoding wild-type FlK exhibited viability similar to that of cultures spotted onto fluoroacetate-free media, which indicates that FlK is competent to confer fluoroacetate resistance in bacteria. As acetyl-CoA is an essential building block in the cell, these results are consistent with the conclusion that FlK demonstrates specificity toward fluoroacetyl-CoA over acetyl-CoA in vivo.
Figure 1.

FlK confers fluoroacetate resistance in vivo. The viability of E. coli BL21(DE3) was assayed by spotting 1:10 serial dilutions of culture onto solid media in the absence (left) or presence (right) of 20 mM fluoroacetate. E. coli harbored no plasmid (top row), empty pET23a-His10-Tev (second row), pET23a-His10-Tev-FlK (third row), or pET23a-His10-Tev-FlK-H76A (fourth row).
Competition with Acetyl-CoA
Previous measurements of FlK activity suggested that this enzyme exhibits remarkable in vitro selectivity for fluoroacetyl-CoA over acetyl-CoA, with no hydrolysis observed at concentrations up to 1 mM acetyl-CoA in the presence of 50 ng of FlK (41). We can envision two major modes for recognition of the fluorinated substrate over the nonfluorinated substrate. One mode of substrate specificity could rely on structural recognition of the fluorine atom, which could be discriminated based on polarity or size. A second possible component of selectivity is the differing chemical reactivities of fluoroacetyl-CoA and acetyl-CoA. The inductively electron-withdrawing fluorine atom activates the thioester C=O in fluoroacetyl-CoA toward nucleophilic attack. Indeed, the pseudo- first-order rate constant for uncatalyzed hydrolysis of fluoroacetyl-CoA was measured to be an order of magnitude faster (1.4 × 10−4 s−1 at pH 7.6) than the hydrolysis of acetyl-CoA (1.3 × 10−5 s−1 at pH 7.6) under the same conditions (Supporting Information Figure S4). Thus, the active site nucleophile of FlK could be tuned toward lower reactivity, which would limit the hydrolysis of acetyl-CoA compared to fluoroacetyl-CoA.
To clarify whether the exclusion of acetyl-CoA binding plays a role in FlK selectivity, we performed a competition experiment in which the fluoroacetyl-CoA concentration was fixed at 20 μM and the rate of hydrolysis was measured in the presence of increasing concentrations of acetyl-CoA (Figure 2). No significant effect on the rate of free CoA release was observed except at the highest concentration (5 mM) of acetyl-CoA, suggesting that the absence of the fluorine modification severely diminishes substrate affinity. This result is surprising in light of previous studies that have demonstrated promiscuous substrate binding in other members of the hot dog-fold thioesterase superfamily, which have been shown to accept a range of diverse acyl-CoA substrates with physiologically relevant kcat/KM values (50–55). Because fluoroacetyl-CoA is larger than acetyl-CoA (estimated total size: C–F, 2.82 Å ; C–H, 2.29 Å (11)), we might have expected that the binding pocket of FlK would also accommodate acetyl-CoA as a substrate, especially considering that several of the substrate-binding interactions with CoA-dependent enzymes involve the carbonyl group and the cofactor itself (50, 54).
Figure 2.
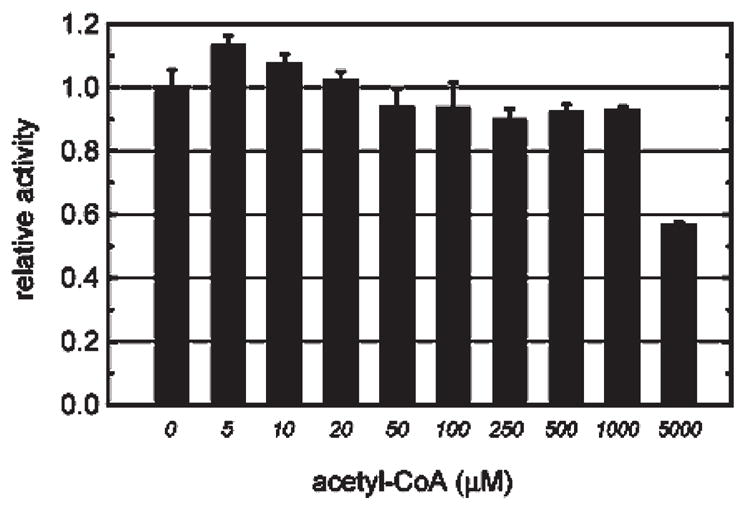
Rates of FlK hydrolysis of fluoroacetyl-CoA in the presence of varying concentrations of acetyl-CoA. The ability of acetyl-CoA to bind to FlK was tested using a competition assay in which the rate of hydrolysis of fluoroacetyl-CoA (fixed at 20 μM) was measured in the presence of increasing concentrations of acetyl-CoA. Data are mean ± SE (n= 3).
Based on the results of the competition experiment, it seemed likely that the KM for acetyl-CoA is above 1 mM, the highest concentration tested in previous studies (41). After both enzyme and substrate concentrations were increased, acetyl-CoA turnover was detectable with a rate well above the nonenzymatic hydrolysis rate (Supporting Information Figure S4). Under these conditions, we measured a kcat of 0.06 ± 0.001 s−1 and a kcat/KM of 30 M−1 s−1 for acetyl-CoA, which corresponds to a 106-fold decrease in catalytic efficiency compared to the fluoroacetyl-CoA substrate (kcat=390 ± 20 s−1; kcat/KM=5 × 107 M−1 s−1) (Table 1). The combination of the slow rate of turnover and low pseudo-second-order rate constant with acetyl-CoA indicates that the nonfluorinated substrate binds poorly and, potentially in unproductive conformations, at the FlK active site. In contrast, the kinetic parameters measured for fluoroacetyl-CoA are similar to the values observed for other members of this superfamily for their substrates (kcat, 0.56–190 s−1; kcat/KM, 104–107 M−1 s−1) (50, 51, 53–58). Taken together, the kinetic data for fluoroacetyl-CoA and acetyl-CoA are consistent with selection against acetyl-CoA as a substrate. Given that the magnitude of the discrimination exhibited by FlK is outside the window expected of single C–H to C–F substitutions that do not perturb ligand conformation or local pKas (8, 9, 11), we turned to structural studies of FlK and FlK mutants in order to further elucidate the interactions at the active site that might promote binding and hydrolysis of the fluorinated substrate while excluding its nonfluorinated congener.
Table 1.
Kinetic Constants Measured for Fluoroacetyl-CoA and Acetyl-CoA Hydrolysis by Wild-Type and Mutant FlKsa
| enzyme | fluoroacetyl-CoA
|
acetyl-CoA
|
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (M−1 s−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1 s−1) | |
| wild type | (3.9 ± 0.2) × 102 | 8 ± 1 | (5 ± 1) × 107 | (6 ± 1) × 10−2 | (2.1 ± 0.5) × 103 | (3 ± 1) × 101 |
| E50Q | (1.3 ± 0.1) × 10−1 | (1.4 ± 0.1) × 101 | (9 ± 1) × 103 | |||
| H76A | (3.0 ± 0.3) × 10−3 | (2.0 ± 0.7) × 101 | (1.5 ± 0.5) × 102 | |||
| T42A | (4.5 ± 0.9) × 10−1 | (1.1 ± 0.4) × 102 | (4 ± 2) × 103 | |||
| T42S | (1.9 ± 0.4) × 101 | (6 ± 2) × 101 | (3 ± 1) × 105 | (1.0 ± 0.1) × 10−2 | (6.1 ± 0.9) × 101 | (1.6 ± 0.3) × 102 |
| T42C | (1.1 ± 0.2) × 101 | (3.6 ± 1) × 102 | (3 ± 1) × 104 | (1.0 ± 0.1) × 10−2 | (3.9 ± 0.9) × 102 | (1.2 ± 0.6) × 101 |
| F33A | (2.7 ± 0.4) × 101 | (1.5 ± 0.4) × 103 | (1.8 ± 0.5) × 104 | ndb | nd | |
| F36A | (2.7 ± 0.2) × 102 | (7.1 ± 0.8) × 102 | (3.7 ± 0.5) × 105 | (1.0 ± 0.2) × 10−1 | (3 ± 1) × 103 | (2.9 ± 0.9) × 101 |
| V23A | (1.5 ± 0.3) × 102 | (3.0 ± 1.5) × 102 | (5 ± 3) × 105 | (1.0 ± 0.1) × 10−2 | (9 ± 1) × 102 | (1.1 ± 0.2) × 101 |
| L26A | 6.7 ± 0.1 | (1.2 ± 0.2) × 102 | (5.7 ± 0.8) × 104 | nd | nd | |
Data are mean ± SE (n=3) as determined from nonlinear curve fitting. Error in the kcat/KM parameter was obtained from propagation of error from the individual kinetic terms.
No detectable activity in the presence of up to 10 μM enzyme and up to 2.5 mM acetyl-CoA.
Crystal Structure of FlK
A set of FlK structures were recently reported (42), with FlK found to be arranged as a dimer in a typical hot dog-fold thioesterase topology (50, 51, 54, 58). We have also determined the crystal structure of the wild-type enzyme at 1.9 Å resolution by molecular replacement using a polyalanine model of T. thermophilus hypothetical protein TTHA0967 (PDB ID 2CWZ) as a search model (Figure 3A, Table 2). The asymmetric unit contained two copies of the FlK monomer, in agreement with the apparent molecular weight measured by size-exclusion chromatography (Supporting Information Figure S2). A BLAST search (59) of FlK followed by multiple sequence alignment using MUSCLE (60) suggests that the FlK active site is composed of Thr 42, Glu 50, and His 76. Indeed, these residues are found at the dimer interface and arranged with a topology similar to the Arthrobacter clade of hot dog-fold thioesterases (Figure 3B) (50). Although FlK adopts the fold that defines the superfamily, the structural data reveal some unique features not observed in structures of other superfamily members. The most striking difference is a “lid” structure formed by short helical segments (α1 and α2, residues 23–26 and 31–34, respectively) connected by flexible linkers, which is positioned directly over the active site of FlK and constrains the size of the active site pocket available for substrate binding (Figure 3C). Tertiary contacts within the lid are mediated by an edge–face interaction between Phe 33 and Phe 36 and by close packing of the side chains of Val 23, Leu 26, Val 39, and Phe 36. The surface of the lid that faces the substrate-binding cavity is made up primarily of hydrophobic side chains (Val 23, Leu 26, Tyr 27, Phe 33, Phe 36, and Val 39). In combination with the side chains of Val 46 and Val 54 from helix α3 and Ile 72 from strand β2, the lid creates a chemical environment within the active site that is largely hydrophobic with the exception of the residues involved in nucleophilic/general base catalysis or water activation. The hydrophobic residues of the lid and those lining the bottom of the active site are well conserved in uncharacterized FlK homologues identified by sequence alignment (Supporting Information Figure S5), although one group of less closely related homologues lacks helix α2 and the second loop of the lid.
Figure 3.

Crystal structure of FlK. (A) Cartoon representation of the FlK crystal structure looking down at the active site and lid structure formed by α1 and α2 (residues 23–26 and 31–34, repectively). Chain A is colored in blue, and chain B is colored in gray (α3, residues 42–58; α4, residues 125–135). (B) View of the hydrogen- bonding network at the putative active site showing Thr 42, His 76, and Glu 50 site water. (C) Stereoview of the active site and the hydrophobic lid (carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow).
Table 2.
Data Collection and Refinement Statistics for Wild-Type FlK Structures
| apo-FlK | FlK-FAc | FlK open | |
|---|---|---|---|
| data collection | |||
| X-ray source | ALS 8.2.2 | ALS 8.3.1 | ALS 8.3.1 |
| space group | C2 | C2 | C2 |
| cell dimensions | |||
| a, b, c (Å) | 48.4, 90.9, 62.0 | 62.3, 92.4, 50.3 | 62.8, 90.7, 52.3 |
| α, β, γ (deg) | 90.0, 100.1, 90.0 | 90.0, 102.0, 90.0 | 90.0, 104.5, 90.0 |
| wavelength | 0.9901 | 1.116 | 1.116 |
| resolutiona | 29.96–1.85 (1.90–1.85) | 19.44–2.46 (2.50–2.46) | 19.77–1.95 (2.00–1.95) |
| Rmerge (%)a | 9.0 (60.1) | 10.6 (70.3) | 8.5 (60.4) |
| I/σIa | 12.25 (2.31) | 12.94 (2.14) | 14.01 (2.19) |
| completeness (%)a | 99.7 (99.8) | 99.5 (100) | 99.7 (99.7) |
| redundancya | 4.1 (4.1) | 4.1 (4.2) | 3.7 (3.7) |
| refinement | |||
| resolution | 29.96–1.85 | 19.44–2.46 | 19.77–1.95 |
| no. of reflections | 20926 | 10125 | 20623 |
| Rwork/Rfree | 0.201/0.231 | 0.194/0.240 | 0.188/0.220 |
| no. of atoms | 2306 | 2195 | 2182 |
| nonsolvent | 2065 | 2103 | 2068 |
| solvent | 241 | 92 | 114 |
| B-factors | |||
| protein | 29.2 | 38.3 | 23.4 |
| water | 43.4 | 39.0 | 30.2 |
| rmsd | |||
| bond lengths (Å) | 0.014 | 0.005 | 0.013 |
| bond angles (deg) | 1.24 | 0.97 | 1.58 |
Values in parentheses are for the highest resolution shell.
Structural alignment of FlK to other structurally characterized thioesterases with the same active site topology revealed the conspicuous absence of a conserved Asn/Gln whose position in the tertiary structure of FlK approximately corresponds to Val 23 (Supporting Information Figure S6). In these other thioesterases, the carboxamide of the Asn/Gln is proposed to function in orientation and polarization of the thioester C=O and to assist in departure of the thiolate leaving group (50, 51, 54). Based on the crystal structure of FlK, there does not appear to be another hydrogen bond donor to compensate for this function. The absence of the Asn/Gln–carbonyl oxygen interaction in FlK could contribute to the lowered catalytic efficiency for acetyl-CoA compared to fluoroacetyl-CoA both by decreasing binding interactions with a shared structural motif and by selecting against hydrolysis of the less reactive acetyl-CoA substrate.
Mutagenesis Studies of the Catalytic Residues of FlK
Although the detailed catalytic mechanism for the hot dog-fold thioesterases still requires further elucidation, Ser/Thr, Asp/Glu, His catalytic triads have been proposed and characterized by mutagenesis for many members of this superfamily (50, 54, 57, 58). From their sequence conservation (Supporting Information Figure S5) and location in the active site pocket of FlK (Figure 3B), FlK appears to contain a similar catalytic grouping comprised by Thr 42, Glu 50, and His 76. A previous report has excluded Glu 50 as a catalytic residue based on its location in the active site pocket as well as an increase in kcat observed for the E50A mutant, leading to a proposal of an alternative catalytic triad comprised of Thr 42, His 76, and an active site water (42). However, the positioning of Glu 50 on the third turn of the hot dog helix (α3) is consistent with the position of the Asp/Glu in hTHEM2, 4-HBA-CoA thioesterase, and PaaI from E. coli (51) (Supporting Information Figure S6) and is within hydrogen-bonding distance of the catalytic Thr.
To test whether Glu 50, His 76, and Thr 42, are involved in FlK catalysis, we measured the kinetic constants for the E50Q, H76A, T42A, T42S, and T42C mutants (Table 1). On the basis of mutagenesis studies on several members of the superfamily, the Asp/Glu and Ser/Thr residues have both been implicated as potentially involved in assisting water attack at the carbonyl (50, 54, 57, 58). Mutation of Glu 50 in FlK to Gln resulted in a significant 3000-fold decrease in kcat, which is consistent with an important role for Glu 50 in catalysis. However, the rate of hydrolysis of fluoroacetyl-CoA catalyzed by FlK-E50Q remains over 2000-fold higher than that of the pseudo-first-order rate of the uncatalyzed reaction at pH 7.6. We should note at this time that the E50Q mutant could only be solubly expressed as aMBP fusion protein. FlK-E50Q remained soluble after removal of the MBP tag; however, a relatively large proportion (~60%) of the mutant eluted from a size-exclusion column in the void volume, suggesting the formation of a soluble aggregate. After protein concentration, we verified that the fraction corresponding to the dimer peak did not revert to soluble aggregate by size-exclusion chromatography after the steady-state kinetic studies were completed (Supporting Information Figure S7). Although we performed our kinetic measurements using only the dimeric form of the enzyme, we cannot rule out the possibility that some fraction of this sample was not correctly folded.
Replacement of His 76 with Ala resulted in a substantial 105-fold decrease in kcat to a rate acceleration of only 20-fold over the pseudo-first-order rate for the uncatalyzed reaction. In comparison, alanine replacement of His 65 in 4-HBA-CoA thioesterase (50) and of His 56 in hTHEM2 (54) resulted in only 4000-fold and 2-fold reductions in kcat, respectively. The effect of this mutation on the overall rate of the reaction is much greater than the effect of the E50Q mutation and may point toward a more important role for His 76 in FlK than in other characterized members of the superfamily. Consistent with the observed defect in kcat, FlK-H76A was not competent to confer fluoroacetate resistance in a heterologous expression system (Figure 1). As discussed previously, His 76 is located at the dimer interface, but it is not within hydrogen-bonding distance of Glu 50 (Figure 3B). Based on a homology model of the FlK-substrate complex (Supporting Information Figure S8), His 76 appears to be positioned closer to the thioester sulfur atom. Thus, it is possible that His 76 helps to replace the function of the conserved Asn/Gln found in other hot dog-fold thioesterases by assisting departure of the thiolate leaving group with less carbonyl polarization in FlK. Because fluoroacetyl-CoA is more activated toward nucleophilic attack, this change in mechanism could contribute to the large observed difference in the kcat parameter between fluoroacetyl-CoA and acetyl-CoA.
We next turned our attention to examining the role of Thr 42 in catalysis by substitution with Ala, which led to a 900-fold reduction in kcat. Although the kcat/KM value measured for the T42A mutant is similar to the E50Q mutant, the 15-fold increase in KM may indicate that this residue participates in both catalysis and recognition of the substrate. Due to speculation that FlK might achieve its selectivity by using a secondary alcohol nucleophile (41, 42), we also characterized the T42S and T42C mutants to further probe the role of nucleophilicity or basicity in the FlK reaction. The T42S and T42C mutants show 20- and 35-fold decreases in kcat, respectively, but differ with regard to their response in kcat/KM. As kcat/KM is an order of magnitude lower for the T42C mutant, it is likely that the structural perturbations at the active site are greater in this mutant and do not allow us to conclusively determine the trend in reactivity for Thr 42 in catalysis.
To further probe the role of Thr 42, we compared the kinetic parameters with respect to both fluoroacetyl-CoA and acetyl-CoA for the T42S and T42C mutants (Table 1). In , the relative decrease in kcat for acetyl-CoA was smaller than the decrease in kcat observed with respect to fluoroacetyl-CoA. However, the most striking effect of these mutations is that neither the T42S nor the T42C mutant shows a substantial difference between fluoroacetyl-CoA and acetyl-CoA in the KM parameter. Interestingly, the KM value for acetyl-CoA in the T42S mutant is within range of other characterized hot dog-fold thioesterases for their substrates. If we assume that the same kinetic mechanism is valid for both fluoroacetyl-CoA and acetyl-CoA, comparison of KM values implies that discrimination in substrate binding has been lost in both the T42S and T42C mutants. In terms of kcat/KM, the discrimination between the fluorinated and nonfluorinated substrates is decreased from 106-fold in the wildtype enzyme to 200-fold for the T42S mutant and to 250-fold for the T42C mutant, and the observed discrimination is accounted for almost entirely by the difference in kcat between the two substrates. One possible explanation for this is that the removal of steric bulk at this position in the T42S and T42C mutants could allow for structural rearrangements that introduce new interactions with acetyl-CoA, enhancing FlK’s affinity for this substrate. In support of this hypothesis, it is interesting to note that the recently reported structure of FlK-T42S in complex with acetyl-CoA shows increased flexibility at the active site as determined by the observed population of two different rotamers of Ser 42 in the crystal structure (42). Although no new interactions with acetyl-CoA were found in the structure, the observed flexibility raises the possibility that a second hydrogen bond donor is accessible to the acetyl-CoA carbonyl group that could account for the observed decrease in KM.
Mutagenesis Studies of the Putative Substrate-Binding Pocket of FlK
We next turned our attention to examining the role of the hydrophobic lid residues identified in the putative substrate-binding pocket (Figure 3C). Based on structural alignments with ligand-bound forms of 4-HBA-CoA thioesterase from Arthrobacter sp. (50) and hTHEM2 (54) (Supporting Information Figure S6), the fluorine atom would be located in a largely hydrophobic environment surrounded by Phe 33, Phe 36, Val 23, Leu 26, Val 54, and Pro 37 (Supporting Information Figure S8), which are all conserved or conservatively substituted with bulky, hydrophobic residues in closely related FlK homologues of unknown function (Supporting Information Figure S5). Mutation of Phe 36 to alanine led to a 100-fold decrease in kcat/KM with very little perturbation of kcat for fluoroacetyl-CoA (Table 1). For the F33A, V23A, and L26A mutants, the magnitude in the decrease of kcat/KM (F33A, 2800-fold; V23A, 100-fold; L26A, 900-fold) was larger than the impact on kcat for fluoroacetyl-CoA (F33A, 14-fold decrease; V23A, 2.5-fold decrease; L26, 58-fold decrease). Interestingly, the kinetic constants with respect to acetyl-CoA for both the F36A and V23A mutants remained relatively unchanged, which is consistent with an important function for these residues in discrimination between the fluorinated and nonfluorinated substrates. Notably, all of these residues are part of the FlK-specific lid structure. Although the lid residues have been found to be conserved in several uncharacterized homologues, Phe 36 is unique and not found in other sequences published to date (Supporting Information Figure S5).
We next looked to characterizing the role of hydrogen-bonding and dipolar interactions in the substrate-binding pocket. Arg 120 is the only polar residue found in the active site pocket besides the catalytic triad and was previously proposed to function as a hydrogen bond donor to the fluorine atom (42). Mutation of Arg 120 to Ala, Gln, and Lys all led to formation of highly insoluble protein with no dimeric protein observed by size-exclusion chromatography, which implies that the 1-point hydrogen bond that this highly conserved residue shares with Glu 50 in apo FlK is important to the structural integrity of the dimer. Except for the conservative E50Q mutation, all other mutants tested of Glu 50 and Arg 120 resulted in insoluble protein that could not be purified in dimeric form. In addition to any enthalpic contributions of C–F · · · H–N dipolar interactions, it is also possible that only substrates that can replace the hydrogen-bonding interaction of Arg 120 with Glu 50, such as fluoroacetyl-CoA, can maintain the dimer interface and bind well in the active site pocket. Attempts to engineer a hydrogen bond donor for the carbonyl oxygen of the acyl-CoA substrate as observed in other members of the superfamily by replacement of Val 23 with Asn and Gln yielded protein with little or no activity toward either fluoroacetyl-CoA or acetyl-CoA. These results suggest that the specifics of active site packing and configuration may have diverged between FlK and other superfamily members without allowing us to directly address the role of the putative missing hydrogen bond.
In light of the kinetic constants measured for fluoroacetyl-CoA and acetyl-CoA for the F36A, F33A, and V23A mutants, a trend emerges in which removal of hydrophobic groups near the substrate-binding pocket decreases the kcat/KM for fluoroacetyl-CoA by 2–3 orders of magnitude while leaving the kcat/KM for acetyl-CoA relatively unchanged. The magnitude of this decrease is consistent with both the difference in KM with respect to fluoroacetyl-CoA and acetyl-CoA measured for wild-type FlK (Table 1) and the results from competitive inhibition studies (Figure 2). Although these data suggest that the hydrophobic groups are involved in specific recognition of the fluorine atom by FlK, a survey of the chemical environments of the fluorine atom in protein crystal structures with a bound fluorinated ligand has previously shown that binding of a fluorine atom in a lipophilic pocket does not typically confer advantages in terms of binding affinity on this scale (8). It has been observed, however, that fluorinated compounds often have negative entropies of aqueous solvation due to the tendency of water molecules to order around the hydrophobic C–F unit (11). In particular, monofluorination at the α-carbon of carbonyl-containing compounds typically increases their lipophilicity as determined by octanol–water and nonanol–water partition coefficients (7). The hydrophobicity of the fluoroacetyl-CoA binding pocket could thus promote fluoroacetyl-CoA binding over acetyl-CoA binding by providing a more lipophilic environment into which fluoroacetyl-CoA can partition, resulting in the release of water and an entropic driving force for binding. Furthermore, any enthalpic dipolar interactions with the C–F unit would be favored in a more apolar environment because organic fluorine does not compete well with stronger hydrogen bond acceptors. As it therefore seems inconsistent with previous studies that these residues are utilized mostly to make enthalpically favorable interactions with the fluorine substituent in the FlK binding pocket, we propose that the a polar, aromatic nature of the substrate-binding pocket provides an entropic driving force for substrate binding by promoting release of ordered water molecules from the C–F unit.
Crystal Structure of the FlK-Fluoroacetate Complex
We solved the structure of the FlK-fluoroacetate complex at 2.3 Å resolution to further investigate fluoroacetyl-CoA binding to FlK (Table 2). Seven fluoroacetate molecules were trapped in the structure (Figure 4), all located either in the putative active site or along the channel where CoA is expected to bind based on homology (Supporting Information Figure S8). The fluoroacetate molecules bound closest to the putative catalytic residues are bound in different orientations at the two active sites, suggesting that their binding may differ from how the fluoroacetyl unit is bound in the substrate complex. Strikingly, a large conformational change occurs in the lid region of one active site in the product complex, involving the swinging aside of the unique Phe 36 and twisting of Phe 33 to open a channel in the protein between the active site and solvent that is blocked by Phe 36 in the apo structure (Figure 4). These movements are consistent with recently reported structures of the wild-type FlK bound to fluoroacetate and pantothenate-based substrate analogues (42). Our structure is distinctive, however, in that we observe a 3.3 Å interaction between a fluoroacetate fluorine atom and Phe 33, suggesting a possible mechanism for disruption of the edge–face interaction between Phe 33 and Phe 36 and gate opening. The orientation of the fluoroacetyl group that leads to the disruption would not be possible in the substrate complex as it would force the CoA chain to occupy the same space as helix α3 from either chain A or chain B. This observation raises the possibility that the Phe 33-Phe 36 gate could be relevant not only for maintaining the hydrophobicity of the active site for fluoroacetyl-CoA binding but also potentially for release of fluoroacetate. The movement of Phe 33 and Phe 36 could facilitate fluoroacetate release either by opening up a channel though which fluoroacetate could exit or by allowing the entrance of water into the active site and the displacement and hydration of fluoroacetate. We were also able to determine the structure of a fluoroacetyl-CoA-soaked crystal at 2.0 Å resolution. Although electron density was clearly observed in the active site, it could not be unambiguously assigned to either substrate or products. The same “open gate” conformation was trapped in this structure, which supports the hypothesis that the observed conformational changes are relevant to the normal catalytic cycle of FlK.
Figure 4.

Conformational changes in the fluoroacetate complex of FlK. Chain A of both the unliganded and fluoroacetate-bound structures is shown in blue, and chain B of both structures is shown in gray. Phe 36 in shown in light blue for the closed conformation and dark blue for the open conformation. A total of seven fluoroacetate molecules were found in the active site (carbon, gray; oxygen, red; fluorine, light green).
Crystal Structures of FlK-F36A and of the FlK-F36A Product Complex
To explore the role of ordered water and Phe 36 in substrate discrimination, we solved the crystal structures of apo-FlK-F36A at 2.3 Å resolution and of the FlK-F36A product complex at 2.0 Å resolution (Table 3). The overall folds of both structures do not appear to be significantly perturbed by the F36A mutation and are in good agreement with the corresponding wild-type structures with a core rmsd of 0.5 Å for both apo- FlK-F36A and the product complex of FlK-F36A. Notably, the channel that is normally occluded by Phe 36 is unblocked in the apo FlK-F36A mutant structure and correspondingly appears to have a more highly aquated active site compared to wild-type enzyme (Figure 5). In both apo structures, water molecules occupy the space that would typically be occupied with the substrate based on homology. However, additional ordered water molecules were observed at the active site of FlK-F36A and found to form a continuous hydrogen-bonding network with the external waters that are usually blocked from the active site by Phe 36. One of these internal waters makes a 2.9 Å hydrogen bond with Arg 120, potentially competing with a favorable interaction with the fluorine atom. In contrast, three ordered water molecules were observed just outside of the Phe 36 “gate” in the wild-type FlK structure that were unable to interact with water molecules within the active site. We propose that the exclusion of water from the active site of the wild-type enzyme prevents solvation of residues that would otherwise make favorable interactions with the fluorine atom and promotes the release of ordered water molecules from the C–F unit, providing an entropic driving force for fluoroacetyl-CoA binding. The greater extent of the water network at the active site of FlK-F36A compared to wild type supports the hypothesis that its large KM defect with respect to fluoroacetyl-CoA arises from a diminished ability to exclude water from the active site.
Table 3.
Data Collection and Refinement Statistics for FlK-F36A Structures
| FlK-F36A | FlK-F36A products | |
|---|---|---|
| data collection | ||
| X-ray source | ALS 8.3.1 | ALS 8.3.1 |
| space group | C2 | C2 |
| cell dimensions | ||
| a, b, c (Å) | 141.9, 89.3, 71.4 | 141.0, 88.6, 70.9 |
| α, β, γ (deg) | 90.0, 118.0, 90.0 | 90.0, 118.0, 90.0 |
| wavelength | 1.116 | 1.116 |
| resolutiona | 19.75–2.30 (2.48–2.30) | 72.48–1.90 (1.95–1.90) |
| Rmerge (%)a | 12.6 (40.3)b | 8.9 (66.3) |
| I/σIa | 6.0 (2.5) | 13.37 (2.22) |
| completeness (%)a | 99.7 (99.4) | 95.9 (90.9) |
| redundancya | 4.1 (4.2) | 4.3 (4.1) |
| refinement | ||
| resolution | 19.75–2.30 | 72.48–2.00 |
| no. of reflections | 34432 | 48041 |
| Rwork/Rfree | 0.220/0.247 | 0.218/0.251 |
| no. of atoms | 6660 | 6351 |
| nonsolvent | 6092 | 6081 |
| solvent | 563 | 270 |
| B-factors | ||
| protein | 33.7 | 32.6 |
| water | 38.2 | 40.1 |
| rmsd | ||
| bond lengths (Å) | 0.018 | 0.006 |
| bond angles (deg) | 1.64 | 1.03 |
Values in parentheses are for the highest resolution shell.
Due to high noncrystallographic symmetry, Rpim is reported.
Figure 5.

Accessibility of the active site to water in FlK and FlK-F36A. Electron density maps are shown in light blue, chain A is colored in blue, chain B is colored in gray, and water molecules are shown as red spheres. (A) 2Fo − Fc map contoured at 0.8σ for water molecules at the active site of wild-type FlK. (B) 2Fo − Fc map contoured at 0.8σ for water molecules at the active site of FlK-F36A. (C) Fo − Fc solvent omit map contoured at 2.5σ for wild-type FlK overlaid with the refined FlK structure. (D) Fo − Fc solvent omit map contoured at 2.5σ for FlK-F36A overlaid with the refined FlK-F36A structure.
We also solved the crystal structure of the FlK-F36A product complex in order to further examine the role of solvation of the fluorine atom itself in the increased kcat/KM of the F36A mutant (Figure 6). A 2.5σ peak was observed in the Fo − Fc solvent/ligand omit electron density map for fluoroacetate and for the first eight atoms of the CoA chain. The orientation and position of the ligands differ significantly from the positions of pantothenate- derived fluoroacetyl-CoA analogues bound in two recently reported structures of FlK (42). Dias et al. have speculated that the ligands are bound in positions that are irrelevant to catalysis due to the absence of the nucleotide moiety of CoA, which is believed to be important in correct binding of the substrate. However, the positions of fluoroacetate and CoA in our structure are in agreement with the positioning of substrates and products in structural homologues of FlK (Supporting Information Figure S6) and confirm the location of the fluorine atom in the hydrophobic pocket, which is consistent with kinetic data implicating these lid residues in substrate recognition. The product-bound structure also reveals that the fluorine atom is indeed within 3.1 Å of the Arg 120 side chain and within 3.7 Å of the Gly 69 backbone amide, in agreement with the computational model generated by Dias et al. (42). Consistent with previously identified fluorine–arginine interactions, the plane of the C–F bond is oriented perpendicular to the plane of the guanidinium side chain in the product structure rather than in a linear hydrogen bond geometry. While hydrogen bonding with organic fluorine is considered rare or weak (8, 61, 62), these specific dipolar C–F · · · H–N interactions are abundant in protein structures with bound fluorinated ligands and are also thought to explain the “fluorophilic” character of arginine (8, 63). Indeed, 32 structures were found in a search of the Protein Data Bank for C–F interactions with the guanidinium side chain (8). As no analogous interactions would be possible for acetyl-CoA, these fluorine-specific interactions could make enthalpic contributions to the observed substrate selectivity. In addition to these interactions, the product structure reveals a 3.3 Å hydrogen bond between the backbone NH of Thr 42 and the thioester C=O and also shows that the second hydrogen bond equivalent to that provided by an Asn/Gln residue in structural homologues is indeed missing. In this structure, the fluorine atom appears to be solvated as it is within 2 Å of a chain of hydrogen-bonded water molecules that would likely be excluded from the substrate-binding pocket by Phe 36 in the closed conformation of FlK, potentially explaining the large observed defect in KM for this mutant. The product complex structure is thus consistent with a model in which the active site lid and Phe 36 in particular aid in discrimination between fluoroacetyl-CoA and acetyl-CoA by actively excluding water from the active site.
Figure 6.

FlK-F36A product complex. Chain A is colored in blue, and chain B is colored in gray. For clarity, only the fluoroacetate and CoA ligands are colored by atom (carbon, gray; nitrogen, blue; oxygen, red; fluorine, light green; sulfur, yellow). (A) A view of the product bound at the FlK-F36A active site shown in the context of the overall fold. (B) Fo − Fc solvent/ligand omit map contoured at 2.5σ overlaid with the refined FlK-F36A product complex structure. (C) Chem Draw figure showing interactions observed between FlK-F36A and the products.
CONCLUSIONS
We have demonstrated that FlK is a fluoroacetyl-CoA-specific thioesterase that exhibits a 106-fold higher catalytic efficiency for fluoroacetyl-CoA compared to acetyl-CoA. Based on experiments in a heterologous E. coli host, the in vitro selectivity of FlK also appears to be relevant in vivo and is consistent with the estimated intracellular concentration of acetyl-CoA (64), which is lower than the measured KM for acetyl-CoA. Based on kinetic measurements, the remarkable discrimination between the fluorinated and nonfluorinated substrates by FlK arises from both differences in substrate recognition (kcat/KM) and chemical reactivity (kcat) related to the fluorine substituent. We hypothesize that the more significant part of substrate selectivity is derived from mechanistic differences between FlK and other members of the superfamily that serve to select against reaction with unactivated acyl-CoAs, such as acetyl-CoA, because of the larger defect in the kcat (~104) compared to the KM (~102) parameter. Using X-ray crystallographic and biochemical studies on wild-type and mutant FlKs, we have identified several components of this selectivity and explored their role in preferential hydrolysis of fluoroacetyl-CoA.
We propose that FlK has evolved a mode of substrate recognition that is distinct from the more promiscuous hot dog-fold thioesterases by minimizing interactions with the carbonyl unit common to all acyl-CoAs to select against acetyl-CoA while recouping the resulting decrease in binding energy through C–F specific properties of fluoroacetyl-CoA. This working hypothesis is supported by structural information that indicates that a key Asn/Gln hydrogen bond to the thioester carbonyl is missing in conjunction with biochemical data comparing fluoroacetyl-CoA and acetyl-CoA hydrolysis in the T42S and T42C mutants. Our data suggest that the specificity of FlK in binding fluoroacetyl-CoA is aided in part by hydrophobic interactions with the lipophilic binding pocket and by C–F · · · H–N dipolar interactions between the fluorine atom and Arg 120. Because neither of these enthalpic contributions would be expected to account for the observed 106-fold selectivity between the fluorinated and nonfluorinated substrates given the existing literature on protein/organofluorine binding affinities, we propose that entropic contributions related to ordered water play a larger role in fluorine-based substrate discrimination. In support of this model, we have biochemically and structurally characterized a mutant of a unique phenylanine (Phe 36) on the hydrophobic “lid” of FlK and have shown that defects in water exclusion from the active site lead to a 100-fold decrease in fluoroacetyl-CoA binding affinity without affecting kcat. We have also identified key active site residues and propose an alternative catalytic triad consisting of Thr 42, Glu 50, and His 76. Based on the low kcat/kuncat measured for the H67A mutant, we further hypothesize that general acid catalysis by His 76 may be sufficient to assist CoA departure in the more activated fluoroacetyl-CoA substrate, but not acetyl-CoA, in the absence of typical interactions that serve to polarize the thioester carbonyl.
Supplementary Material
Acknowledgments
We thank Dr. Dipali Sashital for protein purification and crystallization advice and George Meigs (Beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory) for assistance with X-ray data collection.
Footnotes
This work was supported by the University of California, Berkeley, the Arnold and Mabel Beckman Foundation, and a National Science Foundation Graduate Research Fellowship (to A.M.W.). A.M.W. acknowledges the support of a National Institutes of Health NRSA Training Grant (1 T32 GMO66698). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
The atomic coordinates and structure factors for the structures reported herein have been deposited in the Protein Data Bank (accession numbers 3P2Q, 3P2R, 3P2S, 3P3F, and 3P3I).
Abbreviations: CoA, coenzyme A; TCA cycle, tricarboxylic acid cycle; S. cattleya, Streptomyces cattleya; 4-HBA-CoA, 4-hydroxybenzoyl- CoA; hTHEM2, human thioesterase superfamily member 2; PaaI, phenylacetyl-CoA thioesterase; MBP, maltose binding protein.
SUPPORTING INFORMATION AVAILABLE
Sequences of oligonucleotides, SDS–PAGE, size-exclusion chromatograms, sequence and structural alignments, and nonenzymatic hydrolysis data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gribble GW. The diversity of naturally produced organohalogens. Handb Environ Chem. 2003;3:1–15. doi: 10.1016/S0045-6535(03)00207-8. [DOI] [PubMed] [Google Scholar]
- 2.O’Hagan D, Harper D. Fluorine-containing natural products. J Fluorine Chem. 1999;100:127–133. [Google Scholar]
- 3.Gribble G. Naturally occuring organofluorines. Handb Environ Chem. 2002;3:121–136. [Google Scholar]
- 4.Murphy CD, Schaffrath C, O’Hagan D. Fluorinated natural products: The biosynthesis of fluoroacetate and 4-fluorothreonine in Streptomyces cattleya. Chemosphere. 2003;52:455–461. doi: 10.1016/S0045-6535(03)00191-7. [DOI] [PubMed] [Google Scholar]
- 5.Deng H, O’Hagan D, Schaffrath C. Fluorometabolite biosynthesis and the fluorinase from Streptomyces cattleya. Nat Prod Rep. 2004;21:773–784. doi: 10.1039/b415087m. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda K, Tamura T, Segawa Y, Mutaguchi Y, Inagaki K. Enhanced production of the fluorinated nucleoside antibiotic nucleocidin by a rifR-resistant mutant of Streptomyces calvus IFO13200. Actinomycetologica. 2009;23:51–55. [Google Scholar]
- 7.Smart B. Fluorine substituent effects (on bioactivity) J Fluorine Chem. 2001;109:3–11. [Google Scholar]
- 8.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 9.Purser S, Moore PR, Swallow S, Gouverneur V. Fluorine in medicinal chemistry. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 10.O’Hagan D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 11.Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. Chem Bio Chem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 12.Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: Preclinical pharmacology and mechanisms of action. Semin Oncol. 1996;23:3–15. [PubMed] [Google Scholar]
- 13.van der Donk WA, Yu G, Silva DJ, Stubbe J, McCarthy JR, Jarvi ET, Matthews DP, Resvick RJ, Wagner E. Inactivation of ribonucleotide reductase by (E)-2′-fluoromethylene-2′-deoxycytidine-5′-diphosphate: A paradigm for nucleotide mechanism based inhibitors. Biochemistry. 1996;35:8381–8391. doi: 10.1021/bi960190j. [DOI] [PubMed] [Google Scholar]
- 14.Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW, Yu S, Pon RT, Gmeiner WH, Pommier Y. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res. 2002;8:2499–2504. [PubMed] [Google Scholar]
- 15.Parker WB, Cheng YC. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol Ther. 1990;48:381–395. doi: 10.1016/0163-7258(90)90056-8. [DOI] [PubMed] [Google Scholar]
- 16.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 17.Chenoweth MB. Monofluoroacetic acid and related compounds. J Pharmacol Exp Ther. 1949;97:383–423. [PubMed] [Google Scholar]
- 18.Rowinsky EK, Cazenave LA, Donehower RC. Taxol: A novel investigational antimicrotubule agent. J Natl Cancer Inst. 1990;82:1247–1259. doi: 10.1093/jnci/82.15.1247. [DOI] [PubMed] [Google Scholar]
- 19.Buffa P, Peters RA. The in vivo formation of citrate induced by fluoroacetate and its signficance. J Physiol. 1950;110:488–500. doi: 10.1113/jphysiol.1949.sp004456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buffa P, Peters RA, Wakelin RW. Biochemistry of fluoroacetate poisoning. Isolation of an active tricarboxylic acid fraction from poisoned kidney homogenates. Biochem J. 1951;48:467–477. doi: 10.1042/bj0480467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morrison JF, Peters RA. Biochemistry of fluoroacetate poisoning: The effect of fluorocitrate on purified aconitase. Biochem J. 1954;58:473–479. doi: 10.1042/bj0580473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke DD. Fluoroacetate and fluorocitrate: Mechanism of action. Neurochem Res. 1991;16:1055–1058. doi: 10.1007/BF00965850. [DOI] [PubMed] [Google Scholar]
- 23.Brandt M, Evans S, Mendes-Morao J, Chappell B. Fluorocitrate inhibition of aconitate hydratase and the tricarboxylate carrier of rat liver mitochondria. Biochem J. 1973;134:217–224. doi: 10.1042/bj1340217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villafranca JJ, Platus E. Fluorocitrate inhibition of aconitase. Reversibility of the inactivation. Biochem Biophys Res Commun. 1973;55:1197–1207. doi: 10.1016/s0006-291x(73)80021-x. [DOI] [PubMed] [Google Scholar]
- 25.Eanes RZ, Kun E. Inhibition of liver aconitase isozymes by (−)-erythro-fluorocitrate. Mol Pharmacol. 1974;10:130–139. [PubMed] [Google Scholar]
- 26.Kent TA, Emptage MH, Merkle H, Kennedy MC, Beinert H, Munck E. Mossbauer studies of aconitase. Substrate and inhibitor binding, reaction intermediates, and hyperfine interactions of reduced 3Fe and 4Fe clusters. J Biol Chem. 1985;260:6871–6881. [PubMed] [Google Scholar]
- 27.Lauble H, Kennedy MC, Emptage MH, Beinert H, Stout CD. The reaction of fluorocitrate with aconitase and the crystal structure of the enzyme-inhibitor complex. Proc Natl Acad Sci USA. 1996;93:13699–13703. doi: 10.1073/pnas.93.24.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marais J. Isolation of the toxic principle “K cymonate” from “Gifblaar” Dichapetalum cymosum. J Vet Sci Anim Ind. 1943;18:203–206. [Google Scholar]
- 29.Vickery B, Vickery M. Fluoride metabolism in Dichapetalum toxicarium. Phytochemistry. 1972;11:1905–1909. [Google Scholar]
- 30.Hall R. The distribution of organic fluorine in some toxic tropical plants. New Phytol. 1972;71:855–871. [Google Scholar]
- 31.Twigg L, King D. The impact of fluoroacetate-bearing vegetation on native Australian fauna: A review. Oikos. 1991;61:412–430. [Google Scholar]
- 32.Harper DB, O’Hagan D. The fluorinated natural products. Nat Prod Rep. 1994;11:123–133. doi: 10.1039/np9941100123. [DOI] [PubMed] [Google Scholar]
- 33.Sanada M, Miyano T, Iwadare S, Williamson JM, Arison BH, Smith JL, Douglas AW, Liesch JM, Inamine E. Biosynthesis of fluorothreonine and fluoroacetic acid by the thienamycin producer, Streptomyces cattleya. J Antibiot (Tokyo) 1986;39:259–265. doi: 10.7164/antibiotics.39.259. [DOI] [PubMed] [Google Scholar]
- 34.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 35.Benveniste R, Davies J. Aminoglycoside antibiotic in activating enzymes in actinomycetes similar to those present in clinical isolates of antibiotic-resistant bacteria. Proc Natl Acad Sci USA. 1973;70:2276–2280. doi: 10.1073/pnas.70.8.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cundliffe E. Mechanism of resistance to thiostrepton in the producing-organism Streptomyces azureus. Nature. 1978;272:792–795. doi: 10.1038/272792a0. [DOI] [PubMed] [Google Scholar]
- 37.Fierro JF, Hardisson C, Salas JA. Involvement of cell impermeability in resistance to macrolides in some producer streptomycetes. J Antibiot (Tokyo) 1988;41:142–144. doi: 10.7164/antibiotics.41.142. [DOI] [PubMed] [Google Scholar]
- 38.Thiara AS, Cundliffe E. Cloning and characterization of a DNA gyrase B gene from Streptomyces sphaeroides that confers resistance to novobiocin. EMBO J. 1988;7:2255–2259. doi: 10.1002/j.1460-2075.1988.tb03065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cundliffe E. How antibiotic-producing organisms avoid suicide. Annu Rev Microbiol. 1989;43:207–233. doi: 10.1146/annurev.mi.43.100189.001231. [DOI] [PubMed] [Google Scholar]
- 40.Mendez C, Salas JA. ABC transporters in antibiotic-producing actinomycetes. FEMS Microbiol Lett. 1998;158:1–8. doi: 10.1111/j.1574-6968.1998.tb12792.x. [DOI] [PubMed] [Google Scholar]
- 41.Huang F, Haydock SF, Spiteller D, Mironenko T, Li TL, O’Hagan D, Leadlay PF, Spencer JB. The gene cluster for fluorometabolite biosynthesis in Streptomyces cattleya: A thioesterase confers resistance to fluoroacetyl-coenzyme A. Chem Biol. 2006;13:475–484. doi: 10.1016/j.chembiol.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 42.Dias MV, Huang F, Chirgadze DY, Tosin M, Spiteller D, Dry EF, Leadlay PF, Spencer JB, Blundell TL. Structural basis for the activity and substrate specificity of fluoroacetyl-CoA thioesterase FlK. J Biol Chem. 2010;285:22495–22504. doi: 10.1074/jbc.M110.107177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouillard JM, Lee W, Truan G, Gao X, Zhou X, Gulari E. Gene2Oligo: Oligonucleotide design for in vitro gene synthesis. Nucleic Acids Res. 2004;32:W176–180. doi: 10.1093/nar/gkh401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tropea JE, Cherry S, Waugh DS. Expression and purification of soluble His(6)-tagged TEV protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;212:916–924. [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstsleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, Grosse-Kunstsleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 48.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 49.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 50.Thoden JB, Zhuang Z, Dunaway-Mariano D, Holden HM. The structure of 4-hydroxybenzoyl-CoA thioesterase from Arthrobacter sp. strain SU. J Biol Chem. 2003;278:43709–43716. doi: 10.1074/jbc.M308198200. [DOI] [PubMed] [Google Scholar]
- 51.Song F, Zhuang Z, Finci L, Dunaway-Mariano D, Kniewel R, Buglino JA, Solorzano V, Wu J, Lima CD. Structure, function, and mechanism of the phenylacetate pathway hot dog-fold thioesterase PaaI. J Biol Chem. 2006;281:11028–11038. doi: 10.1074/jbc.M513896200. [DOI] [PubMed] [Google Scholar]
- 52.Cheng Z, Song F, Shan X, Wei Z, Wang Y, Dunaway-Mariano D, Gong W. Crystal structure of human thioesterase superfamily member 2. Biochem Biophys Res Commun. 2006;349:172–177. doi: 10.1016/j.bbrc.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 53.Zhuang Z, Song F, Zhao H, Li L, Cao J, Eisenstein E, Herzberg O, Dunaway-Mariano D. Divergence of function in the hot dog fold enzyme superfamily: The bacterial thioesterase YciA. Biochemistry. 2008;47:2789–2796. doi: 10.1021/bi702334h. [DOI] [PubMed] [Google Scholar]
- 54.Cao J, Xu H, Zhao H, Gong W, Dunaway-Mariano D. The mechanisms of human hotdog-fold thioesterase 2 (hTHEM2) substrate recognition and catalysis illuminated by a structure and function based analysis. Biochemistry. 2009;48:1293–1304. doi: 10.1021/bi801879z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen D, Wu R, Bryan TL, Dunaway-Mariano D. In vitro kinetic analysis of substrate specificity in enterobactin biosynthetic lower pathway enzymes provides insight into the biochemical function of the hot dog-fold thioesterase EntH. Biochemistry. 2009;48:511–513. doi: 10.1021/bi802207t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunishima N, Asada Y, Sugahara M, Ishijima J, Nodake Y, Miyano M, Kuramitsu S, Yokoyama S. A novel induced-fit reaction mechanism of asymmetric hot dog thioesterase PAAI. J Mol Biol. 2005;352:212–228. doi: 10.1016/j.jmb.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 57.Song F, Zhuang Z, Dunaway-Mariano D. Structure-activity analysis of base and enzyme-catalyzed 4-hydroxybenzoyl coenzyme A hydrolysis. Bioorg Chem. 2007;35:1–10. doi: 10.1016/j.bioorg.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama T, Choi KJ, Bosch AM, Yeo HJ. Structure and function of a Campylobacter jejuni thioesterase Cj\915, a hexameric hot dog fold enzyme. Biochim Biophys Acta. 2009;1794:1073–1081. doi: 10.1016/j.bbapap.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 60.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howard J, Hoy V, O’Hagan D, Smith G. How good is fluorine as a hydrogen bond acceptor? Tetrahedron. 1996;52:12613–12622. [Google Scholar]
- 62.Dunitz J, Taylor R. Organic fluorine hardly ever accepts hydrogen bonds. Chem—Eur J. 1997;3:89–98. [Google Scholar]
- 63.Zhou P, Zou J, Tian F, Shang Z. Fluorine bonding—How does it work in protein ligand interactions? J Chem Inf Model. 2009;49:2344–2355. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 64.Shimazu M, Vetcher L, Galazzo JL, Licari P, Santi DV. A sensitive and robust method for quantification of intracellular short-chain coenzyme A esters. Anal Biochem. 2004;328:51–59. doi: 10.1016/j.ab.2004.01.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.