

Abstract
Natriuretic peptides (NPs) control natriuresis and normalize changes in blood pressure. Recent studies suggest that NPs are also involved in the regulation of pain sensitivity, although the underlying mechanisms remain essentially unknown. Many biological effects of NPs are mediated by guanylate cyclase (GC)-coupled NP receptors, NPR-A and NPR-B, whereas the third NP receptor, NPR-C, lacks the GC kinase domain and acts as the NP clearance receptor. In addition, NPR-C can couple to specific Gαi–Gβγ-mediated intracellular signaling cascades in numerous cell types. We found that NPR-C is coexpressed in transient receptor potential vanilloid-1 (TRPV1)-expressing mouse dorsal root ganglia (DRG) neurons. NPR-C can be coimmunoprecipitated with Gαi, and C-type natriuretic peptide (CNP) treatment induced translocation of protein kinase Cε (PKCε) to the plasma membrane of these neurons, which was inhibited by pertussis toxin pretreatment. Application of CNP potentiated capsaicin- and proton-activated TRPV1 currents in cultured mouse DRG neurons and increased their firing frequency, an effect that was absent in DRG neurons from TRPV1−/− mice. CNP-induced sensitization of TRPV1 activity was attenuated by pretreatment of DRG neurons with the specific inhibitors of Gβγ, phospholipase C-β (PLCβ), or PKC, but not of protein kinase A, and was abolished by mutations at two PKC phosphorylation sites in TRPV1. Furthermore, CNP injection into mouse hindpaw led to the development of thermal hyperalgesia that was attenuated by administration of specific inhibitors of Gβγ or TRPV1 and was also absent in TRPV1−/− mice. Thus, our work identifies the Gβγ–PLCβ–PKC-dependent potentiation of TRPV1 as a novel signaling cascade recruited by CNP in mouse DRG neurons that can lead to enhanced nociceptor excitability and thermal hypersensitivity.
Introduction
Natriuretic peptides (NPs) mediate natriuresis to provide homeostatic control of body water, Na+ and K+ electrolytes, and fat, as well as playing a critical role in normalizing changes in blood pressure (Suzuki et al., 2001; Pandey, 2005b; Moro and Berlan, 2006; Potter et al., 2009). These events are mediated via downstream cellular signaling events, resulting from the activation of guanylate cyclase (GC)-coupled natriuretic peptide receptors, NPR-A and NPR-B, by atrial-type (ANP), brain-type (NBP), and C-type (CNP) natriuretic peptides (Pandey, 2005b; Potter et al., 2006, 2009). Additionally, NPs have been implicated in sensory nerve fiber sprouting and bifurcation (Kishimoto et al., 2008; Schmidt et al., 2009), as well as regulation of pain sensitivity (Schmidtko et al., 2008a; Zhang et al., 2010; Heine et al., 2011). Activation of protein kinase G (PKG) downstream of BNP/NPR-A in dorsal root ganglia (DRG) neurons has been shown to mediate the negative regulation of nociceptive transmission via modulation of presynaptic BKCa channel activity in the dorsal horn of the spinal cord (Zhang et al., 2010). Conversely, a recent report showed enhanced mechanical hypersensitivity in rats upon intrathecal injection of CNP (Heine et al., 2011). The canonical NPR-A- and NPR-B-mediated downstream cGMP/PKG signaling pathways mediate all these effects of BNP and CNP, respectively (Schmidtko et al., 2008a; Zhang et al., 2010; Heine et al., 2011). NPR-C is an additional NP receptor that, unlike NPR-A and NPR-B, lacks the GC kinase domain and thereby serves as the NP clearance receptor (Pandey, 2005a; Potter et al., 2006). However, NPR-C has also been shown to signal through the pertussis toxin (PTX)-sensitive Gαi protein in a variety of cell types, in which its activation by CNP (and ANP) leads to specific Gαi/o/βγ downstream signaling, including Gβγ-mediated phospholipase C-β (PLCβ) activation, phosphatidylinositol-4,5-biphosphate (PIP2) hydrolysis, and subsequent activation of protein kinase C (PKC) (Levin, 1993; Murthy and Makhlouf, 1999; Murthy et al., 2000; Anand-Srivastava, 2005).
Vascular endothelial cells secrete elevated levels of CNP during stimulation by inflammatory mediators, such as interleukin-1α/β (IL-1α/β), tumor necrosis factor-α (TNF-α), and lipopolysaccharide (Suga et al., 1993). Furthermore, increased serum CNP levels were found in human patients with rheumatoid arthritis and systemic sclerosis (Olewicz-Gawlik et al., 2010a,b). These findings suggest that CNP plays a role in vascular inflammation (Ahluwalia and Hobbs, 2005) and possibly in inflammatory pain sensitization. The transient receptor potential vanilloid-1 (TRPV1) channel on sensory afferents is a key mediator of peripheral sensory detection and transduction of inflammatory pain. Mechanistically, this process involves diverse phosphorylation-dependent sensitization and proton-activation/potentiation of TRPV1 channel activity by a variety of pro/inflammatory mediators (Hucho and Levine, 2007; Patapoutian et al., 2009; Gold and Gebhart, 2010; Kuner, 2010; Ren and Dubner, 2010). Here we demonstrate that CNP induces acute sensitization of TRPV1 channel activity through a non-canonical Gαi–Gβγ-mediated downstream activation of PKC and phosphorylation of the channel protein, resulting in enhanced nociceptor firing. We also show that CNP injection leads to the development of thermal hyperalgesia that is dependent on Gβ/γ signaling and TRPV1.
Materials and Methods
All experiments involving the use of mice and the procedures followed therein were approved by the University of Iowa Institutional Animal Care and Use Committee and in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. Every effort was made to minimize the number of mice used and their suffering.
Primary cultures of mouse DRG neurons.
DRGs were isolated from adult C57BL/6 mice of both TRPV1+/+ and TRPV1−/− genotypes and cultured on poly-l-ornithine- and laminin-coated glass coverslips, as described previously (Schnizler et al., 2008). Briefly, isolated DRGs were digested with collagenase for 20 min followed by neutralization, centrifugation, and trituration before further digestion with pronase for 10 min. Cells were then pelleted by centrifugation and resuspended in DMEM (Invitrogen) supplemented with 10% fetal bovine serum, before additional trituration with fire-polished glass Pasteur pipettes. Neurons were plated onto poly-l-ornithine- and laminin-coated glass coverslips. After 90 min incubation at 37°C in a 5% CO2 incubator, the culture media was changed to TNB media supplemented with protein–lipid complex (Biochrom) and cultured therein, as described previously (Constantin et al., 2008). Cultured DRG neurons were used in electrophysiological, immunocytochemical, and biochemical experiments within 1–4 days of in vitro (DIV) culturing.
Site-directed mutagenesis and transfection of human embryonic kidney 293T cells.
Mutagenesis of rat TRPV1 cDNA was performed on the pcDNA3–rTRPV1 plasmid (generously provided by Dr. David Julius, University of California, San Francisco, San Francisco, CA) using the quick-change site-directed mutagenesis kit (Agilent Technologies). We generated the rTRPV1–S502A, rTRPV1–T704A, rTRPV1–S800A, and rTRPV1–S502A/T704A/S800A triple PKC phosphorylation site mutant constructs using the pcDNA3–rTRPV1-S502A/S800A plasmid as the template (generously provided by Dr. Carla Nau, University of Erlangen–Nuremberg, Nuremberg, Germany). Human embryonic kidney 293T (HEK293T) cells were cultured in DMEM with 1× GlutaMAX (both from Invitrogen) and 10% fetal bovine serum (HyClone) and maintained at 37°C in a humidified incubator with 5% CO2. Cells were transiently transfected with wild-type (WT) or PKC site mutant TRPV1 plasmids (0.5 μg) along with the reporter plasmid (0.5 μg peGFPc1; Clontech), with or without the pCMV–Sport6–hNPR-C plasmid (0.5 μg; Open BioSystems) using Lipofectamine2000 (Invitrogen) reagent, as per the instructions of the manufacturer. Transfected cells were used for electrophysiological experiments within 36–48 h.
Calcium imaging.
Functional Ca2+ imaging on cultured mouse DRG neurons (2–3 DIV) was performed as described previously (Schnizler et al., 2008). Neurons on glass coverslips were incubated at room temperature (22°C) for 30 min with 2 μm of the AM form of the Ca2+-sensitive dye fura-2 (Invitrogen). The coverslip was then placed in the recording chamber mounted on the stage of an inverted IX-71 microscope (Olympus) and washed for 10 min before the experiment began. Fluorescence was alternately excited at 340 and 380 nm (both 12 nm bandpass) using the Polychrome IV monochromator (T.I.L.L. Photonics), via a 10× or 20× objective [numerical aperture (NA) 0.75; Olympus]. Emitted fluorescence was collected at 510 (80) nm using an IMAGO CCD camera (T.I.L.L. Photonics). Pairs of 340/380 nm images were sampled at 0.2 Hz. Bath application of capsaicin (15 s) was performed twice with a 5 min interval, and NPs, prostaglandin E2 (PGE2), and bradykinin (BK) were applied during this interval. The fluorescence ratio (r = F340/F380) was converted to [Ca2+]i, as described previously (Schnizler et al., 2008). Data were processed and analyzed using TILLvisION 4.0.1.2 (T.I.L.L. Photonics) and Origin 7.0 (OriginLab) software and presented as mean ± SEM. All the Ca2+ imaging experiments with or without NP/PGE2/BK treatments were performed in triplicate across three different batches of mouse DRG neuron cultures.
Electrophysiology and data analyses.
Currents were recorded from small/medium-diameter cultured mouse DRG neurons (1–4 DIV) and transfected HEK293T cells at room temperature with whole-cell configuration of the patch-clamp technique. The holding potential was −70 mV. Patch pipettes were pulled from borosilicate glass tubes (TW150F-4; World Precision Instruments) and heat polished at the tip using a microforge (MF200; World Precision Instruments) to give a resistance of 3–6 MΩ when filled with the pipette solution consisting of the following (in mm): 5 NaCl, 140 KCl, 1 MgCl2, 10 HEPES, and 10 EGTA, pH 7.3. Cells were bathed in extracellular buffer consisting of the following (in mm): 140 NaCl, 5 KCl, 0.1 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 (or pH 6.8 or 6.4 for proton-activation experiments). Low extracellular Ca2+ concentration was used to minimize the Ca2+-dependent desensitization of TRPV1 currents (Koplas et al., 1997; Mohapatra et al., 2003). For recordings in neurons, 1 μm tetrodotoxin was added to the extracellular buffer to block fast-activating Nav currents. For extracellular buffers used in experiments with proton-activated currents (pH 5.8, 5.4, and 4.8), HEPES was substituted with MES. All the activators/drugs were diluted in the extracellular buffer, from a stock solution, to achieve final concentrations. All control and agonist/drug-containing extracellular buffers were applied locally onto the cells under recording, with a separate Teflon tubing-connected glass multiple-barrel perfusion system.
Currents were recorded with an Axopatch 200B patch-clamp amplifier connected to a Digidata 1440A data acquisition system (Molecular Devices), with a sample rate of 2 kHz and filtering at 1 kHz. pClamp 10 software (Molecular Devices) was used for the acquisition of currents, and Clampfit 10 (Molecular Devices) and Origin 7.0 (Origin Lab) software were used for the analysis of currents and preparing traces/figures. Data are presented as means ± SEM or fitted value ± SE of the Hill equation fit for the pH dose–response relationship.
Capsaicin- and proton-induced action potential (AP) firings in small/medium-diameter cultured mouse DRG neurons were recorded under the current-clamp mode at room temperature following similar procedures used in cultured hippocampal neurons (Mohapatra et al., 2009). Neurons were bathed in extracellular buffer containing the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.3. The pipette solution contained the following (in mm): 5 NaCl, 140 KCl, 1 CaCl2,1 MgCl2, 10 HEPES, 5 EGTA, 3 Na-ATP, and 0.3 Mg-GTP, pH 7.3. The number of APs elicited on each capsaicin and acidic pH treatment episodes of similar duration were counted to determine the nociceptor firing frequency.
RT-PCR.
Total RNA was isolated from cultured mouse DRG neurons (2–4 DIV) and HEK293T cells using TRIzol reagent (Invitrogen), treated with DNase-I for 30 min at 37°C, followed by inactivation at 65°C, and subsequently reverse transcribed into cDNA using a SuperScript III RT-PCR kit (Invitrogen), as per manufacturer's instructions. Next, individual PCR reactions were performed using these cDNAs and Pfu DNA polymerase (Agilent Technologies) with specific primer sets for NPR-A (forward, 5′-TTCTCCCTCTGGGATATGGATC-3′; and reverse, 5′-ATGCGTTTACGGTTCACACG-3′), NPR-B (forward, 5′-CAGAACACAACCTGAGCTATGC-3′; and reverse, 5′-CAGCAATAAGGTTCATCAGGG-3′), NPR-C (forward, 5′-ACAATTTCGACGAGACCAAAG-3′; and reverse, 5′-CGCCTCTCAATGGTTATTCTG-3′), and GAPDH (forward, 5′-TCAGCAATGCATCCTGCAC-3′; and reverse, 5′-CCAGGAAATGAGCTTGACAAAG-3′) for 30 cycles. The amplified PCR products were size fractionated in 0.7% agarose gel electrophoresis and stained with ethidium bromide, to visualize the DNA bands and verify the respective predicted product sizes.
Assay for intracellular cGMP production in DRG neurons.
Quantification of ANP/BNP/CNP-induced intracellular cGMP production in freshly isolated mouse DRG neurons was performed using a direct ELISA kit (Enzo Life Sciences) following the instructions of the manufacturer. Briefly, freshly isolated and dissociated DRG neurons from adult male mice were treated with individual NPs (15 min at 37°C) and subjected to lysis and subsequent determination of intracellular cGMP concentration. The protein concentrations in cell lysates were determined by the BCA method (Pierce). Intracellular cGMP concentrations were expressed as picomoles of cGMP per microgram of protein. Three independent experimental replicates were generated from three different batches of mouse DRG neurons.
Immunostaining of DRG sections and cultured DRG neurons.
Adult male C57BL/6 mice were killed, and L4–L6 lumbar DRG pairs were then dissected out from these animals and postfixed with 4% paraformaldehyde plus 5% picric acid [in 0.1 m phosphate buffer (PB)] overnight at 4°C before being transferred into 15% sucrose in 0.1 m PB for an additional incubation overnight at 4°C. Twenty-five-micrometer-thick sections of fixed DRG tissue were obtained using a cooled cryostat (CM3050 S; Leica Microsystems), as detailed previously (Shepherd and Mohapatra, 2012). Free-floating DRG tissue sections were washed in a 48-well tissue culture plate with 0.1 m PB, before being incubated with blocking/permeabilization solution (10% goat serum plus 0.3% Triton X-100 in 0.1 m PB) at 4°C for 60 min. The sections were then incubated with rabbit polyclonal anti-NPR-C antibody (10 μg/ml; Abgent) along with mouse monoclonal anti-calcitonin gene-related peptide (CGRP) (1 μg/ml) or mouse monoclonal anti-neurofilament-200 (NF-200) (1 μg/ml; both from Sigma) or mouse monoclonal anti-TRPV1 (5 μg/ml; clone N221/17; NeuroMab) antibody in blocking solution overnight at 4°C. After washing in blocking solution (three times for 10 min each at room temperature with vigorous agitation), the sections were incubated with goat anti-mouse IgG Alexa Fluor-555 and goat anti-rabbit Alexa Fluor-488-conjugated (1:1000 each; Invitrogen) secondary antibodies for 180 min in blocking solution at 4°C protected from light. The sections were subsequently washed first with 10% goat serum in 0.1 m PB for 5 min, with 0.1 m PB for 5 min, and finally with 0.05 m PB for 5 min (all at room temperature with vigorous agitation). Finally, the sections were carefully transferred onto glass slides and covered with glass coverslips with ProLong Gold antifade reagent (Invitrogen), as described previously (Shepherd and Mohapatra, 2012). Confocal fluorescence images of mounted DRG tissue sections were taken using a BX61WI microscope equipped with the Fluoview 300 laser-scanning confocal imaging system (Olympus) with a 10× objective (NA 0.25; Olympus).
Fluorescence immunostaining of cultured mouse DRG neurons were performed following standardized procedures detailed previously (Mohapatra et al., 2008). Control and drug-treated cultured mouse DRG neurons (2–3 DIV) were washed in Dulbecco's PBS, followed by 30 min fixation with 3% paraformaldehyde containing 4% sucrose at 4°C with slow agitation. Neurons were washed three times for 5 min each with Tris-buffered saline (TBS) and then incubated with the blocking solution (4% fat-free milk powder in TBS with 0.1% Triton X-100) at room temperature for 45–60 min on a medium-speed rocking platform. Cells were incubated with rabbit polyclonal anti-NPR-C (5 μg/ml; Santa Cruz Biotechnology) and mouse monoclonal anti-PKCε (5 μg/ml; BD Bioscience) antibodies diluted in blocking solution at room temperature for 45–60 min. After washing with blocking solution (three times for 5 min each at room temperature), neurons were incubated with goat anti-mouse IgG Alexa Fluor-488-conjugated and goat anti-rabbit Alexa Fluor-555-conjugated (1:2000; Life Technologies) secondary antibodies for 45–60 min in blocking solution at room temperature protected from light. Coverslips were subsequently washed with TBS (four times for 5 min each at room temperature) and mounted onto glass slides. Confocal fluorescence images of neurons were captured using a BX61WI microscope equipped with the Fluoview 300 laser-scanning confocal imaging system with a 10× objective (NA 0.25; Olympus). Quantification of cellular PKCε distribution and translocation in neurons was performed using NIH Image J software as described previously (Cesare et al., 1999; Hucho et al., 2005). From the determined PKCε signal intensity across the line on every neuron, the peak intensity in the outer 10% of the cell diameter was taken as the peripheral, and the peak intensity in the inner 90% of the cell diameter was taken as the cytoplasmic localization. Ratio of the peak peripheral versus cytoplasmic intensity in each neuron was calculated, with a higher ratio indicating more peripheral localization of PKCε. In addition, the number of individual small/medium-diameter cultured DRG neurons with PKCε immunostaining (both peripheral translocation and diffused cytoplasmic distribution) were counted for each treatment group (>500 neurons for each group) in a blinded manner. These data were quantified and plotted as percentage of neurons with peripheral PKCε translocation with different drug treatment conditions. All immunostaining experiments were repeated four times on four different batches of mouse DRG neuron cultures.
Immunoprecipitation and immunoblotting.
Coimmunoprecipitation of NPR-B and NPR-C with G-protein α-subunits from cultured mouse DRG neurons was performed following previously standardized protocols (Mohapatra et al., 2008; Schnizler et al., 2008). Briefly, cultured mouse DRG neurons (2–3 DIV) were lysed with ice-cold lysis buffer consisting of 0.02 m Tris-HCl, pH 8.0, 0.15 m NaCl, 0.001 m NaF, 0.002 m EDTA, 0.001 m PMSF, 1× protease inhibitor cocktail (leupeptin, aprotinin, antipain, and benzamidine-HCl), and 1% Triton X-100, for 30 min at 4°C on a rotator, followed by centrifugation at 12,000 × g for 10 min at 4°C to pellet the debris. The lysates were then incubated with rabbit polyclonal anti-NPR-B- or anti-NPR-C- (both Abgent) or rabbit polyclonal anti-HA antibody-tagged recombinant protein A-Sepharose beads (Pierce) suspended in lysis buffer containing BSA (0.2 mg/ml final) at 4°C for 2 h on a rotator. The beads were then centrifuged at 3500 × g for 5 min at 4°C and washed seven times with the lysis buffer containing BSA, and the supernatant was discarded after the final wash. Immunoprecipitated proteins were released from the beads by boiling with an equal bead volume of 0.05% SDS and 2-mercaptoethanol-containing gel-loading buffer for 5 min and subsequently size fractionated on 10% SDS-PAGE gels, followed by transfer onto nitrocellulose membranes (Bio-Rad). Membranes were probed with rabbit polyclonal anti-Gαq (1:200; Santa Cruz Biotechnology) or anti-Gαi antibodies (1:200; Cell Signaling Technology) or mouse monoclonal anti-Gαs antibody (1:200; clone N192/12; NeuroMab) and subsequently with goat anti-mouse or anti-rabbit IgG–HRP secondary antibodies (1:10,000; Antibodies Inc.). Immunoreactive proteins on membranes were developed with enhanced electrochemiluminescence-plus reagent (ECL-Plus; PerkinElmer Life and Analytical Sciences), and the signals were captured on x-ray film (Kodak-Biomax; Carestream Health).
For ERK phosphorylation assays, cultured mouse DRG neurons (2 DIV) were treated with either vehicle or 100 nm CNP, and cell lysates were prepared 30 min after treatment using the same lysis buffer and methods as mentioned above. Lysates were then run on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were probed with either rabbit polyclonal anti-ERK1/2 (1:1000; Cell Signaling Technology) or mouse monoclonal anti-phospho-ERK1/2 (1:500; BD Bioscience) antibody, along with the mouse monoclonal anti-GRP75 antibody (1:1000; clone N52A/42; NeuroMab), and subsequently with goat anti-mouse or anti-rabbit IgG–HRP secondary antibodies (1:10,000; Antibodies Inc.). Immunoreactive proteins on membranes were developed with enhanced ECL-Plus reagent (PerkinElmer Life and Analytical Sciences), and the signals were captured on x-ray film (Kodak-Biomax). All the immunoprecipitation and immunoblotting experiments were repeated three times on three different batches of mouse DRG neuron cultures.
Chemicals and reagents.
Purified recombinant human/rodent ANP, BNP, and CNP, PTX, PGE2, BK, collagenase, and Pronase were purchased from EMD Chemicals and Phoenix Pharmaceuticals; the fura-2 AM was from Invitrogen; 8-Br-cGMP and 8-pCPT-cGMP were from Enzo Life Sciences; capsaicin and the TRPV1 antagonist AMG9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide] were from Sigma; the Gβγ inhibitor gallein was from Acros Organics; PLCβ inhibitor U73122 (1-[6[[(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione), PKC inhibitor bisindoylmelimide-I (BIM), protein kinase A (PKA) inhibitor KT5720 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1 H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid hexyl ester], PKG inhibitor KT5823 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester], and PKC activator phorbol 12-myristate 13-acetate (PMA) were from Tocris Bioscience. All other chemicals used in this study were purchased from Sigma, Bio-Rad, Roche Applied Science, and Thermo Fisher Scientific. All NeuroMab antibodies were purchased from the University of California, Davis/National Institutes of Health NeuroMab Facility through Antibodies Inc.
Behavioral assessment of inflammatory thermal hyperalgesia.
Adult male C57BL6/J mice of both TRPV1+/+ and TRPV1−/− genotypes were housed as five mice per cage and maintained on a 12 h light/dark cycle (dark cycle beginning at 6:00 P.M.) with access to food and water ad libitum. Animals were acclimated to the testing environment for 2 days before testing for 30 min each and on the day of behavioral testing for 30 min by placing them in individual Plexiglas chambers situated on a glass surface maintained at thermo-neutral temperature (IITC Life Science). Nociceptive thermal sensitivity on each hindpaw was measured by focusing a high-intensity beam of light on the plantar surface, and the time required for the mouse to withdraw its hindpaw from the thermal stimulus was recorded using a programmable digital counter (IITC Life Science) and was termed the paw-withdrawal latency (PWL). The beam intensity was adjusted to elicit baseline PWL of 10–12 s. The cutoff time was 20 s, at which time the test was terminated to prevent tissue injury and the mouse paw was assigned this latency. Average PWL from two readings for each hindpaw were taken for analysis.
After baseline PWL measurements, animals were microinjected with either saline or different concentrations of CNP (10 μl of 10 nm, 100 nm, 500 nm, and 10 μm stock) into the plantar surface of the right hindpaw (ipsilateral) via a 33 gauge stainless steel injector needle coupled to a Hamilton syringe, and subsequent PWLs were measured at 30 min, 2 h, 4 h, 6 h, and 24 h after injection. In a separate groups of animals, gallein [100 mg/kg, i.p.; (Lehmann et al., 2008)] or AMG9810 [30 mg/kg, i.p. (Gavva et al., 2005)] were administered 30 min before saline/CNP injections. At least three replicates of each treatment group were performed. The individual performing the behavioral testing was blinded to the site and details of drug injections, as well as to mouse genotype.
Statistical analyses.
Data are presented as mean ± SEM. For Ca2+ imaging, Kruskal–Wallis test with Dunn's correction was used to test the effects of different NPs, PGE2, and BK on potentiation of capsaicin-induced Ca2+ flux. For cGMP assays, Student's t test was used to test the effects of different NPs on cGMP production. For electrophysiological experiments, one-way ANOVA with post hoc Dunnett's correction was performed to test the statistical significance of the effects of different drug treatments on NP-induced potentiation compared with control (vehicle). Statistical analysis of data for PKCε translocation assays, as well as for the behavioral assessments of thermal hyperalgesia, were performed by one-way ANOVA with post hoc Bonferroni's correction to compare the differences between various treatments. p < 0.05 in each set of data comparisons were considered statistically significant. All statistical analyses were conducted using SPSS 19 software (IBM).
Results
NPs potentiate capsaicin responses in mouse DRG neurons
We first examined the effects of NPs on the activity of TRPV1 in small/medium-diameter cultured mouse DRG neurons by monitoring TRPV1-mediated [Ca2+]i responses, as described previously (Schnizler et al., 2008). Two consecutive applications of capsaicin (50 nm for 15 s, with 5 min washing in between) resulted in desensitization/tachyphylaxis of [Ca2+]i for the second application, with a ratio of 0.35 ± 0.1 for second/first capsaicin-induced [Ca2+]i (n = 14; Fig. 1A,B). However, extracellular application of ANP or CNP (50 and 500 nm each for 5 min) to DRG neurons resulted in significant potentiation of [Ca2+]i responses induced by the second capsaicin application, with peak Ca2+ ratios of 4.67 ± 1.17 (n = 17) and 2.22 ± 0.49 (n = 18) for 50 and 500 nm ANP, respectively, and 3.54 ± 0.8 (n = 15) and 17.2 ± 5.66 (n = 9) for 50 and 500 nm CNP, respectively (Fig. 1A,B). Conversely, BNP application (50 and 500 nm for 5 min) did not show any significant potentiation of [Ca2+]i responses to the second capsaicin application [ratios 0.79 ± 0.28 (n = 9) and 0.53 ± 0.1 (n = 12), respectively; Figure 1A,B]. We also compared the potentiating effects of NPs with two major inflammatory mediators, PGE2 and BK. Extracellular application of PGE2 (5 μm) and BK (50 and 500 nm) resulted in significant potentiation of [Ca2+]i responses induced by the second capsaicin application, with peak Ca2+ ratios of 4.23 ± 1.48 (n = 12), 9.52 ± 6.16 (n = 10), and 11.33 ± 6.26 (n = 9) for 5 μm PGE2, 50 nm BK, and 500 nm BK, respectively (Fig. 1B). However, application of 500 nm PGE2 did not lead to any significant potentiation of [Ca2+]i responses to the second capsaicin application [ratio 2.12 ± 0.73 (n = 16), respectively; Fig. 1B].
Figure 1.

Natriuretic peptides ANP and CNP, but not BNP, sensitize TRPV1 channel activity in mouse DRG neurons. A, Representative traces of Ca2+ flux in small/medium-diameter cultured mouse DRG neurons in response to two successive applications of capsaicin (50 nm, 15 s, arrowheads), with the continuous extracellular perfusion of ANP, BNP, and CNP (50 nm each) or vehicle for 5 min between two capsaicin applications. B, Quantification of Ca2+ flux measurements from Ca2+ imaging experiments as shown in A, with extracellular perfusion of 50 and 500 nm ANP, BNP, CNP, or BK, and 500 nm or 5 μm PGE2. Data are presented as mean ± SEM of the ratio of second versus first Ca2+ peak; n values are shown in parentheses for each treatment group. *p < 0.05, **p < 0.01, and ***p < 0.001, significantly different (NS, not significant) compared with vehicle (Kruskal–Wallis test with Dunn's correction). C, Representative traces of whole-cell TRPV1 currents in small/medium-diameter cultured mouse DRG neurons in response to four successive applications of capsaicin (50 nm for 5 s; with 1 min interval), with continuous extracellular perfusion of ANP, BNP, or CNP (10 nm) or vehicle (extracellular buffer) after the second capsaicin application. D, Quantification of capsaicin-activated TRPV1 currents (ICap) for experiments as shown in C. Data are presented as mean ± SEM of fold increase in peak ICap normalized to second ICap; n values are given for each treatment group. *p < 0.05, significantly different compared with the respective ICap for vehicle group (one-way ANOVA with post hoc Dunnett's correction).
Next, we performed whole-cell patch-clamp analysis to verify NP-mediated sensitization of TRPV1 channel activity suggested by our Ca2+ imaging studies. Application of ANP and CNP, but not BNP (10 nm each), led to significant potentiation of capsaicin-activated (50 nm, ∼5 s) inward currents (ICap) in small/medium-diameter cultured mouse DRG neurons. There were 4.31 ± 1.44-fold (n = 9) and 4.87 ± 0.85-fold (n = 16) increases in the third ICap for ANP and CNP treatment groups, respectively, compared with a 1.6 ± 0.53-fold (n = 10) increase in the vehicle treatment group, and a 5.89 ± 1.72- and 4.72 ± 0.67-fold increase in the fourth ICap for ANP and CNP treatment groups, respectively, compared with a 1.14 ± 0.25-fold increase in the vehicle treatment group (Fig. 1C,D). We further determined the NP dose–response relationships for the potentiation of TRPV1 channel activity. Increasing concentrations of ANP and CNP (1, 3, 10, 30, and 100 nm) resulted in enhanced potentiation of ICap, with saturation being achieved at 30 nm, whereas BNP did not induce any significant potentiation for all concentrations (data not shown). Together, these data indicate that ANP and CNP, but not BNP, significantly potentiate TRPV1 channel activity in a dose-dependent manner.
Noncanonical NPR-C/GPCR signaling underlies the NP-induced potentiation of TRPV1 channel activity
Previous reports have shown the expression of NPR-A/B in rodent DRG neurons (Zhang et al., 2010). We also performed RT-PCR with specific primer sets for NPR-A/B/C and show that all three NPR subtypes are expressed in adult mouse DRG neurons (Fig. 2A). Activation of NPR-A by A/BNP and of NPR-B by CNP has been shown to increase intracellular cGMP production and subsequent activation of PKG (Pandey, 2005b; Potter et al., 2006). Indeed, application of ANP, BNP, or CNP (50 nm, 15 min) to mouse DRG neurons led to increased intracellular cGMP production (0.68 ± 0.5, 1.89 ± 0.26, 1.96 ± 0.29, and 2.89 ± 0.71 pmol cGMP/μg protein for untreated, ANP-, BNP-, and CNP-treated neurons, respectively), as determined by ELISA (Fig. 2B). We next determined whether elevated levels of intracellular cGMP in response to NP treatment underlie TRPV1 channel sensitization. Direct perfusion of two membrane-permeable cGMP analogs, 8-Br-cGMP (100 μm) and 8-pCPT-cGMP (200 μm), did not significantly potentiate ICap in DRG neurons [0.82 ± 0.11- and 0.78 ± 0.15-fold increase in third and fourth ICap, respectively, for 8-Br-cGMP treatment group (n = 6), and 1.26 ± 0.05- and 1.25 ± 0.1-fold increase in third and fourth ICap, respectively, for 8-pCPT-cGMP treatment group (n = 4); Fig. 2C]. Furthermore, pretreatment of neurons with a PKG inhibitor (KT5823, 500 nm) did not inhibit CNP-induced potentiation of peak ICap [3.83 ± 0.44- and 4.69 ± 1.21-fold increase in third and fourth ICap, respectively (n = 7); Fig. 2C]. These data suggest that the canonical cGMP–PKG signaling pathway is not involved in CNP-induced sensitization of TRPV1; therefore, we next tested whether other protein kinases are involved in CNP-induced potentiation of ICap. TRPV1 channel protein is a target for direct phosphorylation by PKA and PKC, which results in potentiation of channel activation by different agonists (Premkumar and Ahern, 2000; Bhave et al., 2002, 2003; Numazaki et al., 2002; Mohapatra and Nau, 2003, 2005). Thus, we next tested whether NP modulation of TRPV1 channel activity is mediated by PKA or PKC. Pretreatment of neurons with a PKC inhibitor (BIM, 1 μm), but not with a PKA inhibitor (KT5720, 400 nm), attenuated the CNP-induced potentiation of peak ICap [0.89 ± 0.15- and 0.56 ± 0.19-fold increase in third and fourth ICap, respectively, for CNP + BIM treatment group (n = 4), and 3.87 ± 1.33- and 6.23 ± 2.4-fold increase in third and fourth ICap, respectively, for CNP + KT5720 treatment group (n = 8); Fig. 2C]. These results indicate that CNP potentiates TRPV1 activity via a PKC-dependent pathway, leading us to further explore the CNP signaling through NPR-C that is coupled to the PKC activation in DRG neurons.
Figure 2.

Expression of NP receptors and role of downstream cGMP signaling in mouse DRG neurons underlying CNP modulation of TRPV1 channel activity. A, All three NP receptors (NPR-A, NPR-B, and NPR-C) are expressed in DRG neurons. Representative RT-PCR analysis of NPR expression in cultured mouse DRG neurons with primers specific for NPR-A, NPR-B, and NPR-C, as well as for GAPDH as the housekeeping gene. B, ELISA-based quantification of intracellular cGMP levels in isolated mouse DRG neurons after ANP, BNP, or CNP treatment (100 nm for 30 min). Data are presented as mean ± SEM (n = 3). *p < 0.05, significantly different compared with the vehicle group (Student's t test). C, CNP-induced sensitization of TRPV1 is dependent on PKC, but not cGMP–PKG or PKA, signaling. Top panel shows representative traces of TRPV1 currents in small/medium-diameter cultured mouse DRG neurons in response to four successive applications of capsaicin (50 nm for 5 s; with 1 min interval), with continuous extracellular perfusion of 8-Br-cGMP (100 μm) after the second Icap. Bottom panel shows the quantification of ICap with indicated drug treatments [100 μm 8-Br-cGMP, 200 μm 8-pCPT-cGMP, 10 nm CNP, 10 nm CNP + 500 nm KT5823 (PKG inhibitor), 10 nm CNP + 400 nm KT5720 (PKA inhibitor), and 10 nm CNP + 1 μm BIM (PKC inhibitor)]. Peak ICap amplitudes are normalized to second ICap application of the respective vehicle or treatment groups, and the data are presented as mean ± SEM of fold-increase in ICap compared with second ICap (respective n values are shown in the figure). *p < 0.05, significantly different compared with the vehicle group (one-way ANOVA with post hoc Dunnett's correction).
Although NPR-C has been reported to serve as the clearance receptor for NPs, several reports have suggested that NPR-C can functionally couple to the Gαi subunit, which in turn produces Gβγ-mediated activation of PLCβ, PIP2 hydrolysis, and PKC activation (Levin, 1993; Murthy and Makhlouf, 1999; Murthy et al., 2000; Anand-Srivastava, 2005). Using immunohistochemical staining of adult mouse lumbar DRGs, we found that NPR-C is expressed in most neurons, including those that express the A-fiber marker NF200 and those that express the peptidergic C-fiber marker CGRP (Fig. 3A). Almost all the TRPV1-positive neurons were also positive for NPR-C (Fig. 3A). Next, we determined the specific type of Gα subunits that couple to NPR-C in mouse DRG neurons. Immunoprecipitation of NPR-C, but not NPR-B, specifically coprecipitated the Gαi but not Gαq or Gαs subunits in mouse DRG neurons (Fig. 3B). Activation of a number of Gαi-coupled GPCRs, including NPR-C, has been shown to activate PKC via the dissociation of Gβγ subunits from Gαi during receptor activation and subsequent activation of PLCβ (Murthy and Makhlouf, 1999; Murthy et al., 2000; Shi et al., 2003; Anand-Srivastava, 2005; Chen et al., 2005; Pandey, 2005b; Sabbatini et al., 2007). Activation of Gαi/o-coupled GPCRs also leads to the activation of MAPK/ERK and PLCβ–DAG–PKC signaling cascades. Therefore, we next determined whether CNP was capable of activating these signaling cascades in DRG neurons. CNP treatment of cultured mouse DRG neurons (100 nm, 30 min) increased phosphorylation of ERK1/2, without any change in total ERK1/2 levels (Fig. 3C). Because CNP-induced potentiation of TRPV1 activity was attenuated by inhibition of PKC (Fig. 2C), we next determined whether CNP treatment could activate PKC by monitoring its translocation to the plasma membrane of DRG neurons as a readout of PKC activation (Cesare et al., 1999; Hucho et al., 2005). We focused on PKCε, because this isoform is known to phosphorylate and sensitize TRPV1 upon activation by capsaicin and heat (Cesare et al., 1999; Premkumar and Ahern, 2000; Vellani et al., 2001; Numazaki et al., 2002; Bhave et al., 2003). Under control conditions, PKCε was evenly distributed throughout the cytoplasm of mouse DRG neurons (Fig. 3D). CNP treatment (100 nm, 5 min) led to translocation of the bulk of PKCε immunoreactivity to the cell plasma membrane, as was similarly observed during treatment with the potent PKC activator PMA (200 nm, 2 min; Fig. 4A,C,D). Pretreatment of neurons with PTX (100 ng/ml for ∼18 h), a potent inhibitor of Gαi–Gβγ subunit dissociation during receptor activation, led to the attenuation of PKCε translocation to the cell plasma membrane in response to CNP but not to PMA treatment (Fig. 4B--D). These results suggest that NPR-C, but not NPR-B, couples to Gαi subunit in mouse DRG neurons to activate MAPK and PKC via Gαi–Gβγ-mediated downstream signaling. Because it has been suggested that the affinities of CNP and ANP for Gαi-coupled NPR-C are higher than that of BNP (He et al., 2001; Anand-Srivastava, 2005), it is possible that distinct NP-mediated signaling pathways prevail in different tissues/cells based on the specific expression of individual NP receptor subtypes.
Figure 3.
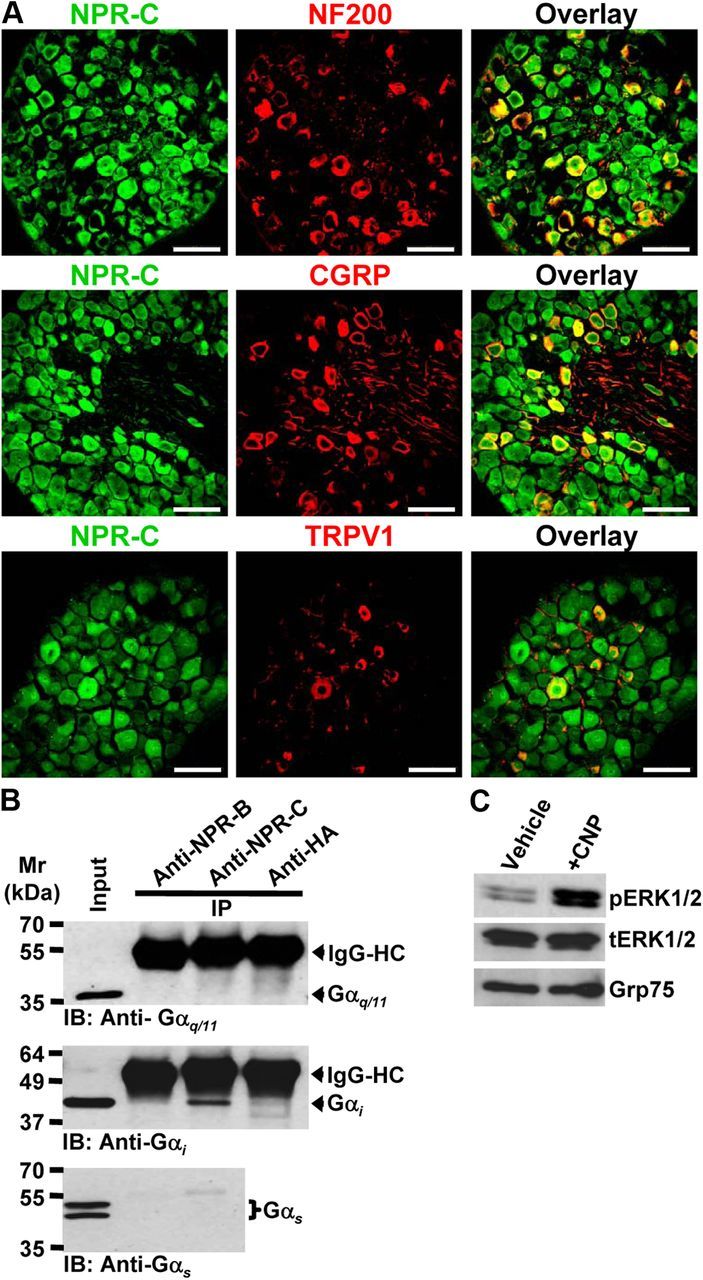
Expression of NPR-C and characterization of downstream intracellular signaling originating from CNP treatment in mouse DRG neurons. A, Representative photomicrographs depicting immunohistochemical staining of mouse L4–L6 DRG sections, indicating NPR-C expression in NF200-positive large-diameter neurons, as well as in CGRP- and TRPV1-positive small/medium-diameter neurons. Scale bar, 50 μm. B, Immunoprecipitation of Gαi subunits, but not Gαq or Gαs subunits, with anti-NPR-C antibody, but not with anti-NPR-B antibody, in cultured mouse DRG neurons. Anti-HA antibody was used as a negative control in immunoprecipitation reactions. The immunoprecipitates, along with the input DRG lysates, were size fractionated in SDS-PAGE and immunoblotted with specific antibodies against Gαi, Gαq, and Gαs subunits. C, Western/immunoblot analysis of ERK1/2 phosphorylation induced by CNP treatment of cultured mouse DRG neurons (100 nm for 30 min). Blots were probed with phospho-ERK1/2 and total ERK1/2, as well as with GRP75 as loading control. IB, Immunoblot; IP, immunoprecipitation.
Figure 4.

CNP treatment leads to translocation of PKCε in mouse DRG neurons. Immunocytochemical analysis of PKCε translocation to the cell periphery in cultured mouse DRG neurons upon treatment with CNP (100 nm for 5 min) and PMA (200 nm for 2 min; as a positive control) compared with vehicle (DMSO) treatment. Representative photomicrographs of neurons immunostained with anti-NPR-C (red) and anti-PKCε (green) antibodies under control (A) and PTX (100 μm, 24 h) (B) treatment conditions. Right panels for each image sets are magnified views of cells marked with respective rectangular white boxes, and the line graphs show the PKCε distribution profile (NIH Image J) within respective cells across the drawn white lines. The high-intensity peak signals near both ends of the path described by the line denote increased PKCε translocation to the cell plasma membrane. C, Quantification of PKCε translocation to the cell periphery under different treatment conditions (for details of analysis, see Materials and Methods). Data are presented at mean ± SEM of the peak intensity ratios (peripheral/cytoplasmic; n = 25 cells for each treatment condition, from four independent cultures). D, Quantification of number of DRG neurons with PKCε translocation to the cell periphery under different treatment conditions. Data are presented at mean ± SEM of the percentage of cells with increased PKCε staining intensity at the cell periphery (n > 500 neurons for each treatment group, from 4 independent cultures). For C, D, *p < 0.05 and ***p < 0.001, significantly different compared with their respective vehicle groups; #p < 0.05 and ###p < 0.001, significantly different compared with CNP treatment in control group (one-way ANOVA with post hoc Bonferroni's correction).
CNP potentiates acid-induced TRPV1 channel activation
Protons (H+) are well-known endogenous activators of TRPV1, and the H+ concentration is substantially increased at sites of tissue injury or inflammation (Dray and Read, 2007; Hucho and Levine, 2007; Basbaum et al., 2009; Gold and Gebhart, 2010; Kuner, 2010). Therefore, we examined whether CNP could potentiate proton-induced inward currents (IpH) in small/medium-diameter mouse DRG neurons. In these experiments, we used a protocol similar to that shown in Figure 1C, except the cells were stimulated with acidic pH extracellular buffer instead of capsaicin. Application of extracellular buffer with pH 6.4 resulted in two types of inward currents in small/medium-diameter cultured mouse DRG neurons. The first type was characterized by a rapidly activating inward current component followed by a sustained inward current (Fig. 5A, left). These neurons also exhibited capsaicin-induced inward currents that were elicited with the application of capsaicin (100 nm, 5 s) after the fourth IpH6.4. The other type displayed rapidly activating and rapidly inactivating inward currents (Fig. 5A, right), and these neurons did not exhibit any response to capsaicin (100 nm, 5 s). CNP application (10 nm) strongly potentiated the peak IpH6.4 [6.58 ± 1.07- and 9.87 ± 2.43-fold increase in third and fourth IpH6.4, respectively (n = 31)] in capsaicin-sensitive neurons but not in capsaicin-insensitive neurons [1.33 ± 0.05- and 1.08 ± 0.14-fold increase in third and fourth IpH6.4, respectively (n = 6); Fig. 5A,C]. These data indicate that CNP potentiates IpH6.4 only in DRG neurons that express TRPV1. To further confirm our findings, we evaluated CNP potentiation of IpH6.4 in cultured small/medium-diameter DRG neurons from TRPV1−/− mice and found that CNP did not potentiate peak IpH6.4 in these neurons [1.43 ± 0.28- and 1.65 ± 0.37-fold increase in third and fourth IpH6.4, respectively (n = 9); Fig. 5B,C]. Similar to CNP, application of ANP also led to the potentiation of IpH6.4 in small/medium-diameter DRG neurons from TRPV1+/+ but not from TRPV1−/− mice (data not shown).
Figure 5.
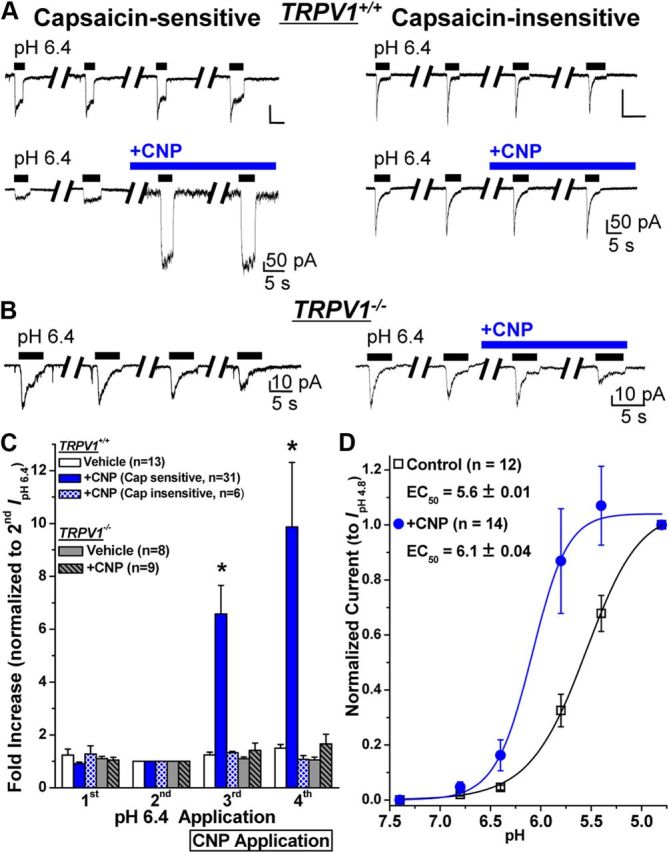
CNP specifically sensitizes TRPV1-mediated acidic pH-induced inward currents in mouse DRG neurons. A, Representative sets of traces of four successive proton (pH 6.4 extracellular buffer for 5 s; with 1 min interval) activated currents (IpH 6.4) in capsaicin-sensitive (left panel sets) and capsaicin-insensitive (right panel sets) small/medium-diameter cultured mouse DRG neurons, with or without continuous extracellular perfusion of CNP (10 nm) after the second IpH 6.4. B, Representative sets of traces of four successive IpH 6.4 recorded from small/medium-diameter cultured mouse DRG neurons obtained from TRPV1−/− mice, with or without continuous extracellular perfusion of CNP (10 nm) after the second IpH 6.4. C, Quantification of IpH 6.4 from experiments as shown in A and B. Data are presented as mean ± SEM of fold increase in peak IpH 6.4 normalized to the second IpH 6.4, and n values are shown in parentheses for each genotype/current/treatment group. *p < 0.05, significantly different compared with the respective IpH 6.4 for the vehicle group (one-way ANOVA with post hoc Dunnett's correction). D, CNP treatment (10 nm for 5 min) led to a leftward shift in the pH dose–response relationship of IpH in capsaicin-sensitive small/medium-diameter cultured mouse DRG neurons. Data are presented as mean ± SEM of inward currents in response to pH 7.4, 6.8, 6.4, 6.8, 5.4, and 4.8, normalized to the peak amplitude of IpH 4.8, and fitted with the Hill equation. The n values and EC50 pH values are mentioned in the panel for vehicle and CNP treatment groups.
We next determined the effect of CNP on the H+ dose–response relationship of TRPV1-mediated IpH activation in small/medium-diameter DRG neurons that are capsaicin sensitive. CNP application led to a significant leftward shift in the H+ dose–response relationship of TRPV1-mediated IpH [EC50 = pH 5.6 ± 0.02 (n = 12) and pH 6.1 ± 0.04 (n = 14) for vehicle and CNP-treated neurons, respectively; Fig. 5D]. These data suggest that, in the presence of CNP, the pH sensitivity of TRPV1 is significantly shifted, such that it has the potential to be activated by tissue acidosis associated with tissue injury and inflammation.
Given that CNP application to DRG neurons led to the translocation of PKCε to the cell periphery (Fig. 4) and that CNP-induced potentiation of ICap could be completely attenuated by the blockade of PKC activity (Fig. 2C), we next determined the precise intracellular signaling cascades underlying CNP potentiation of TRPV1 activity in response to acidic pH. Pretreatment of DRG neurons with inhibitors of Gβγ-mediated activation of PLCβ (gallein, 100 μm), PLCβ activity (U73122, 5 μm), or PKC activity (BIM, 1 μm) led to the attenuation of CNP-induced potentiation of peak IpH6.4 [1.76 ± 0.3- and 2.07 ± 0.4-fold increase in third and fourth IpH6.4, respectively, for gallein group (n = 11); 2.08 ± 0.43- and 4.11 ± 1.82-fold increase in third and fourth IpH6.4, respectively, for PLCβ group (n = 8); and 1.08 ± 0.08- and 1.35 ± 0.09-fold increase in third and fourth IpH6.4, respectively, for BIM group (n = 11); Fig. 6A]. However, as observed for peak ICap (Fig. 2C), pretreatment of neurons with the PKA inhibitor (KT5720, 400 nm) did not attenuate CNP-induced potentiation of peak IpH6.4 [5.89 ± 0.79- and 9.79 ± 1.87-fold increase in third and fourth IpH6.4, respectively; Fig. 6A]. Together, these data suggest that CNP potentiates TRPV1-mediated proton-activated inward currents in mouse DRG neurons via the Gβγ–PLCβ–PKC signaling cascade (Fig. 6B), although it is possible that other second-messenger signaling pathways might be involved in the CNP potentiation of TRPV1 currents.
Figure 6.
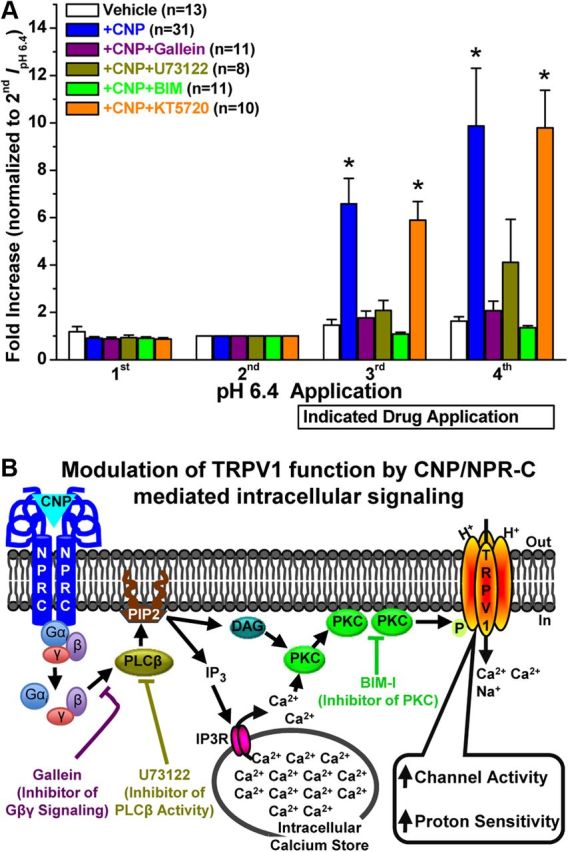
Gβγ/PLCβ/PKC-mediated intracellular signaling underlies CNP-induced sensitization of TRPV1 in mouse DRG neurons. A, Quantified data are shown from whole-cell voltage-clamp experiments with four successive IpH 6.4, with continuous extracellular application of CNP (10 nm), with or without pharmacological modulators after the second IpH 6.4 in capsaicin-sensitive small/medium-diameter cultured mouse DRG neurons, as in experiments shown in Figure 5, A and C. Data for vehicle and CNP treatment groups from Figure 4C are replotted here for comparison. All the data are presented as mean ± SEM of fold increase in peak IpH 6.4 normalized to the second IpH 6.4, and n values are given for each treatment group. *p < 0.05, significantly different compared with the respective IpH 6.4 for the vehicle group (one-way ANOVA with post hoc Dunnett's correction). The CNP-induced potentiation of IpH 6.4 was significantly reduced by pretreatment with the inhibitors of Gβγ-mediated activation of PLCβ (gallein, 100 μm), PLCβ activity (U73122, 5 μm), and PKC activity (BIM, 1 μm), but not by the inhibitor of PKA activity (KT5720, 400 nm). B, A schematic model of CNP-induced sensitization of TRPV1 channel activity via Gβγ–PLCβ–PKC signaling in mouse DRG neurons, as verified experimentally in A.
It has been shown that PKC-induced potentiation of TRPV1 channel activity is governed by direct phosphorylation of the channel protein at three putative Ser/Thr residues S502, T704, and S800 (Numazaki et al., 2002; Bhave et al., 2003). To verify whether these three PKC phosphorylation sites in TRPV1 are essential for the CNP potentiation of channel activity, we performed whole-cell voltage-clamp analysis of TRPV1 channel modulation in HEK293T cells heterologously expressing rTRPV1 and hNPR-C. CNP application (10 nm) led to the potentiation of peak ICap in HEK293T cells coexpressing rTRPV1 and NPR-C [3.08 ± 1.28- and 8.82 ± 2.8-fold increase in third and fourth ICap, respectively (n = 11); Fig. 7A,B]. CNP (10 nm) also potentiated peak ICap in HEK293T cells expressing only rTRPV1, without the cotransfection of the plasmid containing NPR-C cDNA; however, with a time delay [1.22 ± 0.27- and 12.5 ± 4.1-fold increase in third and fourth ICap, respectively (n = 9); Fig. 7A,B]. This led us to suspect that NPR-C is endogenously expressed in HEK293T cells. Indeed, RT-PCR analysis of total mRNA from HEK293T cells showed expression of all three NPRs, NPR-A/B/C, in this cell line (Fig. 6C). The delay in CNP-induced potentiation of TRPV1 currents in HEK293T cells compared with mouse DRG neurons could presumably be attributable to an inefficient Gαi coupling to NPR-C or availability of significantly lesser pool of Gαi-coupled NPR-C in these cells. As such, we used this cell line to affirm the involvement of PKC in the potentiation of TRPV1 channel activity by CNP, by transfecting single, double, and triple Ala mutant TRPV1 channels at three putative PKC phosphorylation sites, S502, T704, and S800, in HEK293T cells. We found that CNP potentiation of ICap was reduced in S502A and S800A, but not T704A, single mutants and almost abolished in cells expressing TRPV1–S502A/S800A double and TRPV1–S502A/T704A/S800A triple mutants compared with cells expressing rTRPV1–WT [22.4 ± 4.6-, 9.5 ± 5.6-, 23.1 ± 12.3-, 8.6 ± 1.9-, 3.4 ± 0.7-, and 3.8 ± 1.2-fold increases in fourth ICap (normalized to second ICap) for, respectively, WT (n = 12), S502A (n = 10), T704A (n = 6), S800A (n = 12), S502A/S800A (n = 14), and S502A/T704A/S800A (n = 13) channels; Fig. 7D]. Collectively, these results suggest that CNP-induced strong potentiation of TRPV1 channel activity is mediated via PKC activation and subsequent modification of TRPV1 protein primarily at two amino acid residues: S502 and S800.
Figure 7.

CNP/NPR-C/PKC-induced sensitization of TRPV1 activity is dependent on two PKC phosphorylation sites in the channel protein. A, Representative traces of TRPV1 currents in HEK293T cells either coexpressing rTRPV1 and hNPR-C (top) or expressing rTRPV1 alone (bottom) in response to four successive applications of capsaicin (20 nm for 5 s; with 2 min interval), with the continuous extracellular perfusion of CNP (10 nm) after the second ICap. B, Quantification of ICap for experiments as shown in A. Data are presented as mean ± SEM of fold increase in peak ICap normalized to second ICap; n values are given for each expression group. *p < 0.05, significantly different compared with respective second ICap currents (one-way ANOVA with post hoc Dunnett's correction). C, Representative RT-PCR analysis of endogenous NPR expression in HEK293T cells with primers specific for NPR-A, NPR-B, and NPR-C, as well as for GAPDH as a housekeeping control. Amplicons of RT-PCR reactions from isolated mouse DRG neuron RNAs are loaded for comparison. D, Quantification of fold increase in ICap for experiments as shown in A, from HEK293T cells expressing rTRPV1 wild-type channel (TRPV1–WT), or single, double, and triple PKC phosphorylation site mutant rTRPV1 channels. Data are presented as mean ± SEM of fold increase in peak fourth ICap normalized to second ICap; n values are shown in parentheses for each expression group. *p < 0.05, significantly different compared with TRPV1–WT (one-way ANOVA with post hoc Dunnett's correction).
CNP increases the frequency of AP firing in DRG neurons
Because CNP potentiates TRPV1 channel activity, we next evaluated whether sensitization of TRPV1 by CNP alters the excitability of DRG neurons, by whole-cell current-clamp recordings in cultured mouse DRG neurons. Application of capsaicin (50 nm, ∼5 s) induced AP firings; however, APs were mostly absent or at low frequency during the second capsaicin application (50 nm, ∼5 s) after a 1 min wash period (Fig. 8A), likely as a result of Ca2+-dependent desensitization of the TRPV1 channel (Koplas et al., 1997). In contrast, treatment with CNP (10 nm, 1 min) between the capsaicin applications led to a marked increase in the frequency of second capsaicin-induced AP firing (Fig. 8A). CNP application (10 nm, 1 min) also led to increased AP firing frequency during extracellular pH 6.8 application, specifically in capsaicin-sensitive, but not in capsaicin-insensitive, small/medium-diameter cultured mouse DRG neurons (Fig. 8B). Furthermore, CNP application did not lead to any increase in pH 6.8-induced AP firing frequency in small/medium-diameter DRG neurons from TRPV1−/− mice (Fig. 8C). Similar to CNP, application of ANP to small/medium-diameter DRG neurons also led to increased AP firing frequency during capsaicin and pH 6.4 applications (data not shown). These data further confirm our hypothesis that CNP-induced potentiation of TRPV1 activity leads to increased sensory neuron firing in response to mild extracellular acidic conditions.
Figure 8.

CNP sensitizes AP firing in response to TRPV1 activation in mouse DRG neurons. Representative traces of AP firings induced by two successive applications of 50 nm capsaicin (A) or pH 6.8 extracellular buffer (B, C), with or without the continuous extracellular application of CNP (10 nm for 1 min) between the agonist applications obtained from small/medium-diameter cultured DRG neurons from TRPV1+/+ (A, B) and TRPV1−/− (C) mice. Note that CNP-mediated sensitization of pH 6.8-induced AP firing was observed only in capsaicin-sensitive (left panels in B) but not in capsaicin-insensitive (right panels in B) small/medium-diameter cultured DRG neurons from TRPV1+/+ mice. No CNP-induced sensitization of pH 6.8-induced AP firing is observed in small/medium-diameter DRG neurons from TRPV1−/− mice (C).
CNP injection leads to the development of acute thermal hyperalgesia and is dependent on Gβγ–TRPV1 signaling
Given the ability of CNP to sensitize TRPV1 channel activity and strongly potentiate nociceptor firing, we next tested whether CNP administration could lead to the development of thermal hyperalgesia in mice. Intraplantar injection of different concentrations of CNP (10 μl of 500 nm and 10 μm in saline) to mice led to the development of thermal hyperalgesia ipsilateral to the injected hindpaw (quantified by a significant decrease in the PWL to a focused high-intensity beam of light on the plantar surface) compared with saline injection. CNP-induced thermal hyperalgesia was observed within 30 min of injection, which persisted for at least 6 h after the injection but subsequently recovered to baseline-level PWLs after 24 h (Fig. 9A). No significant decreases in the PWLs of contralateral paws were observed in response to CNP injection (Fig. 9A). Injection of 10 and 100 nm CNP displayed reduced PWLs, with significant decreases at the 4 h time point after 100 nm CNP injection, compared with the saline injection group (Fig. 9A). We then evaluated the role of the CNP–Gβγ–TRPV1 signaling axis in the development of acute thermal hyperalgesia. Intraperitoneal injection of gallein (100 mg/kg), a specific inhibitor of Gβγ-mediated activation of PLCβ, 30 min before intraplantar CNP injection led to significant attenuation of CNP-induced acute thermal hyperalgesia (Fig. 9B). The magnitude of gallein-mediated attenuation of thermal hyperalgesia at 6 h after injection was abolished compared with 30 min, 2 h, and 4 h time points, possibly attributable to the rapid urinary elimination of gallein. Furthermore, intraperitoneal injection of AMG9810 (30 mg/kg), a specific inhibitor of TRPV1, 30 min before intraplantar CNP injection led to the blockade of the development of thermal hyperalgesia (Fig. 9B). No significant decreases in the PWLs of contralateral paws were observed in response to gallein and CNP or AMG9810 and CNP injections (Fig. 9B), as well as in response to gallein or AMG9810 injection alone (Fig. 9B). Furthermore, to genetically verify the key role of TRPV1 in CNP-induced development of thermal hyperalgesia, we performed intraplantar injection of CNP into TRPV1−/− mice and observed no significant decrease in the PWLs in both ipsilateral and contralateral hindpaws compared with saline-injected controls (Fig. 9C). Collectively, these results suggest that peripheral administration of CNP recruits a noncanonical Gβγ–PLCβ–PKC signaling pathway to sensitize TRPV1 channel activity and nociceptor firing to induce the development of thermal hyperalgesia.
Figure 9.
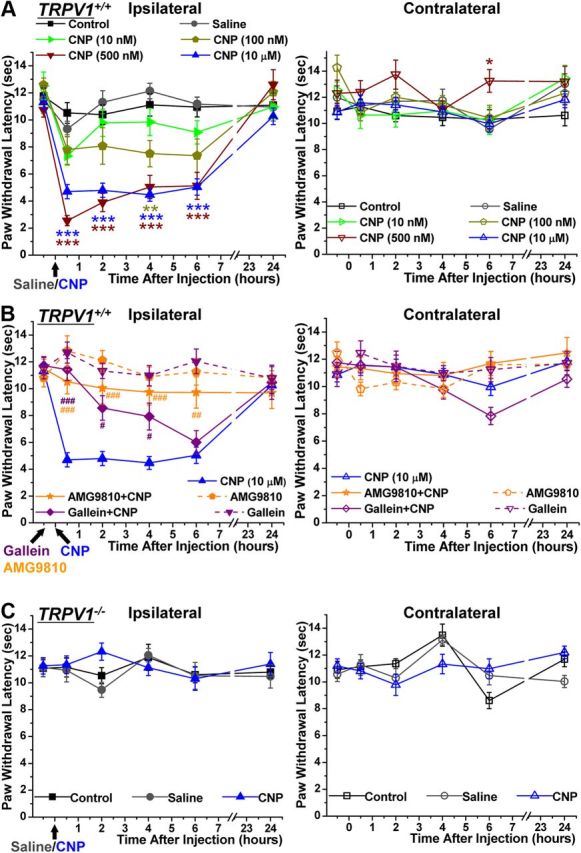
CNP injection in mouse hindpaw leads to the development of thermal hyperalgesia in a Gβγ/TRPV1-dependent manner. A, Mice (TRPV1+/+) were injected with either saline or different concentrations of CNP (10 nm, 100 nm, 500 nm, and 10 μm) in their right hindpaw after baseline assessment of PWLs to a focused high-intensity beam of light on the plantar surface in a Hargreaves plantar analgesia meter, followed by the determination of PWLs after 30 min, 2 h, 4 h, 6 h, and 24 h of CNP injection. Data are presented as mean ± SEM PWLs of ipsilateral (left) and contralateral (right) hindpaws of uninjected (control), saline-injected, and CNP-injected mice (n = 6 for each group, with the exception of 10 μm CNP, for which n = 10). *p < 0.05, **p < 0.01, and ***p < 0.001, significantly different compared with PWLs of saline-injected mice at the respective time points (one-way ANOVA with post hoc Bonferroni's correction). B, The CNP-induced decrease in PWL was significantly attenuated by preinjection of the inhibitor of Gβγ-mediated activation of PLCβ, gallein (100 mg/kg, i.p.), and the specific inhibitor of TRPV1, AMG9810 (30 mg/kg i.p.), administered 30 min before CNP injections. Data are presented as mean ± SEM PWLs of ipsilateral and contralateral hindpaws of gallein + CNP-injected and AMG9810 + CNP-injected mice (n = 6 for each). PWL data of ipsilateral paws for CNP injection group from A are plotted alongside for comparison. #p < 0.05, ##p < 0.01, and ###p < 0.001, significantly different compared with CNP-injected PWLs at the respective time points (one-way ANOVA with post hoc Bonferroni's correction). C, CNP injection (10 μm) did not lead to any significant change in the PWLs of TRPV1−/− mice. Data are presented as mean ± SEM PWLs of ipsilateral and contralateral hindpaws of uninjected, saline-injected, and CNP-injected mice (n = 5 for each).
Discussion
Our study demonstrates that functional NPR-A/B/C are expressed in mouse DRG neurons, in which CNP and ANP, but not BNP, induce sensitization of TRPV1 channel activity that is independent of classical NPR-A/B–cGMP–PKG signaling. Rather, NP sensitization of TRPV1 is mediated through a noncanonical NPR-C–Gβγ–PLCβ–PKC signaling module, as evidenced by the attenuation of CNP-induced modulation of TRPV1 activity by specific pharmacological blockade of these signaling components. Such modulation of TRPV1 channel activity leads to nociceptor sensitization to mild and pathophysiologically relevant acidic conditions and is absent in TRPV1−/− DRG neurons. Furthermore, intraplantar CNP injection induces thermal hyperalgesia in WT mice, but not in TRPV1−/− mice, and is significantly attenuated by systemic administration of Gβγ or TRPV1 inhibitors. Together, these findings suggest that CNP recruits a novel noncanonical Gβγ–PLCβ–PKC–TRPV1 signaling pathway to induce nociceptor sensitization and thermal hyperalgesia, which likely takes place in a variety of tissue injury and inflammatory conditions.
The influence of NPs on inflammatory pain has been suggested recently, with contrasting observations on individual NPs. Intrathecal BNP injection inhibits inflammatory thermal hyperalgesia induced by the injection of complete Freund's adjuvant (CFA) into rat hindpaw, without any significant effect on mechanical hypersensitivity (Zhang et al., 2010). Their study also found that intrathecal BNP injection led to a reduction in formalin-induced nocifensive behaviors. Mechanistically, BNP attenuates glutamate-induced AP firing in small-diameter rat DRG neurons. Furthermore, these effects were attributed to NPR-A–cGMP–PKG signaling, leading to increased BKCa current density (Zhang et al., 2010). Interestingly, BNP alone did not have any influence on AP firing (Zhang et al., 2010). In contrast to these antinociceptive effects of BNP, direct and rapid development of mechanical allodynia was observed in rats during intrathecal injection of CNP and 8-pCPT-cGMP but not ANP (Schmidtko et al., 2008a; Heine et al., 2011). It was suggested that CNP-induced mechanical allodynia is mediated through cysteine-rich protein 2, a downstream effector of increased PKG activity (Schmidtko et al., 2008b). Interestingly, our study found that, in addition to NPR-A/B, mouse DRG neurons also express NPR-C, in which it couples to Gαi. Application of CNP led to PKC activation through Gαi–Gβγ–PLCβ-mediated intracellular signaling. Furthermore, intraplantar injection of CNP led to the development of thermal hyperalgesia that was dependent on Gβγ-mediated signaling, leading to enhanced TRPV1 activity. To this end, TRPV1−/− mice did not exhibit CNP-induced thermal hyperalgesia. Collectively, NPs exert distinct effects on inflammatory thermal and mechanical hypersensitivities. NPR-A/B–cGMP–PKG signaling in the DRG neuron central axons projecting to the spinal cord dorsal horn contributes to the development of mechanical allodynia (Schmidtko et al., 2008a,b; Heine et al., 2011), with the exception of intrathecal BNP, which exerts an analgesic effect on CFA-induced thermal hyperalgesia (Zhang et al., 2010). Our study suggests that CNP–Gβγ–PKC–TRPV1 signaling in the peripheral afferents contributes to the development of thermal hyperalgesia. Furthermore, widespread expression of NPR-B/C in DRG neurons of all sizes and CNP-induced increases in cGMP production in neurons collectively suggest a potential involvement of CNP in sensory afferent sprouting/bifurcation in the periphery and induction of mechanical allodynia/hyperalgesia, which warrants thorough experimental verification of this idea.
Our analysis of NP modulation of TRPV1 in mouse DRG neurons revealed that both ANP and CNP significantly potentiate channel activity and subsequently enhance AP firing frequency. In contrast, BNP did not have such an effect. This is consistent with a previous report in which no direct effects of BNP on AP firing were found in DRG neurons (Zhang et al., 2010). Furthermore, our study shows no involvement of cGMP/PKG activity in NP-induced potentiation of TRPV1 currents, which rules out any modulation of channel activity by NPR-A/B-mediated signaling in DRG neurons. NPR-C has been suggested as the NP clearance receptor (Pandey, 2005a; Potter et al., 2006), yet other reports suggest that NP binding to NPR-C leads to activation of Gαi–Gβγ–PLCβ-mediated intracellular signaling pathways in numerous cell types (Levin, 1993; Anand-Srivastava, 2005). In fact, our results from the coimmunoprecipitation, ERK1/2 phosphorylation, and PKC translocation assays strongly suggest the existence of a functional NPR-C–Gαi–Gβγ–PLCβ–PKC signaling cascade in mouse DRG neurons. It was shown previously that the affinities of CNP and ANP for NPR-C exceed that of BNP (He et al., 2001; Anand-Srivastava, 2005), which provides yet another explanation for the lack of BNP-induced potentiation of TRPV1 currents in our study. As such, our results suggest a novel Gαi–Gβγ–PLCβ–PKC signaling pathway for a GPCR-mediated modulation of TRPV1 and subsequent nociceptor sensitization, which otherwise have been shown to be mediated primarily through Gαq- or Gαs-coupled GPCRs (Basbaum et al., 2009; Patapoutian et al., 2009). High concentrations of morphine were shown to directly activate and potentiate TRPV1 currents in mouse DRG neurons (Forster et al., 2009). Considering the fact that many opioid receptors induce Gαi-coupled downstream signaling (Standifer and Pasternak, 1997; Stein, 2003), it is tempting to speculate that sensitization of TRPV1 channel activity via high doses of morphine (Forster et al., 2009) could be mediated through Gαi–Gβγ–PLCβ–PKC signaling, a hypothesis that needs to be verified experimentally.
Activity and expression of TRPV1 in sensory afferents has been shown to undergo diverse modulations by a large number of proinflammatory mediators, cytokines, chemokines, and bioactive peptides, mainly by influencing the phosphorylation state of the channel protein. The protein kinases PKC, PKA, p38MAPK, and Src are activated downstream as a result of activation of a variety of receptors for inflammatory mediators in sensory neurons/afferents, which subsequently lead to direct phosphorylation-dependent modulation of TRPV1 channel activity and nociceptor sensitization (Bhave et al., 2002, 2003; Ji et al., 2002; Mohapatra and Nau, 2005; Hucho and Levine, 2007; Patapoutian et al., 2009). Activation of PKC and subsequent phosphorylation-dependent modulation of TRPV1 channel activity has been shown to be a convergent mechanism underlying nociceptor sensitization by several mediators (Bhave and Gereau, 2004; Hucho and Levine, 2007; Basbaum et al., 2009; Patapoutian et al., 2009; Gold and Gebhart, 2010; Kuner, 2010). PKC directly phosphorylates TRPV1 at amino acid residues S502, T704, S774, and S800 (Numazaki et al., 2002; Bhave et al., 2003). Indeed, our results demonstrated that substitution mutation of two of these residues, S502A and S800A, abolished CNP-induced potentiation of TRPV1 channel activity. Phosphorylation of these residues in TRPV1 by PKC has been shown to potentiate the heat activation of the channel at temperatures below body temperature and under conditions of mild acidosis (Vellani et al., 2001; Numazaki et al., 2002; Bhave et al., 2003). This phenomenon is manifested in our in vivo observations showing that CNP induction of thermal hyperalgesia is dependent on Gβγ signaling and TRPV1, although the involvement of other sensory ion channels, such as TRPA1, TRPV4, and Nav channels, cannot be ruled out. Based on the role of NPR-B–cGMP–PKG signaling in neurite bifurcation (Schmidt et al., 2009), along with our observation on CNP-induced cGMP production in DRG neurons, it is plausible that CNP might also influence peripheral mechanical hypersensitivity. Our future studies will focus on a thorough investigation along these lines.
Pathological modification/activation of TRPV1 and subsequent nociceptor sensitization by mildly acidic conditions has been proposed as one of the principal mechanisms underlying the development of inflammatory thermal hyperalgesia (Hucho and Levine, 2007; Basbaum et al., 2009; Gold and Gebhart, 2010; Kuner, 2010). Our results clearly show that CNP treatment shifts the H+ dose–response relationship of TRPV1 activation, such that the channel is activated in mildly acidic pH ranges. To this end, our results provide evidence of TRPV1-dependent sensitization of nociceptor firing at pH 6.8 in response to CNP that is within a tissue acidosis range associated with injury and inflammatory conditions, including arthritic joints (Dray and Read, 2007; Hucho and Levine, 2007; Gold and Gebhart, 2010; Kuner, 2010). In view of pain associated with inflamed arthritic joints, TRPV1 modulation in sensory afferents is suggested to play a key role in both thermal and mechanical pain sensitization associated with rheumatoid arthritis and osteoarthritis (Dray and Read, 2007; Premkumar and Sikand, 2008; Gold and Gebhart, 2010; Kuner, 2010). Elevated levels (approximately fivefold) of CNP have been detected in the serum of rheumatoid arthritis patients (Olewicz-Gawlik et al., 2010a), and CNP has also been shown to be secreted at elevated levels by vascular endothelial cells during stimulation by inflammatory mediators such as IL-1α/β and TNF-α (Suga et al., 1993). Studies aimed at determining the tissue levels of CNP at the site of injury and inflammation are needed to precisely understand the role of CNP-induced nociceptor sensitization in a wide variety of inflammatory conditions. Our comparison of CNP with other major inflammatory mediators, such as PGE2 and BK, revealed that CNP is a potent modulator of TRPV1 and nociceptor sensitization at relatively low concentrations (nanomolar range), which is also supported by dose-dependent induction of cutaneous thermal hyperalgesia in mice. Therefore, it is plausible that CNP-induced sensitization of TRPV1 activity at mildly acidic pH and constitutive nociceptor sensitization constitutes one of the mechanisms underlying the induction of thermal hyperalgesia in a wide variety of acute and chronic inflammatory conditions. In conclusion, our study demonstrates a novel noncanonical NP signaling pathway that induces thermal hyperalgesia via phosphorylation-dependent modulation of TRPV1 channel activation by protons and nociceptor sensitization.
Footnotes
This work was supported by Future Leaders in Pain Grant FLP1485 from the American Pain Society (D.P.M.), National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) Grants NS069898 (D.P.M.), and NS054614 and NS072432 (Y.M.U.). A.D.M. was supported by a predoctoral fellowship through NIH/NINDS Institutional T32 Grant NS045549 for “Interdisciplinary Training Program in Pain Research.” We thank Drs. Donna Hammond, Songhai Chen, Christopher Benson, Timothy Brennan, and Stefan Strack for their constructive suggestions on this work and critical comments on this manuscript.
References
- Ahluwalia A, Hobbs AJ. Endothelium-derived C-type natriuretic peptide: more than just a hyperpolarizing factor. Trends Pharmacol Sci. 2005;26:162–167. doi: 10.1016/j.tips.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Anand-Srivastava MB. Natriuretic peptide receptor-C signaling and regulation. Peptides. 2005;26:1044–1059. doi: 10.1016/j.peptides.2004.09.023. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhave G, Gereau RW., 4th Posttranslational mechanisms of peripheral sensitization. J Neurobiol. 2004;61:88–106. doi: 10.1002/neu.20083. [DOI] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci U S A. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chen XK, Wang LC, Zhou Y, Cai Q, Prakriya M, Duan KL, Sheng ZH, Lingle C, Zhou Z. Activation of GPCRs modulates quantal size in chromaffin cells through G(betagamma) and PKC. Nat Neurosci. 2005;8:1160–1168. doi: 10.1038/nn1529. [DOI] [PubMed] [Google Scholar]
- Constantin CE, Mair N, Sailer CA, Andratsch M, Xu ZZ, Blumer MJ, Scherbakov N, Davis JB, Bluethmann H, Ji RR, Kress M. Endogenous tumor necrosis factor α (TNFα) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008;28:5072–5081. doi: 10.1523/JNEUROSCI.4476-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray A, Read SJ. Arthritis and pain. Future targets to control osteoarthritis pain. Arthritis Res Ther. 2007;9:212. doi: 10.1186/ar2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster AB, Reeh PW, Messlinger K, Fischer MJ. High concentrations of morphine sensitize and activate mouse dorsal root ganglia via TRPV1 and TRPA1 receptors. Mol Pain. 2009;5:17. doi: 10.1186/1744-8069-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XI, Chow DC, Martick MM, Garcia KC. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science. 2001;293:1657–1662. doi: 10.1126/science.1062246. [DOI] [PubMed] [Google Scholar]
- Heine S, Michalakis S, Kallenborn-Gerhardt W, Lu R, Lim HY, Weiland J, Del Turco D, Deller T, Tegeder I, Biel M, Geisslinger G, Schmidtko A. CNGA3: a target of spinal nitric oxide/cGMP signaling and modulator of inflammatory pain hypersensitivity. J Neurosci. 2011;31:11184–11192. doi: 10.1523/JNEUROSCI.6159-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4+ neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Kishimoto I, Tokudome T, Horio T, Soeki T, Chusho H, Nakao K, Kangawa K. C-type natriuretic peptide is a Schwann cell-derived factor for development and function of sensory neurones. J Neuroendocrinol. 2008;20:1213–1223. doi: 10.1111/j.1365-2826.2008.01778.x. [DOI] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ER. Natriuretic peptide C-receptor: more than a clearance receptor. Am J Physiol. 1993;264:E483–E489. doi: 10.1152/ajpendo.1993.264.4.E483. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Desensitization of capsaicin-activated currents in the vanilloid receptor TRPV1 is decreased by the cyclic AMP-dependent protein kinase pathway. J Biol Chem. 2003;278:50080–50090. doi: 10.1074/jbc.M306619200. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Nau C. Regulation of Ca2+-dependent desensitization in the vanilloid receptor TRPV1 by calcineurin and cAMP-dependent protein kinase. J Biol Chem. 2005;280:13424–13432. doi: 10.1074/jbc.M410917200. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Wang SY, Wang GK, Nau C. A tyrosine residue in TM6 of the Vanilloid Receptor TRPV1 involved in desensitization and calcium permeability of capsaicin-activated currents. Mol Cell Neurosci. 2003;23:314–324. doi: 10.1016/s1044-7431(03)00054-x. [DOI] [PubMed] [Google Scholar]
- Mohapatra DP, Siino DF, Trimmer JS. Interdomain cytoplasmic interactions govern the intracellular trafficking, gating, and modulation of the Kv2.1 channel. J Neurosci. 2008;28:4982–4994. doi: 10.1523/JNEUROSCI.0186-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra DP, Misonou H, Pan SJ, Held JE, Surmeier DJ, Trimmer JS. Regulation of intrinsic excitability in hippocampal neurons by activity-dependent modulation of the KV2.1 potassium channel. Channels. 2009;3:46–56. doi: 10.4161/chan.3.1.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro C, Berlan M. Cardiovascular and metabolic effects of natriuretic peptides. Fundam Clin Pharmacol. 2006;20:41–49. doi: 10.1111/j.1472-8206.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- Murthy KS, Makhlouf GM. Identification of the G protein-activating domain of the natriuretic peptide clearance receptor (NPR-C) J Biol Chem. 1999;274:17587–17592. doi: 10.1074/jbc.274.25.17587. [DOI] [PubMed] [Google Scholar]
- Murthy KS, Teng BQ, Zhou H, Jin JG, Grider JR, Makhlouf GM. G(i-1)/G(i-2)-dependent signaling by single-transmembrane natriuretic peptide clearance receptor. Am J Physiol Gastrointest Liver Physiol. 2000;278:G974–G980. doi: 10.1152/ajpgi.2000.278.6.G974. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Olewicz-Gawlik A, Trzybulska D, Grala P, Hrycaj P. Blood serum levels of amino-terminal pro-C-type natriuretic peptide in patients with rheumatoid arthritis. Adv Med Sci. 2010a;55:261–265. doi: 10.2478/v10039-010-0036-1. [DOI] [PubMed] [Google Scholar]
- Olewicz-Gawlik A, Danczak-Pazdrowska A, Klama K, Silny W, Prokop J, Mackiewicz S, Grala P, Hrycaj P. Blood serum levels of amino-terminal pro-C-type natriuretic peptide in patients with systemic sclerosis. Connect Tissue Res. 2010b;51:83–87. doi: 10.3109/03008200903056168. [DOI] [PubMed] [Google Scholar]
- Pandey KN. Natriuretic peptides and their receptors. Peptides. 2005a;26:899–900. doi: 10.1016/j.peptides.2005.03.055. [DOI] [PubMed] [Google Scholar]
- Pandey KN. Biology of natriuretic peptides and their receptors. Peptides. 2005b;26:901–932. doi: 10.1016/j.peptides.2004.09.024. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Tate S, Woolf CJ. Transient receptor potential channels: targeting pain at the source. Nat Rev Drug Discov. 2009;8:55–68. doi: 10.1038/nrd2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27:47–72. doi: 10.1210/er.2005-0014. [DOI] [PubMed] [Google Scholar]
- Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009:341–366. doi: 10.1007/978-3-540-68964-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Sikand P. TRPV1: a target for next generation analgesics. Curr Neuropharmacol. 2008;6:151–163. doi: 10.2174/157015908784533888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbatini ME, Rodríguez M, di Carlo MB, Davio CA, Vatta MS, Bianciotti LG. C-type natriuretic peptide enhances amylase release through NPR-C receptors in the exocrine pancreas. Am J Physiol Gastrointest Liver Physiol. 2007;293:G987–G994. doi: 10.1152/ajpgi.00268.2007. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Stonkute A, Jüttner R, Koesling D, Friebe A, Rathjen FG. C-type natriuretic peptide (CNP) is a bifurcation factor for sensory neurons. Proc Natl Acad Sci U S A. 2009;106:16847–16852. doi: 10.1073/pnas.0906571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, König P, Heine S, Motterlini R, Ruth P, Schlossmann J, Koesling D, Niederberger E, Tegeder I, Friebe A, Geisslinger G. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neurosci. 2008a;28:8568–8576. doi: 10.1523/JNEUROSCI.2128-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008b;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnizler K, Shutov LP, Van Kanegan MJ, Merrill MA, Nichols B, McKnight GS, Strack S, Hell JW, Usachev YM. Protein kinase A anchoring via AKAP150 is essential for TRPV1 modulation by forskolin and prostaglandin E2 in mouse sensory neurons. J Neurosci. 2008;28:4904–4917. doi: 10.1523/JNEUROSCI.0233-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Mohapatra DP. Tissue preparation and immunostaining of mouse sensory nerve fibers innervating skin and limb bones. J Vis Exp. 2012;59:e3485. doi: 10.3791/3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Szczesniak A, Mao L, Jollimore C, Coca-Prados M, Hung O, Kelly ME. A3 adenosine and CB1 receptors activate a PKC-sensitive Cl− current in human nonpigmented ciliary epithelial cells via a G beta gamma-coupled MAPK signaling pathway. Br J Pharmacol. 2003;139:475–486. doi: 10.1038/sj.bjp.0705266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standifer KM, Pasternak GW. G proteins and opioid receptor-mediated signalling. Cell Signal. 1997;9:237–248. doi: 10.1016/s0898-6568(96)00174-x. [DOI] [PubMed] [Google Scholar]
- Stein C. Opioid receptors on peripheral sensory neurons. Adv Exp Med Biol. 2003;521:69–76. [PubMed] [Google Scholar]
- Suga S, Itoh H, Komatsu Y, Ogawa Y, Hama N, Yoshimasa T, Nakao K. Cytokine-induced C-type natriuretic peptide (CNP) secretion from vascular endothelial cells—evidence for CNP as a novel autocrine/paracrine regulator from endothelial cells. Endocrinology. 1993;133:3038–3041. doi: 10.1210/endo.133.6.8243333. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Yamazaki T, Yazaki Y. The role of the natriuretic peptides in the cardiovascular system. Cardiovasc Res. 2001;51:489–494. doi: 10.1016/s0008-6363(01)00238-3. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FX, Liu XJ, Gong LQ, Yao JR, Li KC, Li ZY, Lin LB, Lu YJ, Xiao HS, Bao L, Zhang XH, Zhang X. Inhibition of inflammatory pain by activating B-type natriuretic peptide signal pathway in nociceptive sensory neurons. J Neurosci. 2010;30:10927–10938. doi: 10.1523/JNEUROSCI.0657-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]