

Abstract
Interactions between danger-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) and pattern recognition receptors such as Toll-like receptors (TLRs) are critical for the regulation of the inflammatory process via activation of nuclear factor-κB (NF-κB) and cytokine secretion. In this report, we investigated the capacity of lipopolysaccharide (LPS) -free S100A9 (DAMP) protein to activate human and mouse cells compared with lipoprotein-free LPS (PAMP). First, we showed that LPS and S100A9 were able to increase NF-κB activity followed by increased cytokine and nitric oxide (NO) secretion both in human THP-1 cells and in mouse bone marrow-derived dendritic cells. Surprisingly, although S100A9 triggered a weaker cytokine response than LPS, we found that S100A9 more potently induced IκBα degradation and hence NF-κB activation. Both the S100A9-induced response and the LPS-induced response were completely absent in TLR4 knockout mice, whereas it was only slightly affected in RAGE knockout mice. Also, we showed that LPS and S100A9 NF-κB induction were strongly reduced in the presence of specific inhibitors of TLR-signalling. Chloroquine reduced S100A9 but not LPS signalling, indicating that S100A9 may need to be internalized to be fully active as a TLR4 inducer. This was confirmed using A488-labelled S100A9 that was internalized in THP-1 cells, showing a raise in fluorescence after 30 min at 37°. Chloroquine treatment significantly reduced the fluorescence. In summary, our data indicate that both human and mouse S100A9 are TLR4 agonists. Importantly, S100A9 induced stronger NF-κB activation albeit weaker cytokine secretion than LPS, suggesting that S100A9 and LPS activated NF-κB in a qualitatively distinct manner.
Keywords: DAMP, inflammation, nuclear factor-κB, pathogen-associated molecular patterns, THP-1, Toll-like receptor 4
Introduction
Inflammation is a key event in host defence against extracellular pathogens, tissue damage and several diseases such as cancer,1 rheumatoid arthritis,2 systemic lupus erythematosus3 and cystic fibrosis.4,5 The main function of inflammation is to resolve the infection and repair the damage to return to a state of homeostasis.6 A critical step to initiate the inflammatory cascade is represented by the recognition of specific molecules by pattern recognition receptors, such as the Toll-like receptors (TLRs).7,8
Toll-like receptors are a class of transmembrane proteins that play an important role in the innate immune response. Eleven different members of TLRs have been found in mammals; TLRs are involved in the recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs).7 The prototypical PAMP molecule lipopolysaccharide (LPS) is an endotoxin that is the major component of the outer membrane of Gram-negative bacteria. Upon interaction with TLR4 receptor, LPS initiates a signal cascade that leads to the activation of the transcription factor nuclear factor-κB (NF-κB) and, consequently, induces cytokine expression and secretion, causing a strong pro-inflammatory response.9
During the last few years, several studies have demonstrated that S100 proteins can function as DAMP molecules.10,11 An increasing amount of evidence also indicates that members of this protein family, and in particular S100A8 and S100A9, may represent novel markers for inflammation and autoimmune diseases.13–15 S100A9, a small protein with molecular weight 14 000, is constitutively expressed in neutrophils and monocytes.18,19 S100A9 has a central domain flanked by two EF-hand Ca2+ binding-motifs and interacts with S100A8 forming a complex called calprotectin,12 the pro-inflammatory function of which has been well characterized.16–20 In particular, calprotectin triggers NF-κB activation and cytokine secretion,21–24 promotes chemotaxis of neutrophils at the site of inflammation,25,26 induces apoptosis of numerous cell lines27 and has anti-microbial activity.28 Despite this progress, the possible pro-inflammatory effects of S100A9 itself remain elusive.
In this work, we set out to investigate possible pro-inflammatory effects of human and mouse S100A9 on monocytes. More specifically, we have compared the activities of S100A9 and LPS to determine whether PAMP and DAMP molecules would induce distinct responses in target cells.
Materials and methods
Cell culture
The human monocytic leukaemia cell line THP-1 (purchased from American Type Culture Collection, Manassas, VA) was grown in RPMI-1640 culture medium (Invitrogen, Stockholm, Sweden) supplemented with 10% fetal bovine serum (Invitrogen), 2 mm glutamine (Sigma-Aldrich, St Louis, MO), 1 mm sodium pyruvate, 10 mm HEPES, 100 U/ml penicillin and 100 μg/ml streptomycin (P/S; Invitrogen), at 37° in 5% CO2. All the experiments were performed with a cell density of 0·2 × 106 in 96-well plates or 1 × 106 in 24-well plates.
Bone-marrow-derived dendritic cells (BM-DC) were obtained from bone marrow cells of 15- to 20-week-old mice. Bone marrow cells were withdrawn from the femurs and tibias of the mice and cultured for 7 days in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 1 mm sodium pyruvate, 10 mm HEPES and 10% supernatant collected from granulocyte–macrophage colony-stimulating factor gene transfected J558L cell line. The purity of the BM-DC population was assessed by flow cytometry after CD11c labelling.
Mice
Fifteen- to 20-week-old C57BL/6 wild-type and C57BL/6 TLR4 knockout (KO) mice (both bought from TACONIC, Hudson, NY) and C57BL/6 RAGE-KO mice (produced in the laboratory of J. Roth) were used for the experiments. The mice were kept in the animal facility at the Biomedical Centre at Lund University. The experiments were approved by the local ethics committee for use of animals in research.
Endotoxin-free S100A9 protein purification
BL21 (DE3)/pET1120 Escherichia coli cells were treated with isopropyl-β-d-1-thiogalactopyranoside for some hours at 37° to induce h-S100A9 expression. Cells were collected and lysed with lysis buffer (50 mm Tris–HCl, 1 mm EDTA, 25% saccharose, pH 8·0) and sonicated three times for 15 seconds each time. Thereafter, 10 mm MgCl2, 1 mm MnCl2, 10 μg/ml DNaseI were added and lysed cells were incubated for 30 min at room temperature. Then, 20 mm Tris–HCl, pH 7·5, 2 mm EDTA, 1% Nonidet P-40 were added to the solution together with a protease inhibitor tablet (Roche, Mannheim, Germany). The solution was centrifuged, the pellet was resuspended in 0·5% Triton X-100, 1 mm EDTA and sonicated three times for 15 seconds each time. The last centrifugation–sonication step was repeated five times. The final pellet was resuspended in 8 m urea, 40 mm DTT, 500 mm NaH2PO4 pH 1·8 and centrifuged at 10 000 g for 25 min at 4°. Subsequently, five different dialyses were performed on the supernatant as follow: (i) 5 l 50 mm NaH2PO4 buffer, 1·5 mm DTT, pH 2 for 6 hr; (ii) 5 l 10 mm sodium acetate buffer, 150 mm NaCl, 1·5 mm DTT, pH 4 for 15 hr; (iii) 5 l 10 mm sodium acetate buffer, 150 mm NaCl, 1·5 mm DTT, pH 4 for 8 hr; (iv) 5 l 10 mm sodium acetate buffer, 150 mm NaCl, 1·5 mm DTT, pH 4 for 8 hr; 5) 5 l 20 mm Tris–HCl, 1 mm EDTA, 1 mm EGTA, 1·5 mm DTT, pH 8·5 for 6 hr. The last dialysis was centrifuged and the pellet was stored at −20°. The DTT was added to the supernatant to a final concentration of 1·5 mm. Anion-exchange chromatography on a HiPrep Q FF 16/10 column was run at a flow-rate of 1·5 ml/min using a 0–1 m NaCl gradient in 20 mm Tris–HCl, 1 mm EDTA, 1 mm EGTA, 1·5 mm DTT, pH 8·5 for elution of proteins. The same buffer, without NaCl, was used for equilibration and washing before elution. The pooled fractions containing h-S100A9 were concentrated to 1·5 ml using Centriprep YM-3 (Millipore, Solna, Sweden). All chromatography columns and resins were purchased from GE HealthCare, Uppsala, Sweden, and run on an ÄKTA explorer 100 (GE HealthCare). The size-exclusion chromatography on a Superdex 75 16/790 column was run at a flow-rate of 0·5 ml/min using an HBS-N buffer, 10 mm HEPES, 150 mm NaCl, pH 7·4 supplemented with 10 mm DTT. Fractions containing h-S100A9 were pooled and concentrated to approximately 1 ml in Centriprep YM-3. A PD-10 was run for buffer exchange to 10 mm HEPES, 150 mm NaCl, pH 7·5. The same purification procedure was used for mouse S100A9.
Removal of endotoxins was achieved by a Detoxi-Gel endotoxin removing gel. Detoxi-Gel endotoxin removing gel was regenerated in 5 ml 1% sodium deoxycholate in sterilized water and washed with 5 ml ready-made Biacore buffer (10 mm HEPES, 150 mm NaCl, pH 7·5) before the concentrated h-S100A9 sample was added. The h-S100A9 protein was eluted, after 10 min holding time, using the same buffer and gravity-flow and was collected in 0·5-ml fractions. Protein concentration was determined and the positive fractions were collected and stored at −80°. Endotoxin content was tested using LAL Chromogenic Endpoint Assay (Hycult Biotechnology, Uden, the Netherlands).
Measurement of NF-κB activity
THP-1 XBlue cells (InvivoGen, San Diego, CA) are THP-1 cells stably transfected with a reporter construct, expressing a secreted embryonic alkaline phosphatase (SEAP) gene under the control of a promoter inducible by the transcription factors NF-κB and activator protein-1. Upon TLR stimulation, NF-κB and activator protein-1 are activated and subsequently the secretion of SEAP is promoted. THP-1 XBlue cells were stimulated with LPS 100 ng/ml or h-S100A9 20 μg/ml for 4 hr or 48 hr at 37°. Levels of SEAP were detected spectrophotometrically (optical density at 650 nm; SpectraMax340pc; Molecular Devices, Sunnyvale, CA) after 4 hr incubation of supernatants with Quanti-Blue medium (InvivoGen, Vienna, Austria). In some experiments, THP-1 XBlue cells were treated with 50 μg/ml polymyxin B together with S100A9 or LPS at the concentration stated above.
Cytokine secretion
Cytokine concentration in culture supernatants was determined using a Human and Mouse inflammatory cytokine CBA kit (BD Bioscience, San Jose, CA) for simultaneous detection of six cytokines in human THP-1 cells [interleukin-1β (IL-1β), IL-6, IL-8, IL-10, IL-12p70, tumour necrosis factor-α (TNF-α)] and three cytokines in mouse BM-DC (IL-6, IL-1β, TNF-α) according to the manufacturer's instructions. Data were acquired with a BD FACS LSRII flow cytometer (BD Bioscience).
In some experiments THP-1 cells were pre-incubated with proper inhibitors for 30 min at 37° and thereafter stimulated as indicated. Measurements of TNF-α secretion were performed as described above. The following inhibitors were purchased from Merck (Darmstadt, Germany) and used at the indicated concentrations: 10 μm BAY11-7082, 1 μm SB203580, 5 μm MG132, 5 μm PD98059, 10 μm chloroquine. The final concentration of DMSO present in the cultures was < 0·05% of the total culture volume for each inhibitor.
NO measurement
Supernatants were collected, and nitrite content was determined as follows: cell culture supernatants or sodium nitrite standards (0–100 nm) were mixed with an equal volume of freshly prepared Griess reagent (a mixture of 0·1% (weight/volume) N-(1-naphthyl)-ethylenediamine dihydrochloride and 1% (weight/volume) sulphanilamide in 5% (volume/volume) phosphoric acid). After 30 min incubation at room temperature, the absorbance at 550 nm was measured using a plate reader (Spectramax340pc; Molecular Devices).
Western blot
Cells were collected and cytoplasmic/nuclear extracts were isolated as follow: cells were washed twice in TBS (50 mm Tris–HCl pH 7·4, 150 mm NaCl) and incubated for 15 min on ice with buffer A (10 mm HEPES pH 7·9, 10 mm KCl, 0·1 mm EDTA, 0·1 mm EGTA, 1 mm DTT, protease inhibitor cocktail Complete; Roche). Then, 1% NP-40 was added to each sample and the samples were centrifuged briefly. Supernatants were collected (cytoplasmic extract), the pellet was incubated in buffer C (20 mm HEPES pH 7·9, 400 mm NaCl, 1 mm EGTA, 1 mm EDTA, 1 mm DTT, protease inhibitor cocktail Complete), shaken vigorously for 15 min at 4° and thereafter briefly centrifuged. Supernatants were collected, divided into aliquots and stored at −80° (nuclear extract). For Western blot, 10 μg cytoplasmic extract/sample was loaded onto 12% polyacrylamide gels (C.B.S. Scientific, San Diego, CA). Proteins were subsequently transferred to PVDF membrane (Roche), which was saturated with 1% dry milk in PBS. Thereafter, the membranes were incubated with the appropriate primary antibody and secondary antibodies and filters were finally developed using an enhanced chemiluminescence kit (GE Healthcare, Uppsala, Sweden). The primary antibodies used for the Western blot were the following: IκBα, IKK-α/β and p50/p105 (Santa Cruz, Heidelberg, Germany).
Alternatively, whole cell extract was collected and incubated with lysis buffer (50 mutes Tris–HCl pH 6·8, 2% SDS, 5% glycerol, 1% 2-mercaptoethenol, Complete protease inhibitor cocktail from Roche) for 30 min at room temperature. 10 μg of proteins/sample were used to perform Western blot for h-S100A9 detection as described above (1C10 anti-human S100A9 antibody diluted 1 : 1000 was purchased from Novus Biologicals Inc., Cambridge, UK).
TransAM NF-κB transcription factor ELISA kit
Nuclear extracts were isolated as described above. The assay was performed following the manufacturer's instructions. The optical density at 650 nm was determined using a SPECTROSTARnano plate reader (BMG Labtech, Ortenburg, Germany).
Surface biotinylation and A488-labelled h-S100A9 internalization
A488-labelled h-S100A9 was incubated for 30 min at 37° with THP-1 cells. Thereafter, the cell surfaces were biotinylated using an EZ-Link Sulfo-NHS-LC-LC-Biotin kit (Pierce, Rockford, IL) following the instructions of the manufacturer. At the end of the incubation, biotinylated plasma membranes were isolated from cytoplasm using streptavidin beads included in the kit and fluorescence (on a Gemini™ Spectra max Microplate Reader; Molecular Devices, Biberach an der Riss, Germany) of cytosolic and membrane fractions was measured (excitation 484 nm and emission 525 nm). In some experiments, THP-1 cells were pre-treated with 10 μm chloroquine for 30 min.
Statistical analysis
Statistical analysis was performed using Student's t-test or, when data were normalized as fold of control, using one-way analysis of variance test: *P < 0·05; **P < 0·01; ***P < 0·005.
Results
S100A9 stimulates NF-κB activity
Knowing that human S100A9 (h-S100A9) is a TRL4 ligand,44 we wanted to determine whether h-S100A9 could induce NF-κB activity similarly to LPS.9 For this purpose we stimulated CD14+ THP-1 XBlue cells for 48 hr with increasing concentrations of highly purified human recombinant S100A9 (1, 15 and 40 μg/ml). In these conditions, h-S100A9 stimulated NF-κB activity in a dose-dependent way, showing almost no effect at the lowest concentration (see Supplementary material, Fig. S1a). Based on the result of this assay, we decided to keep the human and mouse S100A9 concentrations at 20 μg/ml for future experiments (both proteins were provided by Active Biotech AB, Lund, Sweden). Then, we monitored the capacity of h-S100A9 and lipoprotein-free LPS capacity to stimulate NF-κB activity in CD14+ THP-1 XBlue cells in a time-dependent way. The results in Fig. 1 show that S100A9 induced NF-κB activity already after 4 hr as potently as 100 ng/ml LPS. After 48 hr, however, h-S100A9-stimulated cells showed almost no further increment in NF-κB activity, but LPS-stimulated cells had further increased NF-κB activity at 48 hr of stimulation (Fig. 1).
Figure 1.
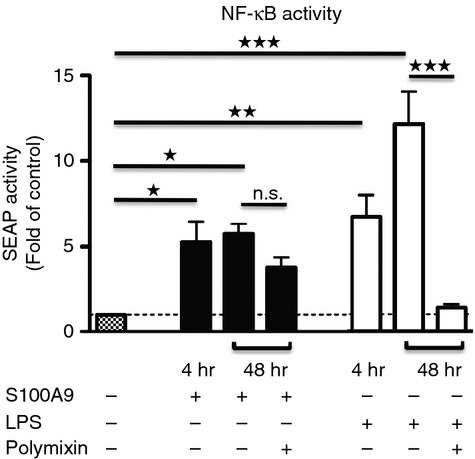
h-S100A9 protein induces nuclear factor- κB (NF- κB) activation in THP-1 cells. THP-1 XBlue cells were stimulated with 20 μg/ml h-S100A9 or 100 ng/ml ultra pure lipopolysaccharide (LPS) for 4 hr or 48 hr at 37°, in the presence or absence of 50 μg/ml polymyxin B. Relative expression levels of SEAP (reflecting NF-κB activity) in cell-free supernatants were determined spectrophotometrically at 650 nm. The data represent mean values of four independent experiments, were normalized against non-stimulated cells and analysed with one-way analysis of variance using Bonferroni post hoc test: *P < 0·05; **P < 0·01; ***P < 0·005.
During the past few years, emerging evidence showed that at least part of the effects claimed for S100A9 protein might have been influenced by LPS contamination.29,30 To avoid this problem, we ensured that highly purified recombinant human and mouse S100A9 protein was used. To confirm that the h-S100A9 protein was LPS free, we stimulated THP-1 XBlue cells with h-S100A9 or LPS in the presence of 50 μg/ml polymyxin B. After 48 hr of incubation, the h-S100A9 effect was only slightly inhibited by polymyxin B, whereas the LPS effect was completely absent (Fig. 1). The partial inhibition of the h-S100A9 effect by polymyxin B might be the result of an effect of polymyxin B on cell signalling. To address this possibility, we incubated THP-1 XBlue cells with 1 ng/ml TNF-α with or without polymyxin B and also here, we found a slight reduction of NF-&kgr;B activation (see Supplementary material, Fig. S1c), indicating that part of the inhibition mediated by polymyxin B might not be the result of LPS contaminants. Furthermore, we also treated h-S100A9 and LPS at 80° for 30 min and observed that the h-S100A9 effect was almost completely abolished, whereas the LPS effect remained intact (see Supplementary material, Fig. S1b). From these results we could conclude that h-S100A9 induced NF-κB activity directly.
S100A9 and LPS differentially stimulate cytokine and NO secretion in THP-1 cells and mouse BM-DC
We next investigated whether h-S100A9 would induce a similar cytokine response as did LPS. After 4 hr stimulation, both molecules induced elevated secretion of IL-1β, TNF-α, IL-6, IL-8 and IL-10. However, despite the similar NF-κB activation after 4 hr of stimulation, h-S100A9 induced less potent cytokine secretion than LPS. After 48 hr of stimulation, LPS was still able to stimulate IL-6, IL-8 and above all IL-1β secretion, whereas h-S100A9 stimulation only induced secretion of IL-8 at this time-point (Fig. 2).
Figure 2.
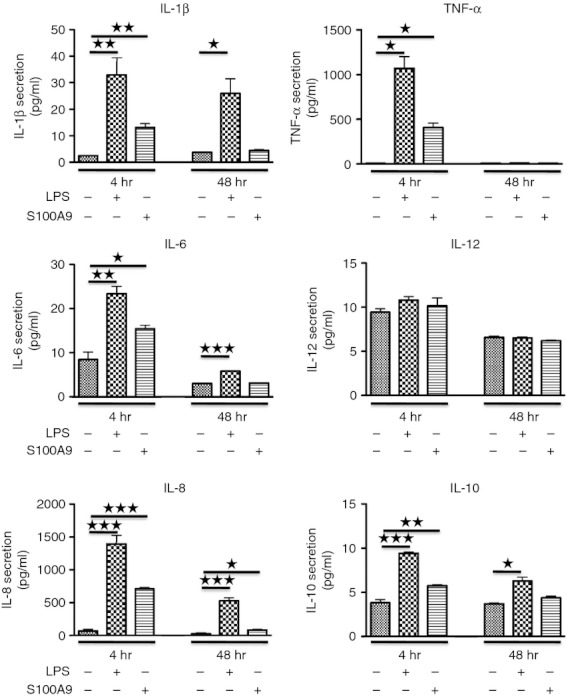
Lipopolysaccharide (LPS) and h-S100A9 induce distinct cytokine responses in THP-1 cells. THP-1 cells were stimulated either with 100 ng/ml LPS or 20 μg/ml h-S100A9 and cytokine secretion was analysed after 4 and 48 hr stimulation. Data of three independent experiments made in duplicate were compiled and analysed using Student's t-test: *P < 0·05; **P < 0·01; ***P < 0·005.
We confirmed our findings using mouse BM-DC stimulated with murine S100A9 (m-S100A9) or LPS under the same conditions as human THP-1 cells (Fig. 3a). Mouse BM-DC are considered a good model of mouse monocytes48 and the name ‘dendritic cells’ refers mainly to their shape, which resembles dendritic cells. In this model, we chose to study cytokine secretion after 48 hr stimulation when the cytokine concentration reached the plateau even if we could observe cytokine secretion already at 4 hr (data not shown). Once again, the m-S100A9 effect was less potent than LPS. The BM-DC remained a heterogeneous population of granulocytes, macrophages and DC even after the incubation with granulocyte–macrophage colony-stimulating factor for 7 days. Hence, we confirmed our findings with isolated CD11c+ BM-DC.
Figure 3.
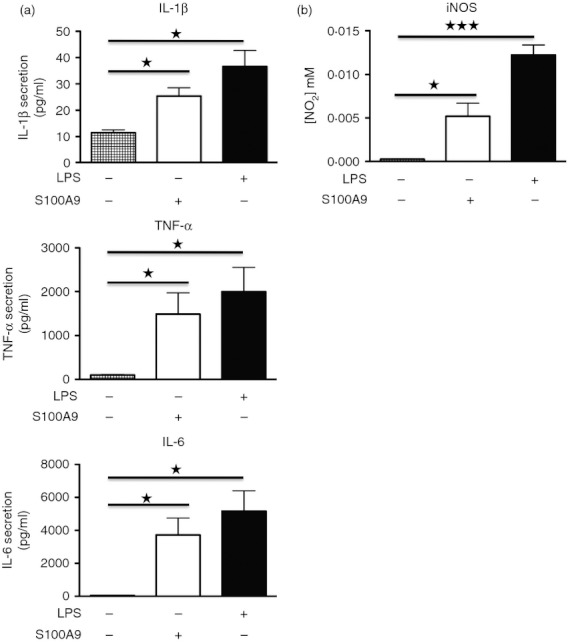
Lipopolysaccharide (LPS) and m-S100A9 induce cytokine and nitric oxide (NO) secretion in bone marrow-derived dendritic cells (BM-DC). The BM-DC were stimulated with or without 100 ng/ml LPS or 20 μg/ml m-S100A9 protein. Supernatants were collected after 48 hr and cytokine concentration (a) and NO (b) were determined. Data of three independent experiments made in duplicate were compiled and analysed using Student's t-test: *P < 0·05; ***P < 0·005.
To further confirm the pro-inflammatory activity of S100A9, we analysed whether m-S100A9 was able to trigger inducible NO synthase activity in mouse BM-DC. As shown in Fig. 3(b) both m-S100A9 and LPS stimulated NO production, again with LPS as the more potent inducer. These results further supported the pro-inflammatory activity of S100A9.
S100A9 and LPS activate NF-κB through the same signalling pathway
Our next step was to determine whether h-S100A9 would exert its effects on NF-κB activation through the same or a different signalling pathway than LPS. Hence, we pre-incubated THP-1 cells with selected inhibitors to block key steps in the main pathway involved in NF-κB activation and then stimulated the cells and measured TNF-α secretion. Figure 4 shows that BAY11-7082, which reduces IκBα phosphorylation,31 effectively blocked both the LPS-induced and h-S100A9-induced response. Further, PD98059 and SB203580, which are inhibitors of MEK133 and p38,32 respectively, strongly inhibited the TNF-α response triggered both by LPS and h-S100A9, suggesting that mitogen-activated protein kinase proteins were involved both in the LPS and h-S100A9-induced signalling pathways. The inhibitor of proteasome activity MG132,34 which blocks IκBα degradation, inhibited TNF-α responses almost completely, suggesting that IκBα could be involved in the h-S100A9 signalling pathway. For all the inhibitors tested, we could observe more than 50% inhibition of LPS-mediated and h-S100A9-mediated TNF-α secretion. The above-mentioned inhibitors did not significantly affect cell viability (see Supplementary material, Fig. S2a).
Figure 4.
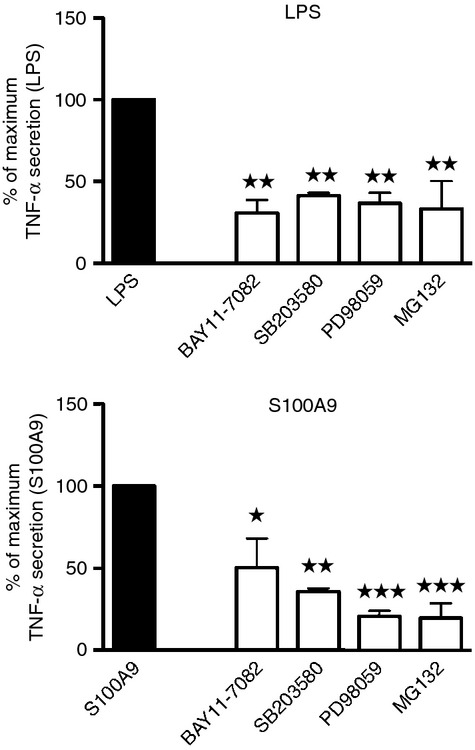
Similar sensitivity of h-S100A9 and lipopolysaccharide (LPS) induced tumour necrosis factor-α (TNF-α) response to nuclear factor-κB (NF-κB) pathway inhibitors. THP-1 were pre-treated with specific inhibitors for 30 min and subsequently stimulated by LPS or h-S100A9 for 4 hr. Data were normalized as percentage of TNF-α triggered by either LPS or h-S100A9: 100% no inhibition; 0 background level. Data of three independent experiments made in duplicate were compiled and analysed with one-way analysis of varaince using Bonferroni post hoc test: *P < 0·05; **P < 0·01; ***P < 0·005.
Taken together, these data indicate that LPS and h-S100A9 exerted their pro-inflammatory effects through basically the same signalling pathway to activate NF-κB.
S100A9 and LPS activate NF-κB in qualitatively different ways
To further confirm the activation of NF-κB by human and mouse S100A9, we monitored IκBα degradation. IαBκ is activated via phosphorylation by IKK proteins upon proper cellular stimulation. In this way, IκBα is targeted for proteasomal degradation and NF-κB subunits are able to interact and form the mature NF-κB dimers.35
As human S100A9 was less potent than LPS in promoting cytokine secretion, we expected to find that h-S100A9 provoked a weaker IκBα degradation. Surprisingly, Western blot analysis revealed the opposite. Hence, h-S100A9-mediated stimulation of THP-1 XBlue cells effectively reduced the IκBα level already after 15 min and it remained reduced for up to 60 min after stimulation. The LPS-induced degradation was significant only at 60 min of stimulation and in this case there was only a slight IκBα degradation (Fig. 5a).
Figure 5.
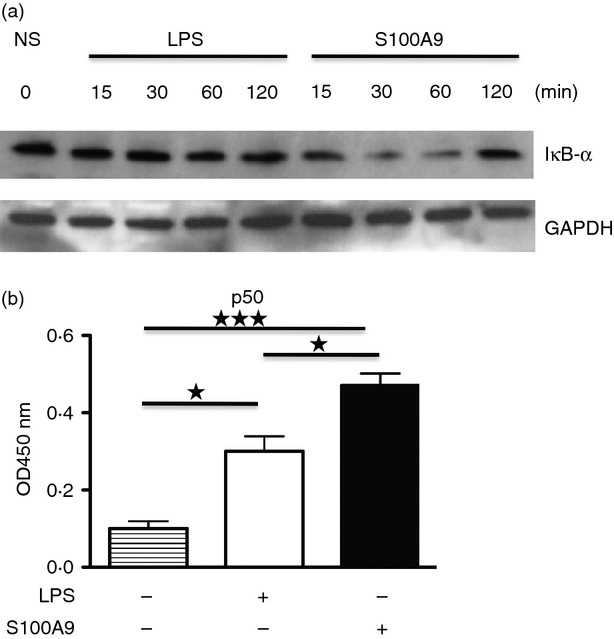
h-S100A9 and lipopolysaccharide (LPS) induces nuclear factor-κB (NF-κB) protein activation in a qualitatively different way. THP-1 Xblue were treated with 20 μg/ml h-S100A9 or 100 ng/ml LPS for 15, 30, 60 and 120 min. Subsequently, cells were collected and cytoplasmic/nuclear extracts were isolated from each sample. Ten micrograms of cytoplasmic extract was used for Western blot and detection of IκBα protein. GAPDH was used as control (a). Three micrograms of nuclear extract was used to perform TransAM transcription factor ELISA for NF-κB binding. The figure shows data from three independent experiments (b). *P < 0·05; ***P < 0·005.
These results further confirmed that h-S100A9 activated the NF-κB transcription factor. Most importantly, the kinetics of the h-S100A9-induced NF-κB activation was more rapid, even though it led to a weaker cytokine response. In contrast, LPS provoked delayed and weaker NF-κB activation but a more potent and sustained cytokine response. These results were in agreement with the pro-inflammatory role of h-S100A9 but in apparent contrast with Fig. 1, which showed that h-S100A9 promoted NF-κB activity in a comparable way to LPS.
To explain this apparent discrepancy, we incubated THP-1 XBlue cells for 2 hr in the presence of h-S100A9 or LPS, we isolated nuclear extracts and the content of NF-κB subunit p50 was determined using a commercially available ELISA kit. In this way, we could show that NF-κB dimers induced by h-S100A9 contained more of the p50 NF-κB isoform, suggesting different NF-κB isoform formation in cells stimulated by h-S100A9 and LPS, respectively (Fig. 5b).
In summary from these data we can conclude that h-S100A9 and LPS exerted their pro-inflammatory effects in a qualitatively different way. We suggest that this may be a result of the formation of different NF-κB isoforms in the stimulated cells.
TLR4 is essential for S100A9-induced NFκB activation, while it is only partially RAGE-dependent
We wanted to determine which cell-surface receptors might contribute to the m-S100A9-induced response. Previous reports have indicated that S100A9 could interact both with RAGE23,36–38 and TLR4.30 To determine whether m-S100A9 would induce cytokine responses via these receptors, we prepared BM-DC from TLR4-KO and RAGE-KO mice and stimulated them with either m-S100A9 or LPS. As shown in Fig. 6(a), the secretion of TNF-α, IL-6 and IL-1β triggered by LPS and by m-S100A9 was completely absent in TLR4-KO BM-DC, whereas IL-1β (> 50%) but not TNF-α secretion was inhibited in RAGE-KO BM-DC. We also observed a weak inhibition of IL-6 secretion in RAGE-KO BM-DC stimulated with both m-S100A9 and LPS.
Figure 6.
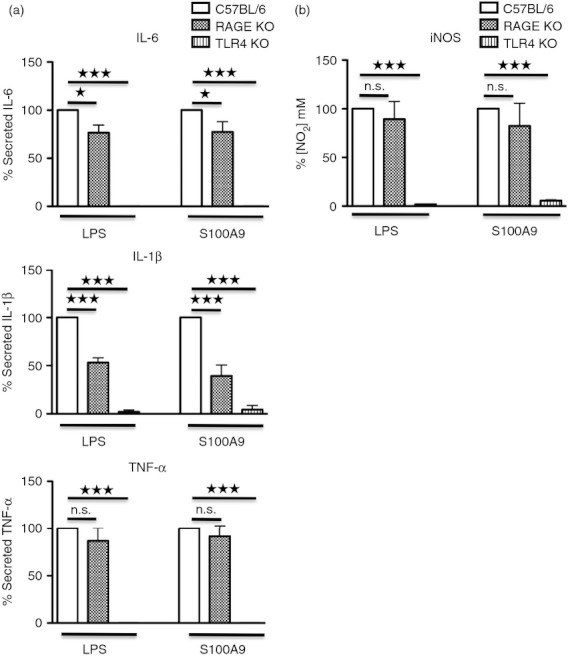
m-S100A9- and lipopolysaccharide (LPS) -induced responses are Toll-like receptor 4 (TLR4) -dependent. Bone marrow-derived dendritic cells (BM-DC) from C57BL/6, TLR4-knockout (KO) and RAGE-KO mice were stimulated with 100 ng/ml LPS or 20 μg/ml m-S100A9 for 48 hr. Secretion of cytokines (a) and nitric oxide (NO) (b) was determined in culture supernatants. Data of three independent experiments were compiled and analysed using Student's t-test: *P < 0·05; ***P < 0·005.
Taken together, these data suggest that m-S100A9 was able to interact with both RAGE and TLR4 receptors. Most importantly, whereas TLR4 seems to be crucial for the induction of all cytokines, RAGE was involved mainly in IL-1β secretion. This result was further confirmed by analysing NO secretion in TLR4-KO and RAGE-KO BM-DC. The NO secretion triggered by m-S100A9 completely disappeared in TLR4-KO BM-DC, but it was not affected in RAGE-KO BM-DC (Fig. 6b).
S100A9 needs to be internalized to signal
It is well established that TLR4 can be internalized in cells upon triggering. The TRIF (TIR-domain-containing adapter-inducing interferon-β)-mediated type-1 interferon stimulation via TLR4-stimulation involves receptor internalization. Recent results also suggested the possibility that even the MyD88-dependent pathway might need TLR4 internalization.39–41 To test whether h-S100A9-mediated stimulation would involve receptor internalization, we tried to inhibit endosomal signalling using chloroquine. This molecule is a weak base, blocking endosome maturation42 and clathrin-mediated internalization.43 Secretion of TNF-α measured after pre-treatment of THP-1 with 10 μm chloroquine was significantly reduced in h-S100A9-stimulated cells but not in LPS-stimulated cells (Fig. 7a).
Figure 7.
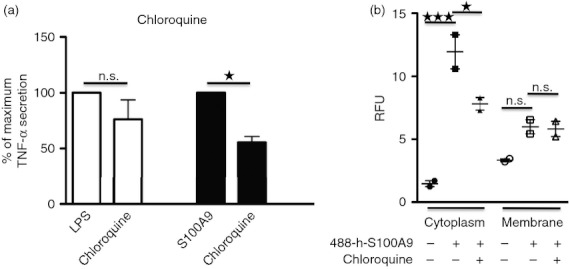
h-S100A9 induced tumour necrosis factor-α (TNF-α) response requires endosomal activity and protein internalization into the cell. (a) THP-1 cells were pre-treated for 30 min with 10 μm chloroquine and subsequently stimulated by 100 ng/ml lipopolysaccharide (LPS) or 20 μg/ml h-S100A9 for 4 hr. Data were normalized as percentage of TNF-α triggered by LPS or h-S100A9: 100% no inhibition at all; 0 background level. The figure shows compiled data of three independent experiments made in duplicate. (b) THP-1 cells were stimulated for 30 min with 488-labelled h-S100A9, with or without chloroquine to inhibit internalization. Thereafter, cell membranes were labelled with biotin, separated from the cytoplasm and the fractions were analysed for fluorescence at excitation 484 nm and emission 525 nm. The figure shows data from three independent experiments. Data were analysed with one-way analysis of variance using Bonferroni post hoc test: *P < 0·05; ***P < 0·005.
These data suggested that h-S100A9-induced triggering, but not LPS-induced triggering, may need receptor internalization to promote cytokine secretion. To corroborate our previous finding, we incubated A488-labelled h-S100A9 for 30 min at 37° with THP-1 cells. Then, after surface biotinylation, we isolated plasma membrane from cytoplasm using streptavidin-conjugated beads and measured the fluorescence of the different fractions.
We observed that A488-labelled h-S100A9 treatment produced an increment of fluorescence in the cytosolic fraction, which was significantly reduced upon chloroquine pre-treatment. To prevent any artefacts caused by h-S100A9 non-specific binding on the cell surface, we measured fluorescence also for the plasma membrane fraction and found only a small increase of fluorescence value, confirming the specificity of the assay.
Discussion
In this study we have investigated the pro-inflammatory effect of murine and human S100A9 protein. Our data show that S100A9 and LPS activated NF-κB and promoted cytokine secretion in qualitatively different ways. However, there were only minor differences between S100A9 and LPS signals regarding induction of the NF-κB signalling pathway.
For this work, it was important to use pure and controlled human and mouse S100A9 and LPS as previous studies have shown that LPS or lipoprotein contaminants could affect the results of the experiments.29,49 As both murine and human S100A9 was purified from bacteria, the proteins must be purified using protocols, which minimize the presence of LPS contaminants. To avoid this problem we used tested LPS-free S100A9 batches in which the highest amount of possible LPS contamination was below 0·1 EU/ml. However, to further confirm the successful removal of LPS contaminants, we added polymyxin B to h-S100A9-stimulated cultures. Under these conditions, we could observe a minor inhibition of the h-S100A9 effect, whereas the LPS response was completely blocked. The inhibition of the h-S100A9 effect could be a result of the polymyxin non-specific effect during the 48 hr incubation because stimulation with 1 ng/ml TNF-α was also slightly inhibited (see Supplementary material, Fig. S1c). The almost complete loss of biological activity after heat-denaturation of h-S100A9 at 80°, compared with the LPS response which was insensitive to heating, provided further evidence that the biological activity of h-S100A9 was not the result of LPS contamination. We used this protocol of heat inactivation because Tsan et al.29 have shown that using heat inactivation at boiling temperatures can also inactivate LPS activity. In addition, because m-S100A9-induced cytokine secretion was abolished in TLR4-KO BM-DC, lipoprotein contamination of the m-S100A9 preparations was unlikely.
Concerning the TLR4 ligand LPS, it was important to exclude lipoprotein contamination, which could potentially activate the TLR2 pathway. In this case, we titrated the activity of a highly purified preparation of lipoprotein-free LPS (InvivoGen) and could observe the following: (i) LPS could induce NF-κB activity showing a plateau at 100 ng/ml (data not showed); (ii) LPS-mediated IκBα degradation was weak (Fig. 5) even at 1 μg/ml (data not showed); (iii) we confirmed that LPS preparation was completely devoid of cytokine-inducing activity in TLR4-KO BM-DC. Importantly, the regular LPS behaved differently, provoking an increment in NF-κB activity without reaching any plateau even at 10 μg/ml. Further, regular LPS provoked a marked IκBα degradation and showed a residual effect in TNF-α secretion from TLR4-KO BM-DC (data not shown).
Using these controlled conditions, we wanted to investigate the role of S100A9 in inflammation. Although the S100A9 effect in association with S100A8 is well characterized,21–26,30 to our knowledge very few reports have focused on the role of S100A9 itself in the inflammation process. Vogl et al.30 showed that S100A8, but not S100A9, was able to stimulate TNF-α secretion from bone marrow cells. In that study the appropriate controls needed to exclude LPS contamination were performed. The apparent discrepancy with our data could be a result of different S100A9 concentrations used in the experiments. Indeed, we titrated h-S100A9 effect in THP-1 XBlue cells for NF-κB activation and we noted that too low h-S100A9 protein concentration (1 μg/ml) had no effect at all, but higher concentrations showed a dose-dependent NF-κB stimulation (see Supplementary material, Fig. S1a).
As it has been demonstrated that S100A9 is a ligand for TLR430 and RAGE,36–38,45 we wanted to investigate whether S100A9-mediated NF-κB stimulation was dependent on both of these receptors. Cytokine secretion was completely absent in m-S100A9-stimulated BM-DC from TLR4-KO mice, proving that TLR4 was essential for the stimulatory activity of m-S100A9. In RAGE-KO mice, instead, there was reduction primarily of IL-1β secretion in both m-S100A9-stimulated cells and LPS-stimulated cells, indicating that RAGE contributed only partially to the m-S100A9-induced and LPS-induced cytokine response. These findings suggest that the main pathways activated by m-S100A9 and LPS might be the same. Furthermore, only BM-DC derived from TLR4-KO mice showed a complete absence of NO secretion. RAGE-KO-derived BM-DC NO secretion was not affected.
Finally, we investigated the signalling pathways promoting NF-κB activation and cytokine secretion in S100A9-activated and LPS-activated cells. We focused on two main pathways that promote NF-κB activation: IκBa-mediated pathway or mitogen-activated protein kinase-mediated pathway. In the IκBa-mediated pathway, IKK proteins are phosphorylated upon interaction between the proper ligand and its receptor. This event leads to IκB phosphorylation and degradation, provoking the release of NF-κB subunits, which are free to interact, forming dimers, entering the nucleus, binding to DNA and promoting transcription of target genes.35 Ulivi et al.46 demonstrated also that NF-κB could be activated by the MEK kinase cascade and hence p38, which was located upstream of NF-κB. We found that both h-S100A9 and LPS pro-inflammatory effects were dependent on both pathways and the potency of the inhibition was equal for both molecules.
Among all the inhibitors tested, it was intriguing to observe that pre-treatment of THP-1 cells with chloroquine significantly inhibited h-S100A9 but not LPS signalling. Chloroquine prevents endosomal acidification and hence can block signalling deriving from receptors that transmit signals from this cellular compartment.47 This result indicated that h-S100A9-induced but not LPS-induced signalling may need internalization of TLR4 into the endosomal compartment. This consideration raised the possibility that h-S100A9 could exert its effect also via receptors other than TLR4, such as TLR7 or TLR9, which are located in endosomes. Interestingly, it has previously been shown that chloroquine could inhibit LPS-mediated TNF-α expression.47 However, this inhibition occurred at 100 μm chloroquine. In our experiments we used only 10 μm chloroquine, which was shown to be ineffective for the LPS-induced response.47
It has been shown that chloroquine is an inhibitor of clathrin-dependent endocytosis.43 To test this hypothesis on h-S100A9 and to further validate our previous finding, we incubated A488-labelled h-S100A9 with THP-1 for 30 min at 37°, followed by cell surface biotinylation and separation of plasma membrane from cytosolic fraction and measured the fluorescence in the different fractions. Upon A488-labelled h-S100A9 incubation with THP-1, we could observe a consistent increased fluorescence in the cytosolic fraction, which was reduced upon chloroquine pre-treatment. As the plasma membrane fraction showed a marginal fluorescence increment upon A488-labelled h-S100A9 incubation, we are confident that the assay performed was specific. Lastly, we tested if A488-labelled h-S100A9 was still able to stimulate NF-κB activity, when no change in protein behaviour and structure had occurred. We therefore performed an NF-κB assay incubating A488-labelled h-S100A9 protein with THP-1 XBlue cells as described in the Materials and methods, and found the same NF-κB stimulation activity as previously observed for the unlabelled h-S100A9 (data not shown), arguing that A488 labelling did not affect the function, and hence the structure, of h-S100A9 protein.
In summary, our work demonstrated a pro-inflammatory role of the human and mouse S100A9 protein. Furthermore, by comparing the pro-inflammatory effects of S100A9 and LPS, we noticed that, even if h-S100A9 could trigger NF-κB activation more rapidly, earlier and more strongly than LPS, the following cytokine response was weaker in potency and duration. Hence, subtle differences between DAMP and PAMP activation of the same receptor can be detected and may result in distinct host responses.
Acknowledgments
This work was supported by grants from the Swedish Research Council, The Swedish Cancer Foundation, Greta och Johan Kocks Stiftelser and Alfred Österlunds Stiftelse.
Disclosures
TL is a part time employee and PB full time employees of Active Biotech that develops S100A9 inhibitors for the treatment of autoimmune diseases and cancer. FI has a research grant from Active Biotech.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. THP-1 XBlue cells were stimulated as follow: (A) increasing concentration of S100A9 (1 μg/ml, 15 μg/ml, 40 μg/ml) for 48h at 37°C (n = 2). (B) 20 μg/ml S100A9 or 100 ng/ml ultra pure LPS for 48h with or without heat pre-treatment made at 80°C for 30 minutes (n = 2). (C) 1 ng/ml TNFα or 100 ng/ml ultra-pure LPS for 48h (n = 2). Relative expression levels of SEAP in cell-free supernatants were determined spectrophotometrically at 650 nm. Data were normalized using one-way ANOVA test: *P < 0·05; **P < 0·01; ***P < 0·005.
Figure S2. THP-1 cells were stimulated using 20 μg/ml S100A9 or 100 ng/ml ultra pure LPS for 4h or 48h with (A) or without (B) inhibitors pre-treatment for 30 minutes. Cells were collected and Trypan Blue assay was performed to count viable cells. The figure represents one of at least two experiments, in which every sample is repeated 6 times.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Immunol. 2007;7:429–42. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 3.Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358:929–39. doi: 10.1056/NEJMra071297. [DOI] [PubMed] [Google Scholar]
- 4.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros. 2005;4:3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Dorin JR, Novak M, Hill RE, Brock DJ, Secher DS, van Heyningen V. A clue to the basic defect in cystic fibrosis from cloning the CF antigen gene. Nature. 1987;326:614–7. doi: 10.1038/326614a0. [DOI] [PubMed] [Google Scholar]
- 6.Bachmann MF, Kopf M. On the role of the innate immunity in autoimmune disease. J Exp Med. 2001;193:F47–50. doi: 10.1084/jem.193.12.f47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barton GM, Kagan JC. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nat Rev Immunol. 2009;9:535–42. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. 2008;118:413–20. doi: 10.1172/JCI34431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant CE, Spring DR, Gangloff M, Gay NJ. The molecular basis of the host response to lipopolysaccharide. Nature. 2010;8:8–14. doi: 10.1038/nrmicro2266. [DOI] [PubMed] [Google Scholar]
- 10.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2006;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 11.Oppenheim JJ, Yang D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005;17:359–65. doi: 10.1016/j.coi.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8, 14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266:7706–13. [PubMed] [Google Scholar]
- 13.Foell D, Frosch M, Sorg C, Roth J. Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta. 2004;344:37–51. doi: 10.1016/j.cccn.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 14.Hermani A, Hess J, De Servi B, Medunjanin S, Grobholz R, Trojan L, Angel P, Mayer D. Calcium-binding proteins S100A8 and S100A9 as novel diagnostic markers in human prostate cancer. Clin Cancer Res. 2005;11:5146–52. doi: 10.1158/1078-0432.CCR-05-0352. [DOI] [PubMed] [Google Scholar]
- 15.Sidler MA, Leach ST, Day AS. Fecal S100A12 and fecal calprotectin as noninvasive markers for inflammatory bowel disease in children. Inflamm Bowel Dis. 2008;14:359–366. doi: 10.1002/ibd.20336. [DOI] [PubMed] [Google Scholar]
- 16.Lagasse E, Clerc RG. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol. 1988;8:2402–10. doi: 10.1128/mcb.8.6.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002;7:d1356–68. doi: 10.2741/A846. [DOI] [PubMed] [Google Scholar]
- 18.Zwadlo G, Bruggen J, Gerhards G, Schlegel R, Sorg C. Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol. 1988;72:510–5. [PMC free article] [PubMed] [Google Scholar]
- 19.Hessian PA, Edgeworth J, Hogg N. MRP-8 and MRP-14, two abundant Ca2+-binding proteins of neutrophils and monocytes. J Leukoc Biol. 1993;53:197–204. [PubMed] [Google Scholar]
- 20.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem Biophys Res Commun. 2004;322:1111–22. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 21.Roth J, Vogl T, Sorg C, Sunderkotter C. Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 2003;24:155–8. doi: 10.1016/s1471-4906(03)00062-0. [DOI] [PubMed] [Google Scholar]
- 22.Benedyk M, Sopalla C, Nacken W, Bode G, Melkonyan H, Banfi B, Kerkhoff C. HaCat keratinocytes overexpressing the S100 proteins S100A8 and S100A9 show increased NADPH oxidase and NF-κB activities. J Invest Dermatol. 2007;127:2001–11. doi: 10.1038/sj.jid.5700820. [DOI] [PubMed] [Google Scholar]
- 23.Sunahori K, Yamamura M, Yamana J, et al. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther. 2006;8:1–12. doi: 10.1186/ar1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181:4666–75. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH. Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol. 2008;166:4678–88. doi: 10.4049/jimmunol.166.7.4678. [DOI] [PubMed] [Google Scholar]
- 26.Robinson M, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2001;277:3658–65. doi: 10.1074/jbc.M102950200. [DOI] [PubMed] [Google Scholar]
- 27.Ghavami S, Kerkhoff C, Los M, Hashemi M, Sorg C, Karami-Tehrani F. Mechanism of apoptosis induced by S1008/A9 in colon cancer cell lines: the role of ROS and the effect of metal ions. J Leukoc Biol. 2004;76:169–75. doi: 10.1189/jlb.0903435. [DOI] [PubMed] [Google Scholar]
- 28.Sohnle PG, Hunter MJ, Hahn B, Chazin WJ. Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14) J Infect Dis. 2000;4:1272–5. doi: 10.1086/315810. [DOI] [PubMed] [Google Scholar]
- 29.Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–9. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- 30.Vogl T, Tenbrock K, Ludwig S, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–9. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 31.Mori N, Yamada Y, Ikeda S, et al. Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-infected T-cell lines and primary adult T-cell leukemia cells. Blood. 2002;100:1828–34. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]
- 32.Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995;364:229–33. doi: 10.1016/0014-5793(95)00357-f. [DOI] [PubMed] [Google Scholar]
- 33.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–94. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 34.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/s0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh S, May MJ, Kopp EB. NF-κB AND REL PROTEINS: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- 36.Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D. S100A8 and S100A9 activate MAP kinase and NF-κB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res. 2006;312:184–97. doi: 10.1016/j.yexcr.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102:1239–46. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 38.Ghavami S, Rashedi I, Dattilo BM, et al. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol. 2008;83:1484–92. doi: 10.1189/jlb.0607397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol. 2008;9:361–8. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Husebye H, Aune MH, Stenvik J, et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity. 2010;33:583–96. doi: 10.1016/j.immuni.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Husebye H, Halaas Ø, Stenmark H, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25:683–92. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yasuda H, Leelahavanichkul A, Tsunoda S, et al. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am J Physiol Renal Physiol. 2008;294:F1050–8. doi: 10.1152/ajprenal.00461.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CL, Hou WH, Liu IH, Hsiao G, Huang SS, Huang JS. Inhibitors of clathrin-dependent endocytosis enhance TGFβ signaling and responses. J Cell Sci. 2009;122:1863–71. doi: 10.1242/jcs.038729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bjork P, Bjork A, Vogl T, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009;7:800–12. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 46.Ulivi V, Giannoni P, Gentili C, Cancedda R, Descalzi F. p38/NF-κB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J Cell Biochem. 2008;104:1393–406. doi: 10.1002/jcb.21717. [DOI] [PubMed] [Google Scholar]
- 47.Karres I, Kremer JP, Dietl I, Steckholzer U, Jochum M, Ertel W. Chloroquine inhibits proinflammatory cytokine release into human whole blood. Am J Physiol. 1998;274:R1058–64. doi: 10.1152/ajpregu.1998.274.4.R1058. [DOI] [PubMed] [Google Scholar]
- 48.Inaba K, Inaba M, Deguchi M, Hagi K, Yasumizu R, Ikehara S, Muramatsu S, Steinman RM. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc Natl Acad Sci USA. 1993;90:3038–42. doi: 10.1073/pnas.90.7.3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor α release by murine macrophages. J Biol Chem. 2002;278:174–9. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.