

Abstract
Peptide amphiphiles (PAs) self-assemble nanostructures with potential applications in drug delivery and tissue engineering. Some PAs share environmentally responsive behavior with their peptide components. Here we report a new type of PAs biologically inspired from human tropoelastin. Above a lower critical solution temperature (LCST), elastin-like polypeptides (ELPs) undergo a reversible inverse phase transition. Similar to other PAs, elastin-like PAs (ELPAs) assemble micelles with fiber-like nanostructures. Similar to ELPs, ELPAs have inverse phase transition behavior. Here we demonstrate control over ELPAs fiber-length and cellular uptake. In addition, we observed that both peptide assembly and nanofiber phase separation are accompanied by a distinctive secondary structure attributed primarily to a type-1 β turn. We also demonstrate increased solubility of hydrophobic Paclitaxel (PAX) in the presence of ELPAs. Due to their biodegradability, biocompatibility, and environmental responsiveness, elastin-inspired biopolymers are an emerging platform for drug and cell delivery; furthermore, the discovery of ELPAs may provide a new and useful approach to engineer these materials into stimuli-responsive gels and drug carriers.
Keywords: Peptide amphiphiles, Elastin like polypeptides, Circular dichroism, Micelles, nanofibers, Lower critical solution temperature, Transmission electron microscopy, Atomic force microscopy
Introduction
Peptide amphiphiles (PAs) have unique properties that arise from the combination of their secondary structure and their ability to self-assemble via hydrophobic interactions1. Like other amphiphiles, PAs have distinct hydrophobic and hydrophilic regions that drive nanostructure formation, similar to those formed by detergents or phospholipids2-5. The nature of the hydrophobic region can be either peptide based or, more commonly, one or two saturated lipid chains. Some of the most interesting observations of peptide amphiphiles include their propensity to form cylindrical nanostructures6, 7. Due to their unique self-assembling properties, PAs are being explored as potential drug carriers and substrates for tissue engineering8-10. Because they are generated using peptides, PAs may be engineered to be biodegradable and bioresponsive.
A host of peptide sequences are known to be responsive to their environment. There are peptide sequences that are thermo-responsive, pH-responsive, reductive-responsive, ion-responsive, and even responsive to analytes11. To develop PAs that are responsive to their microenvironment, we have focused on building PAs using peptides that display an inverse phase transition temperature. For example, elastin-like polypeptides (ELPs) are environmentally responsive peptides derived from the human protein, tropoelastin12. Similar to some synthetic polymers, including pNIPAM, ELPs undergo an inverse phase transition that is analogous to the lower critical solution temperature (LCST) observed for synthetic peptides. ELPs consist of the motif (Val-Pro-Gly-Xaa-Gly)n, where the identity of Xaa and n can be adjusted to control the phase behavior of these peptides12. During the aggregation process, ELPs form additional secondary structures, which have been attributed to β-turn spirals12. The phase transition temperature of an ELP depends strongly on Xaa and n; furthermore, most ELPs with LCST behavior have been reported with n between 20 to 200 repeat units (Fig. 1a).
Figure 1. Design of ELPAs with LCST behavior.
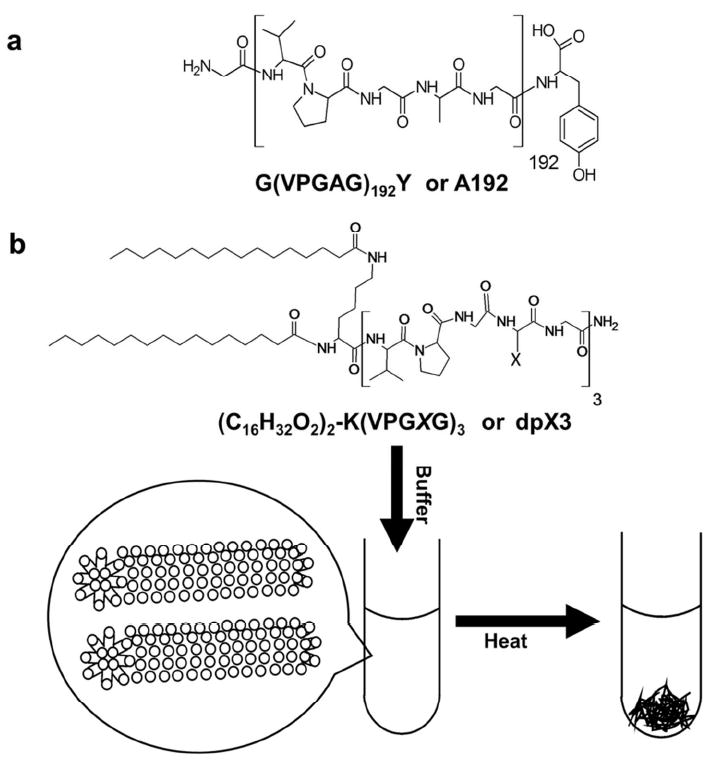
(a) A high molecular weight ELP, A192. (b) A low molecular weight ELPA construct modified with a dialmitoyl lysine, dpA3. The dipalmitoyl lipids drive assembly of cylindrical micelles with inverse phase transition behavior.
Here we describe a new type of elastin-like peptide-amphiphiles (ELPAs) composed from short ELPs (n=3), which are unable to phase separate. Our surprising finding is that the addition of two saturated 16-carbon chains enables these short oligopeptides, dpX3 (Fig. 1b) to phase separate at temperatures similar to their higher molecular weight counterparts, A192 (Fig 1a). Dipalmitoylation (vs. monopalmitoylation) was evaluated to promote co-formulation with traditional phospholipids and to provide a stronger driving force for hydrophobic assembly. To the best of our knowledge, this is the first report of ELPAs; furthermore, this class of peptides have the following unique properties that may promote their in vivo applications: i) they are charge neutral; ii) they display temperature dependent phase separation (Fig. 1b), and iii) their aspect ratio can be controlled by adjusting the ratio of a ‘capping’ lipid. In addition, we present evidence that suggests that both nanofiber assembly and phase separation are accompanied by significant alterations in peptide secondary structure. We also demonstrate formulation of paclitaxel using ELPAs.
Materials & Methods
Materials
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) was purchased from Avanti polar lipids (Birmingham, AL). Rink amide MBHA resin, Fluorenylmethyloxycarbonyl Valine (FMOC-Val), FMOC-Proline (FMOC-Pro), FMOC-Glycine (FMOC-Gly), FMOC-Alanine (FMOC-Ala), FMOC-Isoleucine (FMOC-Ile), O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU), N-Methyl-2-pyrrolidone (NMP), and Acetonitrile (ACN) were purchased from EMD chemicals (Gibbstown, NJ). Kaiser test kit (ninhydrin assay), DiFMOC-Lysine (DiFMOC-Lys), Diethyl ether (Ether), Triisopropyl silane (TIPS), N,N-Diisopropylethylamine (DIPEA), Chloroform (CHCl3), trifluoroacetic acid (TFA), Methanol (MeOH), Ethanol (EtOH), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and Palmitic acid purchased from Sigma Aldrich (St. Louis, MO). 4-Methyl piperidine (MeP) was purchased from fisher chemicals (Tustin, CA). The Waters C-18 and YMC C4 reverse phase semi prep columns were purchased from Waters Inc. (Milford, MA). 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) and 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindocarbocyanine Perchlorate (DiI) were purchased from Invitrogen (Grand Island, NY). Deionized water was obtained using a Barnstead purification system (Asheville, NC) and used for all aqueous buffers.
Synthesis and purification of ELP biomaterials
Short sequences with three pentameric ELP repeats were synthesized using standard FMOC chemistry with Ala (A3), Val (V3) and Ile (I3) as guest residues13. Briefly, 500 mg (0.6 mMole/gm substitution) of Rink amide MBHA resin was allowed to swell in NMP for 30mins. The resin was deprotected using 25% MeP. The amino acid coupling was performed for 45mins with 5 times excess amino acid and HBTU under basic conditions. The generation and consumption of free primary amine is monitored using the ninhydrin test. The deprotection and coupling sequence is continued until the peptide of desired length is obtained. The N-terminal is capped with DiFMOC-Lys (KA3, KV3 and KI3) and the protecting groups are removed. A sample was collected for qualitative analysis.
The deprotected N-termini were treated with molar excess of palmitic acid under basic conditions to give dipalmitoylated ELP sequences (Fig. 1b). The reaction was performed until all primary amines are exhausted. After completion the conjugates (dpA3, dpV3 and dpI3) were cleaved using a cleavage mixture made of 95% TFA in the presence of TIPS for 3 hrs at room temperature. The cleaved product is precipitated using cold ether. The crude product was dissolved in 100% HPLC grade MeOH.
The crude ELP sequences and lipid conjugates were qualitatively analyzed at 214nm on a Perkin Elmer 200 series high performance liquid chromatography (HPLC) system using a Waters C-18 and YMC C-4 reverse phase column respectively. The ELP sequences were run on a H2O:MeOH gradient with MeOH starting from 30% to 70%. The lipid conjugates were run on a H2O:MeOH gradient starting at 80% to 100% MeOH. The purified fractions were collected and mass confirmed using a DECA-LcQ ESI mass spec system (Thermo scientific, Waltham, MA). After confirmation the products were purified and lyophilized. Stocks of the lyophilized samples were made in 100% HPLC grade MeOH.
Determination of critical micellar concentration (CMC)
CMC was calculated by the pyrene assay14. Briefly, 0.6 μmoles of pyrene was added to round bottom test tubes and dried under a stream of air. Increasing concentrations of the ELPA were added to the dried pyrene and vigorously vortexed until pyrene dissolves. The fluorescence intensity of the samples were analyzed at λex at 334 nm (slit width= 8 nm) and λem recorded between 350 and 410 nm (slit width = 2 nm) using a Horiba Fluorolog® 3 spectrofluorometer (Edison, NJ). The ratio of 373/384 (L1/L3) was calculated and plotted against concentration. The concentration at which the slope change is maximum (break point) is the CMC. All ELPA samples were prepared in filtered PBS.
Preparation and particle size of ELPAs
All ELPAs were prepared using film hydration with an aqueous solution. Briefly, the required amounts of ELPA stocks were mixed in a clean glass test tube and evaporated on a Heidolph Laborota 4011 (Schwabach, Germany) to form an even film. For cell binding/uptake studies, 0.2% mol of DiI was added to the film. The film was placed under vacuum for 4 hrs to remove trace organic solvent. The dry film was then hydrated with appropriate buffer and placed in a water bath at 75 °C for 2 mins. The hydrated film was sonicated for 5-10 secs and vortexed gently for an additional 10-15 secs to ensure complete hydration of film. After complete hydration, 50 μl of the sample was analyzed on a Wyatt Dynapro plate reader (Santa Barbara, CA) using a 384 well Greiner bio-one clear bottom plate (Monroe, NC). Samples for light scattering measurements were hydrated in 10 mM HEPES buffered saline.
LCST determination of ELPAs
The temperature dependent phase transition of ELPAs was demonstrated using optical density measurements at 350 nm over a set temperature range. Increasing concentrations of constructs were added to 300 μl Beckman coulter Tm microcells (Brea, CA) and the temperature was ramped at a rate of 1°C/min. The optical density was plotted as a function of temperature, and the maximum first derivative of this curve was defined as the phase transition temperature. All ELPA samples were prepared in filtered PBS
Secondary structure determination using circular dichroism (CD)
CD was performed to determine the secondary structure adopted by the synthesized constructs at different temperatures. Increasing concentrations of the constructs were run on a Jasco J-815 CD spectrometer (Easton, MD) using a quartz cuvette (path length ~ 1 mm). The ellipticity was monitored from 180-300 nm and the spectra of buffer subtracted post run. All the constructs were prepared in filtered diH2O. All plots were exported using Jasco Spectral manager V2 (Easton, MD) to Excel and deconvoluted assuming that the observed molar ellipticity [θ] is a weighted linear sum of the ellipticity for known secondary structures (Eq. 1). The data was fit using nonlinear regression on Microsoft Excel.
| (Eq. 1) |
Transmission electron microscopy (TEM)
A small drop of freshly prepared ELPA was pipetted onto a Ted Pella carbon/formavar grid (Redding, CA). The excess liquid was wicked off using filter paper and a drop of 1 % uranyl acetate was pipetted on. The excess uranyl acetate was wicked off using filter paper and the grid was dried at 37 °C. The grid was carefully placed into a JEOL JEM 2100 laB6 microscope (Tokyo, Japan). All samples were run using an accelerating voltage of 200 kV and processed using ImageJ (NIH, USA). The end to end measurements of the ELPA nanofibers was first made by setting the scale using the scale bar on the raw image. Once the scale was set the length was measured using the ‘straight line’ function on ImageJ.
Atomic force microscopy (AFM)
AFM was used as an alternate approach to control for procedural artifacts on the TEM. Briefly, 2 μl of sample was pipetted onto a freshly stripped flat mica surface and dried under a steady stream of air. The mica plate was then analyzed using a tapping mode Veeco Nanoscope IIIa Atomic Force Microscope (Santa Barbara, CA). Bruker AFM probes (Camarillo, CA) with a spring constant (k) between 20-80 N/m and a frequency (fo) of 320-357 kHz were used. All samples used were prepared in filtered diH2O to avoid formation of salt crystals. After image acquisition the files were exported as jpegs and analyzed using ImageJ (NIH, USA) using previously described procedure.
Cell binding/uptake studies
Cell uptake studies of ELPAs were performed on human embryonic kidney cells (HEK 293) and cervical cancer cells (HeLa). ELPA samples for cell uptake studies were prepared in 0.2 μm sterile-filtered diH2O. Glass coverslips (22mm × 22mm) were seeded with 3 × 105 cells and cultured in DMEM supplemented with 10% FBS until they were >80% confluent. Appropriate quantities of the nanoparticles were added to the cells and incubated at 37°C at 5% CO2 for 5 hrs. DAPI was added 2 hours prior to imaging. After incubation, coverslips were washed with PBS and mounted onto glass slides using Fluoromount® (Sigma Aldrich). The slides were allowed to dry and imaged on a Zeiss LSM 510 confocal microscope using appropriate settings for fluorophore used (DiI λex =543 nm, DAPI λex = 790 nm). The pinhole sizes used for DiI and DAPI channels are 240 μm and 504 μm respectively. The gain for all the images was kept constant at 840 (red channel) and 1100 (blue channel).
Cellular uptake was compared between cells using red pixel area. The pixel area was calculated using ImageJ. Briefly, the red channels are exported as TIFFs and converted to 8-bit images. The image is thresholded for contrast of red puncta. Puncta were selected using the ROI manager and area in (Pixel)2 measured. The measured area is then converted to μm2 based on the dimensions of the original image. The data was compared using ANOVA/Tukey analysis.
Paclitaxel encapsulation and delivery studies
To test optimal loading concentrations 5, 12.5, 25, 50 and 100 μM solutions of Paclitaxel (PAX) in methanol were added to a fixed ELPA concentration (125 μM). The methanolic PAX:ELPAs were dried down to make even films and hydrated as previously discussed. All films were hydrated with sterile diH2O for HPLC analysis. After hydration the solution was spun at 10,000 rpm for 5 mins to remove unencapsulated PAX. The supernatant was then run on an analytical reverse phase C-18 column using a 20% to 100% ACN (0.1% TFA) gradient and encapsulated PAX absorbance was followed using UV-Vis spectrophotometry at 227 nm. The concentration of encapsulated PAX was determined using a standard curve run under identical conditions.
PAX delivery by ELPAs was tested by determining HeLa cell viability. Briefly, 5 × 104 cells were seeded in a 96-well plate and grown for 24hrs or until >80% confluence was achieved. PAX encapsulated nanoparticles were added to the cells in increasing concentrations and incubated for 48 hrs. After incubation the cells media was replaced and cell viability determined. The cell viability was measured by treatment with water soluble MTS based Promega CellTiter® 96 Cell Proliferation kit (Madison, WI) according to the manufacturer’s protocol. The plates were then analyzed at 490 nm using a Biorad Benchmark plus plate reader (Hercules, CA). For cell viability assays all nanoparticles were prepared in sterile filtered diH2O and control PAX dissolved in 20% sterile filtered DMSO. The amount of PAX encapsulated was determined using previously mentioned reverse phase HPLC method before cellular incubation.
Results
Synthesis and purification of ELPAs
Solid phase synthesis yielded a relatively pure crude product, which was further purified using reverse phase chromatography to produce pure ELPAs (~97%). Mass spectrometry was used to characterize the synthesized ELPAs (Table. S1).
ELPAs assemble nanofibers with a low CMC
Using the pyrene fluorescence assay, the CMC of dpA3 was estimated to be 1.7 μM (Fig. 2a,b). In contrast, dpV3 and dpI3 were not soluble as binary mixtures with water; therefore, their CMC was not assessed. Dynamic light scattering was used to characterize the hydrodynamic radius of dpA3. Above the CMC, dpA3 formed particles with a broad size distribution of 94.2 ± 87.6 nm (Fig. 2c) in radius. Their radius and polydispersity suggest that the ELPAs exist as heterogeneous particles in aqueous buffer; furthermore, these properties are not consistent with small, spherical micelles.
Figure 2. ELPAs self-assemble micelles with a low critical micelle concentration (CMC).
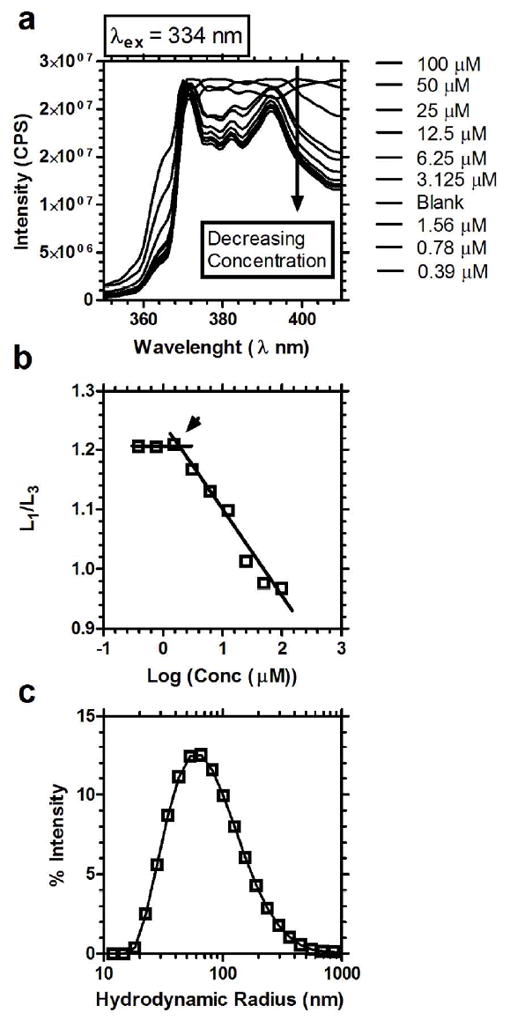
dpA3 was assessed for micelle assembly using pyrene and dynamic light scattering. (a) Emission spectra of pyrene as a function of dpA3 concentration. A decrease in the intensity of spectra was observed with decreasing concentrations of the conjugate. (b) L1/L3 was plotted against log(concentration) of the dpA3 conjugate, where L1 and L3 are the intensity at 373, and 384 nm respectively. The CMC for dpA3 conjugate was determined to be below 1.7 μM at the position indicated by the arrow. (c) Particle size distribution of dpA3 at 1000 μM in 10mM HEPES buffered saline using dynamic light scattering (DLS).
While the > 10 nm hydrodynamic radius provided some evidence that peptide dipalmitoylation produces nanostructures, TEM and AFM were necessary to define the morphology and aspect ratio for these structures (Fig. 3,4). TEM images (Fig. 3a,b) show that dpA3 assembles very long cylindrical micelles 1.7 ± 0.2 μm (approx.) length and 16.0 ± 2.0 nm in width (Fig. 3b,c). AFM provided independent confirmation of the nanofiber assembly. AFM images showed dense networks (Fig. 4a) composed of bundles of cylindrical micelles (Fig. 4b). The average width of each individual strand is 13.9 ± 1.4 nm (Fig. 4c). The control KA3 peptide did not demonstrate structural assembly (Fig. S3). The lengths on the AFM could not be assessed due to imaging limitations that prevented the identification of both ends of a fiber. Regardless, both microscopy techniques confirmed that dpA3 assembles thin fibers with very high aspect ratios.
Figure 3. dpA3 self-assembles cylindrical fibers.
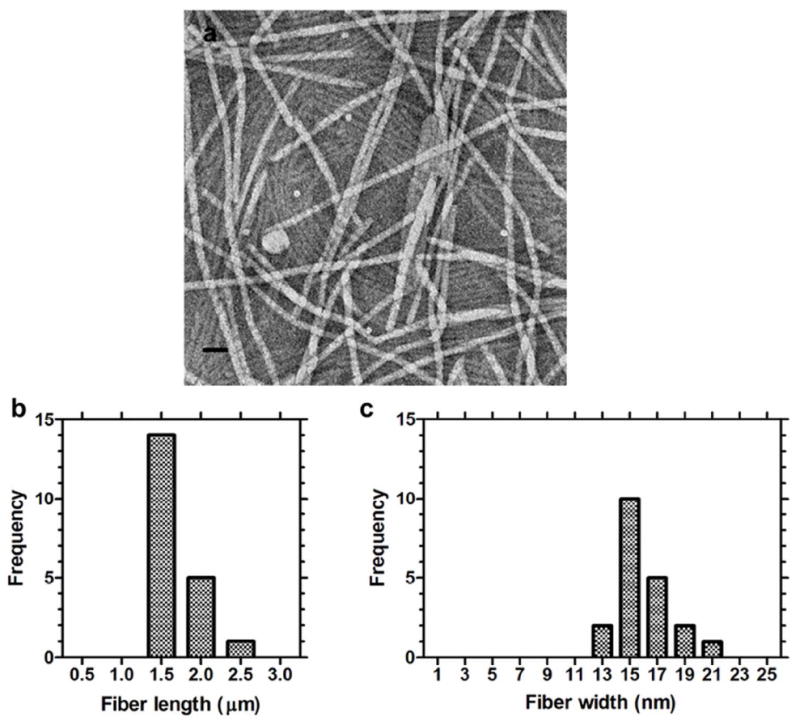
The dpA3 (1000 μM) was stained with 1% uranyl acetate and imaged using transmission electron microscopy (TEM). (a) Numerous fiber-like cylindrical micelle structures 1.7 ± 0.2 nm (mean ± SD, n=3) in length were observed. (b) Distribution of fiber lengths. (c) Distribution of fiber widths. Scale bar = 50 nm.
Figure 4. dpA3 fibers form dense networks.
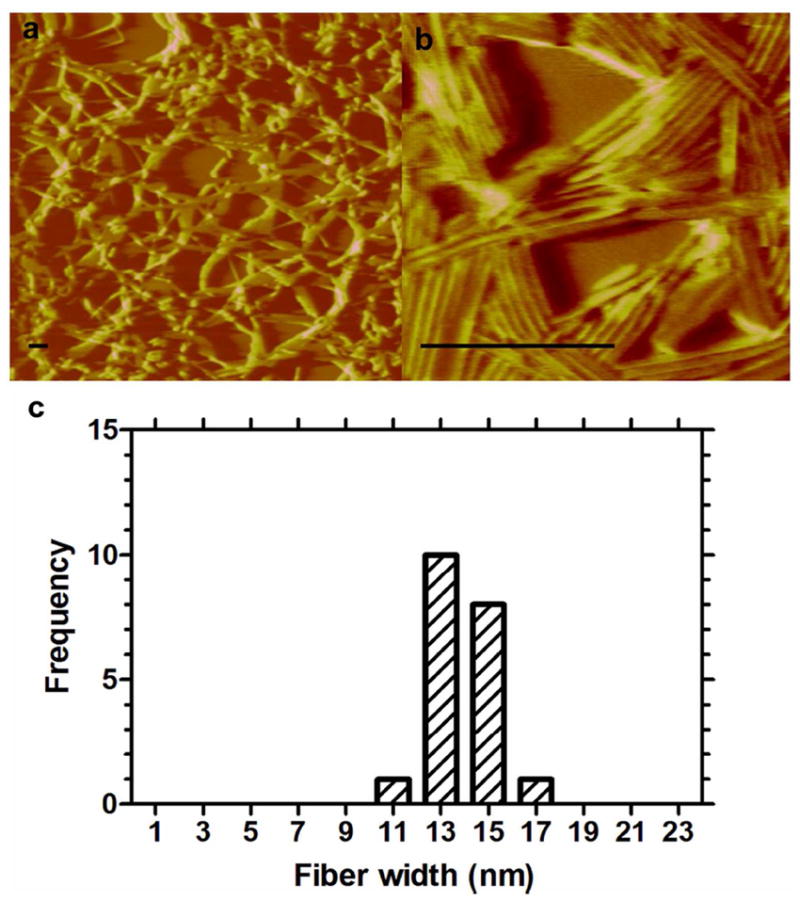
100 μM dpA3 was dried onto a freshly stripped mica surface and imaged using atomic force microscopy (AFM). (a) dpA3 forms a network of bundled fibers. (b) The bundled fibers are composed of parallel cylindrical fibers (c) The widths of the individual fibers are consistent with the measurements obtained from TEM (n=3). Length measurements were not obtained because the fibers extended outside the field of view. Scale bar = 250 nm.
To control the length of the dpA3 nanofibers, phospholipids were mixed with the ELPA to determine if this approach could adjust the average aspect ratio. This attempted to create end-caps to the fibers, under the assumption that the ratio of capping lipids to dpA3 may provide a simple approach to modulate the length, and possibly cellular uptake, for PA nanofibers. As a capping lipid, phospholipid DOPE, was selected because it cannot form stable lipid bilayers in the absence of other lipid components15. This allows for easy identification of ELPA fibers. Since experiments were performed in neutral conditions the adoption of an alpha-II conformation by DOPE is unlikely. It was intended that DOPE would be less likely to form a second population of vesicles, which may have resulted from mixtures with phosphocholine lipids. At ratios of 4:1 dpA3 to DOPE, the size distribution of the ELPAs was brought down to 282.7 ± 68.4 nm. At [1:1] ratios of dpA3 to DOPE, the size of the ELPAs was further reduced to 71.3 ± 13.2 nm (Fig. 5a,b). The addition of capping lipids had no effect on the width of observed nanofibers. The widths by TEM imaging for ELPAs at 4:1 and 1:1 dpA3 to DOPE are 12.5 ± 0.9 nm and 13.2 ± 2.2 nm. Hence, we have successfully developed a method to independently modulate the length for ELPAs, which may have utility in the design of peptide amphiphiles using other peptide sequences.
Figure 5. The length of the ELPA fibers can be controlled using phospholipids.
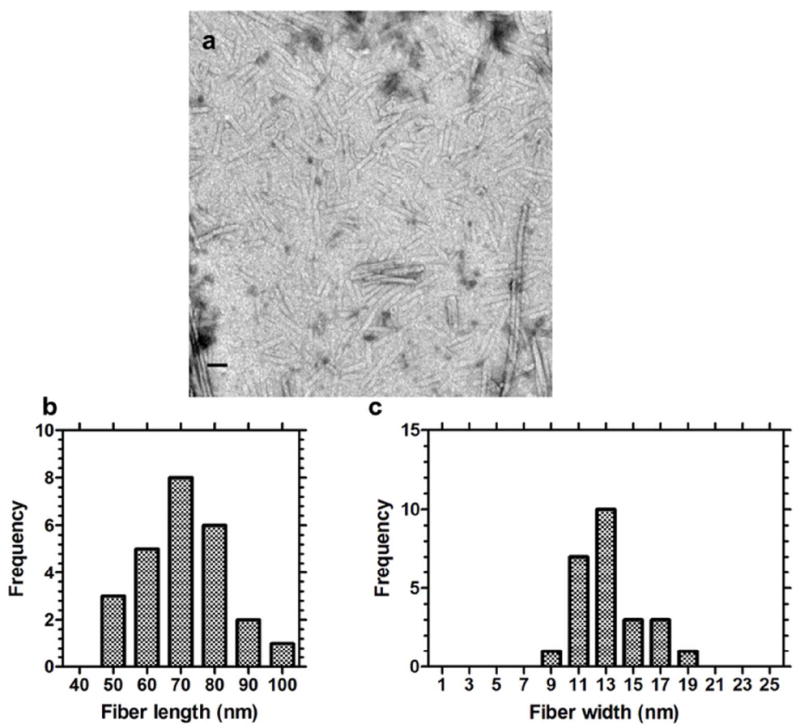
(a) The cylindrical fibers were shortened in length by 10-fold to 71.0 ± 13.2 nm (mean ± SD, n=3) by mixture of equimolar quantities of DOPE:dpA3 [1:1] (b) Distribution of fiber length. (c) The distribution of fiber widths was unaffected by the addition of phospholipid. Scale bar = 50 nm.
ELP nanofibers exhibit LCST behavior
High molecular weight ELPs like A192 (Fig. 6a), phase separate above an inverse phase transition temperature, which is similar to an LCST. This effect is length dependent. Short ELPs like KA3 do not phase separate below 100 °C at atmospheric pressure16. It was anticipated that ELPA self-assembly into fibers would form a dense peptide brush that would have LCST properties. When heated, the ELPA nanofibers did exhibit a concentration dependent change in the transition temperature (Fig. 6b). The phase diagram of dpA3 (2kD) was similar to that for a high molecular weight ELP, A192 (73.2 kD), albeit at slightly higher molar concentrations (Fig. 6c). The plain peptide, KA3, does not phase separate in this temperature range (Fig. S2).
Figure 6. ELPAs (dpA3) and soluble ELPs (A192) both undergo LCST behavior.
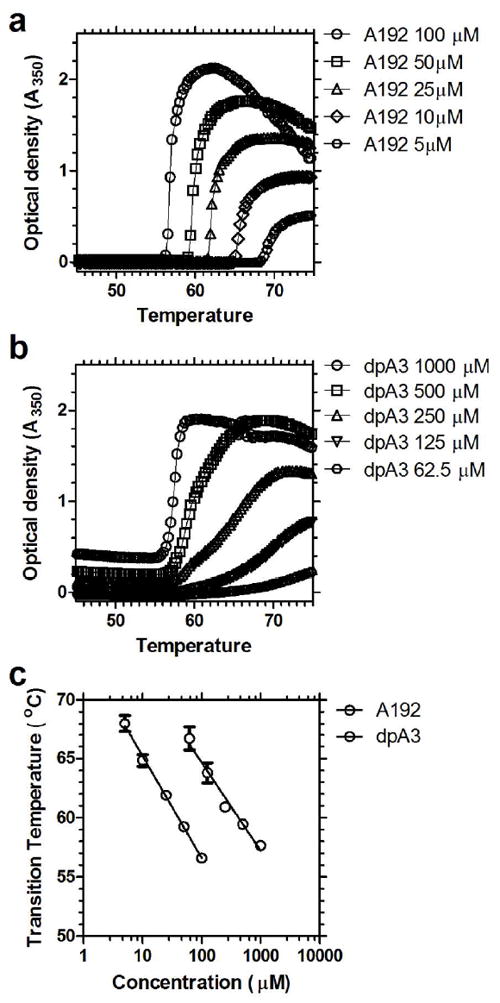
The optical density was monitored over a temperature gradient of 1 °C per min from 45 to 80 °C. (a) A soluble ELP with a high MW (73.2 kD), A192 (100 μM=7.3 mg/ml). (b) ELPA nanofibers composed from the low molecular weight (1.7 kD) dpA3 (1000 μM=1.7mg/ml). (c) The transition temperatures for both constructs are inversely proportional to Log of concentration for both A192 (slope = -8.9 ± 0.8 °C (Log10[μM])-1 (mean ± 95% CI), r2 = 0.9975, p < 0.01) and dpA3 (slope = -7.1 ± 1.8 °C (Log10[μM])-1 (mean ± 95% CI), r2 = 0.98, p < 0.01).
β-turn formation associated with approach of ELPA transition temperature
To investigate the mechanism for ELPA phase separation, CD was used to characterize the peptide secondary structure as a function of concentration, dipalmitoylation, and temperature. The CD spectra were deconvoluted using the standard curves of (a) α-helix17 (b) β-sheet17 (c) Random coil17 (d) β-turn 118 and (e) β-turn 219. Preliminary CD studies of KA3 failed to show any concentration (data not shown) or temperature (Fig. 7) dependent change in structure. In contrast, the addition of two palmitic chains had a very significant impact on the secondary structure of KA3 (Fig. 7a,b). By inducing peptide assembly into nanofibers, the palmitic chains promote order in dpA3. At 25 °C, the majority of dpA3 exists in either a random coil or a β-sheet conformation (Fig. 7c). While approaching the phase transition temperature, there is an increase in the β-type-1 turn content. Above the phase transition temperature (~60 °C), the structure becomes highly disordered like the unmodified peptide. The control KA3 peptide has CD spectra very similar to a random coil, which has been observed previously for ELPs below their transition temperatures20. Deconvolution of the spectra showed that the majority of the KA3 peptide exists in a random coil / β-sheet with a small population in a β-turn 1 conformation, which remains largely unchanged at higher temperatures (Fig. 7d). These data confirm that nanofiber assembly of dpA3, mediated by N-dipalmitoylation, produces a moderate degree of β type-1 turn structures; furthermore, increasing temperature stabilized this structure up until above the bulk phase transition temperature.
Figure 7. dpA3 forms temperature-dependent secondary structure.
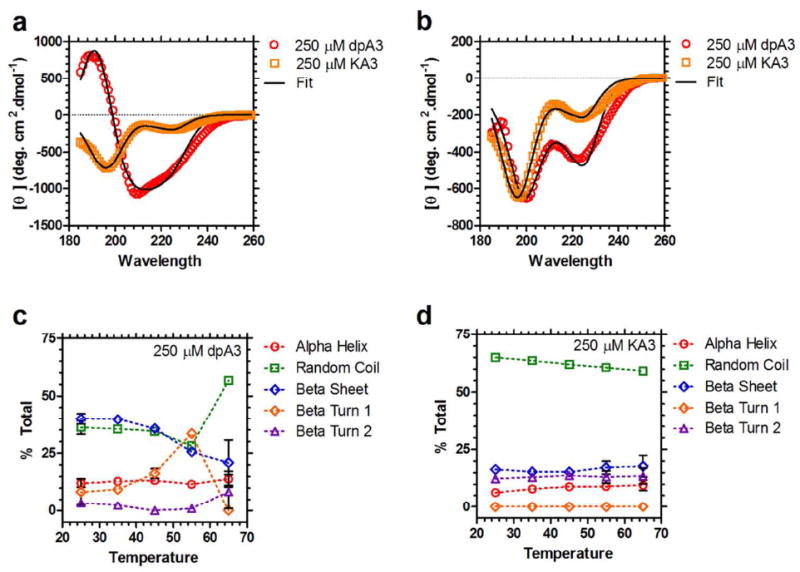
(a) Circular dichroism (CD) spectra for dpA3 (250 μM, red) and KA3 (250 μM, orange) below the transition temperature (T = 55 °C). The spectrum for the unmodified KA3 (R2=0.9930) peptide is considerably different from the dipalmitoylated dpA3 (R2=0.9922), suggesting that nanofiber assembly induces formation of partial secondary structure. (b) Above the transition temperature for dpA3 (T = 65 °C) there is a noticeable change in the molar ellipticities (red, R2=0.9832); however, the unmodified peptide KA3 (orange, R2=0.9913) remains unchanged. (c) At lower temperatures the majority of the dpA3 (250 μM) population adopts either a β-sheet or random coil conformation. On heating there is an increase in the β-turn 1 component suggesting that β-turn 1 formation is necessary for the phase separation of this class of ELPAs. (d) The major conformation of unmodified KA3 at all temperatures is random coil. There is no significant change in the secondary structure at higher temperatures.
Inclusion of DOPE promotes ELPA cell uptake
We next investigated the possibility that the inclusion of other lipids might change the physico-chemico properties of dpA3 nanofibers, with the goal of tuning cellular uptake. To accomplish this, we evaluated multiple phospholipids in mixture with ELPAs, which included phosphocholines (POPC, DPPC) and phosphoenthanolamines (DOPE). Only DOPE formulations produced stable nanofibers with shortened lengths (< 50% by mol lipid); therefore, these formulations were evaluated for their cellular uptake in cultured HEK 293 and HeLa cells. ELPA uptake was studied using confocal microscopy, whereby nanofibers were labeled with 0.2% by mol lipid DiI (red). DiI is a strong, non-exchangeable marker for stable cell membranes and requires a hydrophobic environment to fluoresce. Hence, DiI makes an excellent hydrophobic compound to study ELPA nanofiber-mediated cellular uptake. DiI labeled DOPC liposomes with equal quantities of DOPE were used as negative controls for the nonspecific uptake of neutral liposomes (Fig. S5). The uptake of pure dpA3 nanofibers was minimal in both HEK 293 and HeLa cells (Fig. 8a-c,m-p). In contrast, the addition of DOPE promoted cellular uptake of ELPAs in both cell lines. The inclusion of DOPE had an easily observable effect on cellular binding and uptake. This effect may arise from the negative curvature of DOPE lipids, which promotes fusion of pure phospholipid membranes21. Alternatively, evidence that DOPE shortens the length of ELP nanofibers (Fig. 5) from microns to below 300 nm raises the possibility that ELPA internalization (non-phagocytic) may be tuned by controlling fiber aspect ratio. Interestingly, the addition of DOPE to DOPC liposomes also enhanced the transfer of the DiI label to cells however to a much lesser extent than for ELPA nanofibers (Fig. S5). Unformulated, free DiI showed minimal staining of phospholipid cellular membranes (Fig. S6). Hence the addition of DOPE may mediate uptake through two mechanisms: i) membrane fusion and ii) reduction of nanofiber length. Due to the inability of non-fusogenic phospholipids (DPPC, POPC) to form stable and short ELPA nanofibers, we are currently unable to distinguish between these two mechanisms. Regardless, DOPE incorporation unambiguously modulates the transfer of a model hydrophobic (DiI) compound between ELPA nanofibers and cells.
Figure 8. DOPE addition promotes cellular uptake of ELPAs.
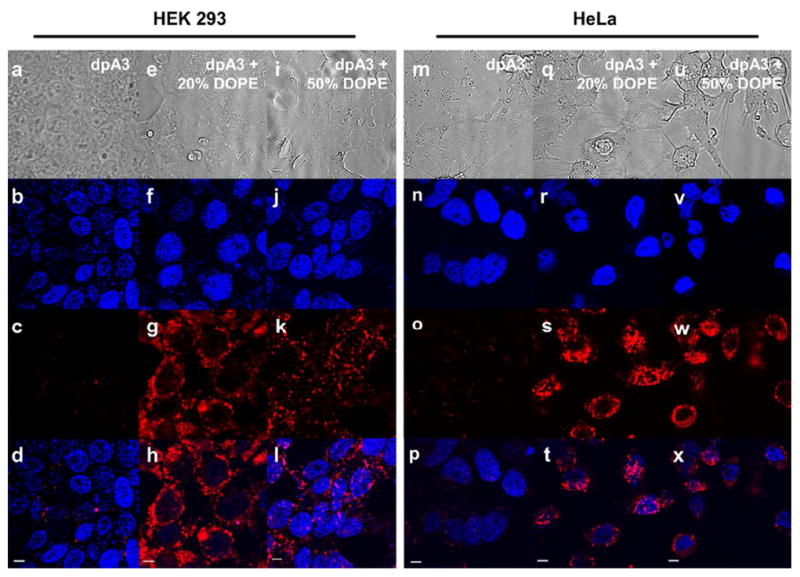
16.7μM of particles were added to HEK293 cells and incubated for 5hrs. DiI was used to label nanoparticles (red). DAPI (Blue) was used to stain the nucleus. All images were obtained with the same microscope settings. Image panels (a) to (d) and (m) to (p) show minimal uptake of dpA3 in both cell lines respectively. Conversely, image panels (e) to (l) and (q) to (x) show uptake of dpA3:DOPE particles which suggests DOPE influence in cellular uptake. Control DOPC liposomes did not show any cellular uptake (Fig. S5). Scale bars represent 5 μm.
Encapsulated PAX reduces tumor cell viability
Film hydration showed no precipitation of PAX, suggesting high efficiency encapsulation. Using dpA3, we have shown a significant increase in PAX solubility in aqueous solvents i.e from <0.1 μg/ml to 85 μg/ml (100μM)22. PAX encapsulation is saturated above a 1:0.8 molar loading ratio of dpA3 to PAX (Fig. 9a). Using encapsulated PAX we demonstrate significant inhibition of HeLa cell growth (Fig. 9b). The inhibition of cell growth is similar to soluble unencapsulated PAX delivered in DMSO. At the concentrations studied, the inclusion of DOPE did not enhance the efficacy of encapsulated PAX (Fig. 9b). The study was not performed at lower dpA3 concentrations due to concerns that below their CMC, the ELPA nanofibers may disassemble. The maximum concentration of DMSO over the cells was 10%. These results suggest that dpA3 nanofibers may have potential as drug delivery vehicles for PAX.
Figure 9. dpA3 Encapsulated PAX reduces HeLa cell viability.
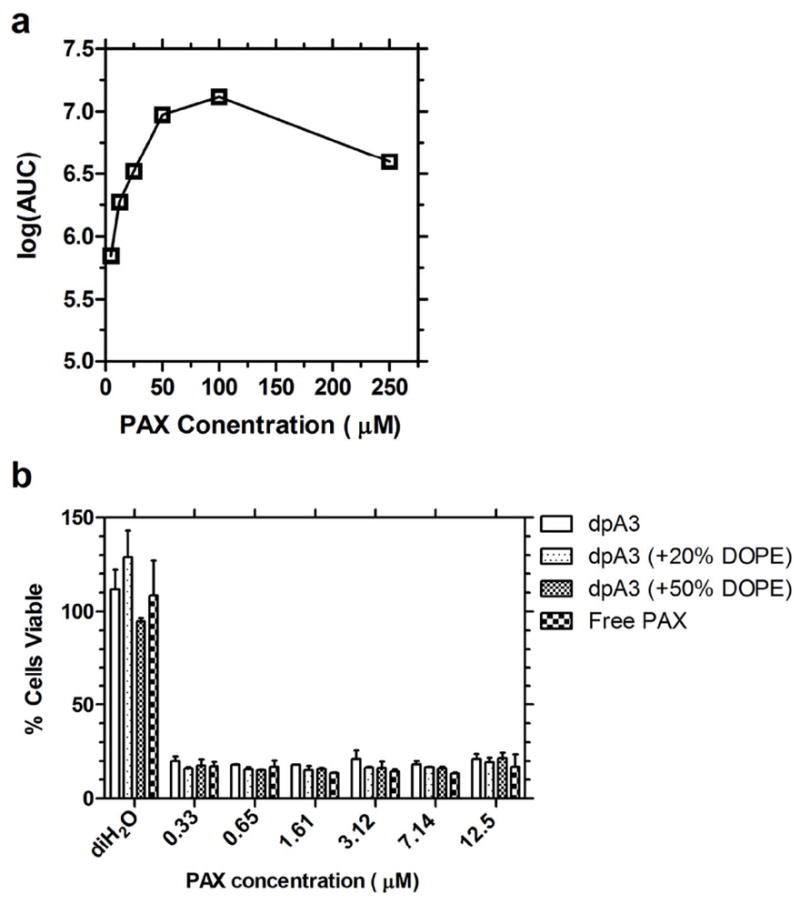
(a) At a constant concentration of dpA3 (125 μM), increasing concentrations of PAX were encapsulated using thin film hydration. Analytical Reverse Phase HPLC was used to quantify PAX, and the area under the curve (AUC) for the appropriate peak is presented. This trace shows saturable encapsulation above 50 μM PAX at dpA3:PAX ratio of 1:0.8; therefore, dpA3:PAX nanoparticles approach ~29% by mass drug loading. (b) A cytotoxity assay was used to determine if dpA3 was able to deliver PAX to cells using the MTS assay. There is no difference in efficacy of encapsulated PAX compared to free PAX solubilized in DMSO.
Discussion
Here we report the formation of peptide amphiphile nanofibers of tunable length with some properties of elastin-like polypeptides. The dipalmitoylation of KA3 yielded a PA with a low CMC, similar to that found for other dipalmitoyl phosphatidylcholine lipids using the same pyrene assay23. A potential advantage of this chemistry is the production of stable amide bonds between the peptide and the lipids, which lack the ester bonds found on phospholipids. The palmitic acid chains reduce the transition temperature to well below 100 °C, allowing this approach to be used in the induction of the phase behavior of short hydrophilic ELPs. In addition to the stability of ELPAs (low CMC, amide linkage), we have identified several key variables that enable tuning of the aspect ratio (capping lipids) and cellular uptake (mixing ELPAs with different fusogenic lipids). We have also uncovered new evidence that the ELPA phase separation is mediated by the formation of distinct secondary structure. At low temperatures, the CD spectra for dpA3 showed sheet like structures that were similar to those reported for other PAs24, 25. CD spectra further revealed that dpA3 phase separation correlates with the maturation of β-type-1 turns during heating. In contrast, the unmodified KA3, remains in a random coil conformation and shows no temperature-dependent change in secondary structure. Intriguingly, above its phase transition temperature (>60 °C), dpA3 (Fig. 7c) forms a random coil conformation; suggesting that an entropic change from a highly ordered secondary structure below the transition temperature to a disordered peptide backbone above the transition temperature may partly contribute to differences in the free energy of mixing responsible for phase separation. While this possibility warrants further study, the ability to probe peptide structure above the bulk phase transition using CD is limited by light scattering from large aggregates.
Similar to other reported PAs, dpA3 formed fiber-like micelles above its CMC26, 27. We confirmed the assembly of these fibers using TEM and AFM. To further modify its assembly we incorporated fusogenic lipids in our PA. Previously, the Hartgernik group demonstrated that phospholipid inclusion modulates the gelation properties of PAs28; however, this approach was never demonstrated to control the length of any PA nanofibers. Here we used the same concept to cap the nanofiber termini and control their lengths. Our proposed mechanism is that the phospholipids arrest the growth of the nanofiber, possibly by local phase separation to the termini with dpA3 occupying the trunk of the nanofiber (Fig. 10). This approach to control aspect ratio is significant because it provides a method to tune the length of peptide amphiphile nanofibers as needed to optimize cellular uptake (Fig. 8), mechanical properties, or pharmacokinetics. At equimolar ratios of capping lipid DOPE to dpA3, length was reduced 100 fold with almost no change in fiber width. To the best of our knowledge, this degree of control over PA nanofiber length has never been reported. By further increasing the capping lipid concentration to above 50% by mol lipid, the fibers were abolished and the major species observed were round structures (Fig. S4)29. Under our proposed mechanism, the cylindrical trunk of the nanofiber is predominantly composed of dpA3 (Fig. 10a). Increasing percentages of the capping lipid, DOPE, reduced the average nanofiber length observed using TEM (Fig. 10b). The addition of DOPE also had significant effect on the cellular uptake of dpA3 (Fig. 10c). The poor uptake of dpA3 may be attributed to its extreme aspect ratio and uncharged nature. By controlling the aspect ratio, lipids like DOPE may shorten ELPA nanofibers into shorter fibers (Fig. 5), which are more suitable for non-phagocytic mechanisms of cellular internalization, Including potocytosis or clathrin-mediated endocytosis. Alternatively, the inclusion of DOPE may promote membrane fusion that mimics intracellular uptake. In either case, this lipid capping approach may also useful for polarized functionalization of ELPA termini with cargo or targeting moieties.
Figure 10. Proposed mechanism for fiber-length by capping phospholipid.
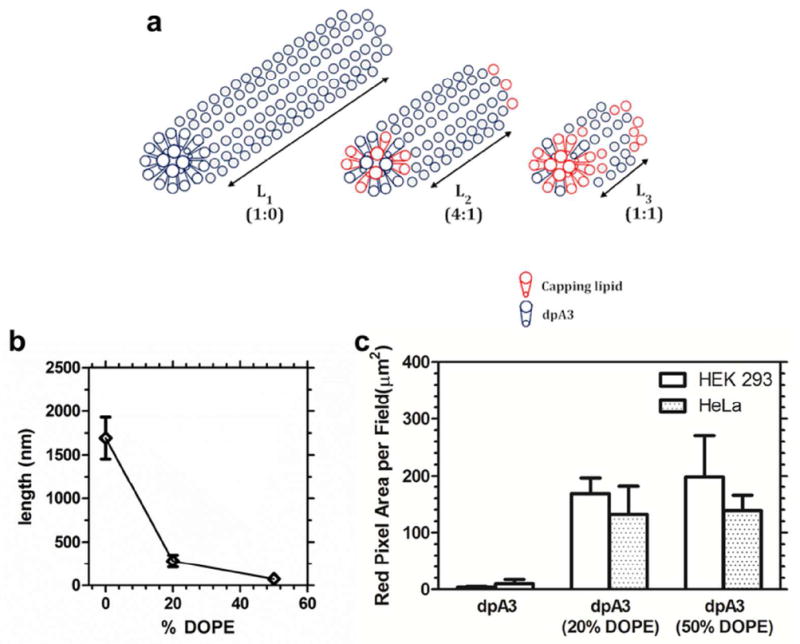
(a) Unsaturated lipid chains of DOPE promote intra-fiber phase separation to the termini of ELPA nanofibers assembled by saturated lipids. (b) The average nanofiber length was plotted as a function of the % of DOPE capping lipid. (c) Plain dpA3 fibers have low propensity for cellular uptake. ANOVA analysis shows a significant difference between the 3 groups (P=0.0188). Tukey’s post hoc analysis shows a significant difference between dpA3 and formulation with 20% & 50% DOPE (P<0.05). No significant difference in uptake between 20% & 50% DOPE was observed. Hence, addition of DOPE increases uptake irrespective of the cell line used. Values indicate the Mean ± SD (n=3)
In addition to the delivery of a model hydrophobic fluorophore, DiI, the hydrophobic core of the ELPA nanofibers demonstrated high efficiency encapsulation of the hydrophobic chemotherapeutic PAX. PAX is commonly marketed as Taxol® and is dissolved in a castor oil based Cremophor EL® system30. Cremophor EL® is a non-ionic surfactant which helps emulsify PAX in aqueous solutions. In addition to severe discomfort during infusion, the Cremophor EL® system is also reported to cause allergic reactions, which can be fatal31. The pure ELPA system can serve as an ideal alternative to such surfactants since it is non-ionic and not easily taken up into cells (as shown in Fig. 8). Further studies may be warranted in an effort to translate ELPA carriers from the bench to the clinic. While genetically engineered ELPs such as A192, have many potential applications32, ELPAs have three potential advantages as drug carriers: i) they have a truly hydrophobic core capable of physical entrapment of hydrophobic drugs like PAX; ii) they are not recombinant products, reducing their cost and complexity; and iii) the observation that they formed nanofibers makes them a substrate with a very different surface compared to large soluble ELPs or spherical ELP nanoparticles.
One potential application of ELPAs will be in the development of nanoparticles that are synergistic with hyperthermia-mediated drug delivery32. In this manuscript, we have focused on developing ELPAs as soluble drug carriers with a tunable fiber length; however, future studies will be required to tune their thermal responses closer to physiological temperature. The most direct approach to modulate ELP transition temperature is to adjust the hydrophobicity of the guest residue from Ala to Val or Ile. In fact, we successfully purified both compounds (Table S1); however, neither dpV3 nor dpI3 formed soluble, stable nanofibers in the presence of water. One plausible explanation is that, similar to dipalmitoyl phospholipids, the dipalmitoylated ELPAs have a lipid melting temperature slightly above body temperature. Furthermore, it is plausible that if the transition temperature of the ELP corona is below that of the lipid core, the ELPAs are unable to reorder themselves into stable nanofibers upon hydration. Any effort to melt the lipid core results in bulk phase separation of the peptide corona. Having now optimized the drug loading and tunability of nanofiber length, future studies will address the lipid core melting temperature and tunability of nanofiber transition temperature to better utilize hyperthermiamediated targeting.
PAs are an emerging nanomaterial with applications ranging from tissue engineering to drug delivery. Dr. Stupp and coworkers have extensively studies PA networks for cell repair and regeneration. For example, they developed PA systems with tunable RGD presentation which have shown promise as cell adhesion scaffolds33. The same group has also shown neuronal regeneration using laminin derived PAs, which have potential for treating spinal cord injuries34. Apart from tissue regeneration, PAs have found use in conventional drug delivery aspects. PAs have been used to encapsulate hydrophobic small molecules (Cisplatin, Nabumetone)35, 36 and as contrast agents37, 38 in MRI imaging. As they are bioresponsive, uncharged, and of tunable length, we propose that elastin-like PAs provide multiple opportunities to facilitate the successful translation of PA-derived therapies such as those above into clinical use.
Conclusion
Herein we describe a new type of peptide amphiphiles prepared by dipalmitoylation of a non-ionic polypeptide derived from human tropoelastin. These ELPAs form cylindrical micelles in solution, which undergo inverse phase transition behavior similar to high molecular weight ELPs. The lengths of these ELPA nanofibers can be tuned by inclusion of other lipids. The addition of lipids promoted cellular uptake of described ELPA nanofibers. We also have preliminary evidence for its application as a drug delivery vehicle. This new class of PAs may be a useful platform to develop nanostructures with potential in targeted drug delivery applications.
Supplementary Material
Acknowledgments
This work was made possible by the University of Southern California, the National Institute of Health R21EB012281, The Wright Foundation, The Stop Cancer Foundation, the USC Ming Hsieh Institute, and the Whittier Foundation to J.A.M., P30DK048522-15 to the USC Research Center for Liver Diseases, P30CA014089 to the Norris Comprehensive Cancer Center and the Translational Research Laboratory at the School of Pharmacy. CD and AFM data were obtained at USC Nanobiophysics core facility. The TEM images were obtained at the USC Cell and Tissue Imaging Core. We extend thanks to S. Louie for access mass spectrometry equipment in the USC Pharmacoanalytical Core Facility.
Abbreviations
- ELPs
Elastin like polypeptides
- PAs
Peptide Amphiphiles
- ELPAs
Elastin like peptide amphiphiles
- CD
Circular Dichroism
- TEM
Transmission electron microscopy
- AFM
Atomic Force Microscopy
- LCST
Lower Critical Solution temperature
- CMC
Critical micellar concentration
- HPLC
High performance liquid chromatography
Footnotes
Supporting Information Available
An additional table and six supporting figures are available as supplementary materials. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Cui H, Webber MJ, Stupp SI. Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers. 2010;94(1):1–18. doi: 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geng Y, Ahmed F, Bhasin N, Discher DE. Visualizing worm micelle dynamics and phase transitions of a charged diblock copolymer in water. J Phys Chem B. 2005;109(9):3772–9. doi: 10.1021/jp0459559. [DOI] [PubMed] [Google Scholar]
- 3.Kim Y, Dalhaimer P, Christian DA, Discher DE. Polymeric worm micelles as nano-carriers for drug delivery. Nanotechnology. 2005;16(7):S484–91. doi: 10.1088/0957-4484/16/7/024. [DOI] [PubMed] [Google Scholar]
- 4.Paramonov SE, Jun H-W, Hartgerink JD. Self-Assembly of Peptide–Amphiphile Nanofibers: The Roles of Hydrogen Bonding and Amphiphilic Packing. Journal of the American Chemical Society. 2006;128(22):7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 5.Simone EA, Dziubla TD, Discher DE, Muzykantov VR. Filamentous polymer nanocarriers of tunable stiffness that encapsulate the therapeutic enzyme catalase. Biomacromolecules. 2009;10(6):1324–30. doi: 10.1021/bm900189x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartgerink JD, Granja JR, Milligan RA, Ghadiri MR. Self-Assembling Peptide Nanotubes. Journal of the American Chemical Society. 1996;118(1):43–50. [Google Scholar]
- 7.Hartgerink JD, Beniash E, Stupp SI. Peptide-amphiphile nanofibers: a versatile scaffold for the preparation of self-assembling materials. Proc Natl Acad Sci U S A. 2002;99(8):5133–8. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu YC, Berndt P, Tirrell M, Fields GB. Self-assembling amphiphiles for construction of protein molecular architecture. Journal of the American Chemical Society. 1996;118(50):12515–12520. [Google Scholar]
- 9.Huang Z, Newcomb CJ, Bringas P, Jr, Stupp SI, Snead ML. Biological synthesis of tooth enamel instructed by an artificial matrix. Biomaterials. 2010;31(35):9202–11. doi: 10.1016/j.biomaterials.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J, Saiz E, Bertozzi CR. A New Approach to Mineralization of Biocompatible Hydrogel Scaffolds: An Efficient Process toward 3-Dimensional Bonelike Composites. Journal of the American Chemical Society. 2003;125(5):1236–1243. doi: 10.1021/ja028559h. [DOI] [PubMed] [Google Scholar]
- 11.Aluri S, Janib SM, Mackay JA. Environmentally responsive peptides as anticancer drug carriers. Adv Drug Deliv Rev. 2009;61(11):940–52. doi: 10.1016/j.addr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urry DW. Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. Journal of Physical Chemistry B. 1997;101(51):11007–11028. [Google Scholar]
- 13.Fields CG, Lloyd DH, Macdonald RL, Otteson KM, Noble RL. HBTU activation for automated Fmoc solid-phase peptide synthesis. Pept Res. 1991;4(2):95–101. [PubMed] [Google Scholar]
- 14.Goddard ED, Turro NJ, Kuo PL, Ananthapadmanabhan KP. Fluorescence probes for critical micelle concentration determination. Langmuir. 1985;1(3):352–355. doi: 10.1021/la00063a015. [DOI] [PubMed] [Google Scholar]
- 15.Yeagle PL, Sen A. Hydration and the lamellar to hexagonal(II) phase transition of phosphatidylethanolamine. Biochemistry. 1986;25(23):7518–7522. doi: 10.1021/bi00371a039. [DOI] [PubMed] [Google Scholar]
- 16.Meyer DE, Chilkoti A. Quantification of the Effects of Chain Length and Concentration on the Thermal Behavior of Elastin-like Polypeptides. Biomacromolecules. 2004;5(3):846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 17.Greenfield N, Fasman GD. Computed circular dichroism spectra for the evaluation of protein conformation. Biochemistry. 1969;8(10):4108–16. doi: 10.1021/bi00838a031. [DOI] [PubMed] [Google Scholar]
- 18.Perczel A, Fasman GD. Quantitative analysis of cyclic beta-turn models. Protein Sci. 1992;1(3):378–95. doi: 10.1002/pro.5560010310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brahms S, Brahms J, Spach G, Brack A. Identification of beta,beta-turns and unordered conformations in polypeptide chains by vacuum ultraviolet circular dichroism. Proc Natl Acad Sci U S A. 1977;74(8):3208–12. doi: 10.1073/pnas.74.8.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klok H-A. Secondary Structure Formation and LCST Behavior of Short Elastin-Like Peptides. Biomacromolecules. 2008;9(10):2755–2763. doi: 10.1021/bm800784y. [DOI] [PubMed] [Google Scholar]
- 21.Guo X, MacKay JA, Szoka FC., Jr Mechanism of pH-triggered collapse of phosphatidylethanolamine liposomes stabilized by an ortho ester polyethyleneglycol lipid. Biophys J. 2003;84(3):1784–95. doi: 10.1016/s0006-3495(03)74986-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konno T, Watanabe J, Ishihara K. Enhanced solubility of paclitaxel using water-soluble and biocompatible 2-methacryloyloxyethyl phosphorylcholine polymers. Journal of Biomedical Materials Research Part A. 2003;65A(2):209–214. doi: 10.1002/jbm.a.10481. [DOI] [PubMed] [Google Scholar]
- 23.Kanamoto R, Wada Y, Miyajima G, Kito M. Phospholipid- phospholipid interaction in soybean oil. Journal of the American Oil Chemists’ Society. 1981;58(12):1050–1053. [Google Scholar]
- 24.Xu XD, Jin Y, Liu Y, Zhang XZ, Zhuo RX. Self-assembly behavior of peptide amphiphiles (PAs) with different length of hydrophobic alkyl tails. Colloids Surf B Biointerfaces. 2010;81(1):329–35. doi: 10.1016/j.colsurfb.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 25.Yokoyama K, Kubo T, Fujii M. AMPHIPHILIC •-SHEET PEPTIDES CAN BIND TO DOUBLE AND TRIPLE STRANDED DNA. Nucleosides, Nucleotides and Nucleic Acids. 2001;20(4-7):1317–1320. doi: 10.1081/NCN-100002545. [DOI] [PubMed] [Google Scholar]
- 26.Hartgerink JD, Beniash E, Stupp SI. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science. 2001;294(5547):1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 27.Chow LW, Wang LJ, Kaufman DB, Stupp SI. Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets. Biomaterials. 2010 doi: 10.1016/j.biomaterials.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paramonov SE, Jun HW, Hartgerink JD. Modulation of peptide-amphiphile nanofibers via phospholipid inclusions. Biomacromolecules. 2006;7(1):24–6. doi: 10.1021/bm050798k. [DOI] [PubMed] [Google Scholar]
- 29.Fluck DJ. Physical studies of phospholipids. 3. Electron microscope studies of some pure fully saturated 2,3-diacyl-DL-phosphatidyl-ethanolamines and phosphatidyl-cholines. The Journal of cell biology. 1966;30(1):1. doi: 10.1083/jcb.30.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowinsky EK, Onetto N, Canetta RM, Arbuck SG. Taxol: the first of the taxanes, an important new class of antitumor agents. Semin Oncol. 1992;19(6):646–62. [PubMed] [Google Scholar]
- 31.Liebmann J, Cook J, Mitchell J. Cremophor EL, solvent for paclitaxel, and toxicity. The Lancet. 1993;342(8884):1428. doi: 10.1016/0140-6736(93)92789-v. [DOI] [PubMed] [Google Scholar]
- 32.Andrew Mackay J, Chilkoti A. Temperature sensitive peptides: Engineering hyperthermia-directed therapeutics. International Journal of Hyperthermia. 2008;24(6):483–495. doi: 10.1080/02656730802149570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Storrie H, Guler MO, Abu-Amara SN, Volberg T, Rao M, Geiger B, Stupp SI. Supramolecular crafting of cell adhesion. Biomaterials. 2007;28(31):4608–18. doi: 10.1016/j.biomaterials.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 34.Tysseling VM, Sahni V, Pashuck ET, Birch D, Hebert A, Czeisler C, Stupp SI, Kessler JA. Self-assembling peptide amphiphile promotes plasticity of serotonergic fibers following spinal cord injury. J Neurosci Res. 2010;88(14):3161–70. doi: 10.1002/jnr.22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JK, Anderson J, Jun HW, Repka MA, Jo S. Self-assembling peptide amphiphile-based nanofiber gel for bioresponsive cisplatin delivery. Mol Pharm. 2009;6(3):978–85. doi: 10.1021/mp900009n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matson JB, Stupp SI. Drug release from hydrazone-containing peptide amphiphiles. Chem Commun (Camb) 2011;47(28):7962–4. doi: 10.1039/c1cc12570b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bull SR, Guler MO, Bras RE, Meade TJ, Stupp SI. Self-assembled peptide amphiphile nanofibers conjugated to MRI contrast agents. Nano Lett. 2005;5(1):1–4. doi: 10.1021/nl0484898. [DOI] [PubMed] [Google Scholar]
- 38.Bull SR, Guler MO, Bras RE, Venkatasubramanian PN, Stupp SI, Meade TJ. Magnetic Resonance Imaging of Self-Assembled Biomaterial Scaffolds. Bioconjugate Chemistry. 2005;16(6):1343–1348. doi: 10.1021/bc050153h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.