

Abstract
The phosphatidylinositol-3-kinase (PI3K)/AKT signaling pathway is critical for cellular growth and metabolism. Correspondingly, loss of function of PTEN, a negative regulator of PI3K, or activating mutations in AKT1, AKT2, or AKT3 have been found in distinct disorders featuring overgrowth or hypoglycemia. We performed exome sequencing of DNA from unaffected and affected cells of a patient with an unclassified syndrome of congenital, progressive segmental overgrowth of fibrous and adipose tissue and bone and identified the cancer-associated p.His1047Leu mutation in PIK3CA, which encodes the p110α catalytic subunit of PI3K, only in affected cells. Sequencing of PIK3CA in 10 further patients with overlapping syndromes identified either p.His1047Leu or a second cancer-associated mutation, p.His1047Arg, in 9 cases. Affected dermal fibroblasts showed enhanced basal and EGF-stimulated phosphatidylinositol-3,4,5-trisphosphate (PIP3) generation and concomitant activation of downstream signaling. Our findings characterize a distinct overgrowth syndrome, biochemically demonstrate activation of PI3K signaling and thereby identify a rational therapeutic target.
Mutations that compromise the regulation of cellular proliferation and death by reducing apoptosis, or by allowing constitutive cell division, cause pathological tissue growth. The most extreme example is cancer, where numerous activating mutations of genes in growth-promoting pathways have been documented. A commonly affected pathway is the receptor tyrosine kinase/phosphatidylinositol-3-kinase/AKT (RTK/PI3K/AKT) pathway, which harbors activating mutations in most solid tumors1.
Given the importance of RTK/PI3K/AKT for cell proliferation and metabolism, germline mutations upregulating the pathway would likely be lethal in embryonic life, and have not been reported. However, mosaic mutations can cause segmental, or patchy, overgrowth disorders. Germline loss-of-function mutations in PTEN, a negative regulator of PI3K signaling , cause overgrowth, and a somatic second hit in PTEN causes the Type II Segmental Cowden Syndrome2. Proteus syndrome, a progressively deforming regional overgrowth syndrome that affects bones, adipose, and other mesenchymal tissues, is caused by a somatic p.Glu17Lys AKT1 mutation3 which constitutively activates PI3K/AKT signaling3,4. These findings proved the hypothesis that segmental overgrowth disorders can be caused by somatic mosaicism for mutations5, and suggested that similar disorders may also be caused by somatic activation of the RTK/PI3K/AKT pathway. We describe here the clinical and molecular characterization of 10 such patients.
Ten patients with a previously uncharacterized progressive segmental overgrowth syndrome were evaluated. Of those for whom records were available, 7 of 8 had congenital overgrowth. Their major manifestation was segmental progressive overgrowth of subcutaneous, muscular, and visceral fibroadipose tissue with skeletal overgrowth (Figures 1 & 2). The overgrowth was predominantly adipose tissue, or a mixture of adipose and fibrous tissues. We have thus designated this as ‘fibroadipose’ tissue. In some affected areas, this tissue was solitary, but in others there was admixture of the fibroadipose tissue with muscle, or investiture within other tissues (e.g., spinal canal). The range of severity and the natural history among the patients was remarkable. Patient C1 had massive overgrowth of her body from the waist down, weighing 117 kg, with leg circumferences of 100-110 cm. Her total adiposity assessed by DXA was 50%, accounted for mostly by her legs. Her overgrowth continued into adulthood. In contrast, C2 had overgrowth limited to an arm and thumb and N110 had overgrowth limited to two rays of one foot (Figure 2 O-P). Patient N7 had an extensive lipoma of the left lower trunk, buttock and leg that necessitated amputation (Figure 1F-G). Patient N45 had fibroadipose overgrowth of the lower body including a pelvic mass that encased the rectum (Figure 2J) with attendant constipation. The overgrowth in this patient continued into adulthood. However, the overgrowth in patient N68 mostly ceased after puberty.
Figure 1. Spectrum of Overgrowth in Patients with Activating PIK3CA Mutations.
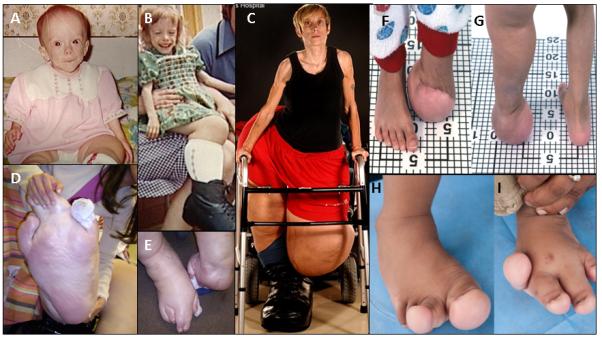
Patient C1 showing (A)&(B) Lower extremity overgrowth with a paucity of facial adipose tissue at ages 12 months and 7 years progressing to (C) Massive leg overgrowth with lack of upper body adipose tissue at 37 years old, by which time left above knee amputation had been undertaken. (D) Right foot overgrowth at 35 years old (E) Feet at 35 years showing overgrowth, cutaneous syndactyly of the left foot and rotational deformity of the right foot .(F) and (G). Patient N7 showing his leg status post debulking surgery and massive overgrowth of the left foot, which was partially amputated. (H) and (I) Patient N99 showing more limited overgrowth confined to several rays of the feet. Informed consent to publish photographs was obtained from the identifiable patient in this figure.
Figure 2. Segmental Skeletal and Fibroadipose Overgrowth in Patients with Activating PIK3CA Mutations.
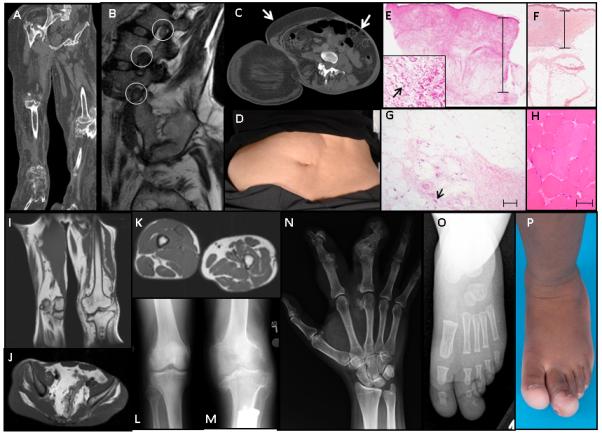
Patient C1 (A) Reformatted coronal CT of the legs, showing bony overgrowth, destructive arthropathy, adipose expansion and relative lack of muscle (B) Sagittal T2-weighted MRI images of lumbar spine depicting enlarged neural structures within the foramina (circled 12-13 mm diameter). Normal nerve roots in this region are up to 5-6 mm and up to 8-9 mm for the dorsal root ganglion31. (C) & (D) transverse CT image and photograph of anterior abdomen showing right-sided adipose overgrowth (arrow) and left-sided lack of adipose tissue (arrowhead) to the level of insertion of erector spinae. (E) Hematoxylin- and eosin-stained left foot skin showing marked dermal thickening (scale bar 15 μm). Inset - myofibroblasts with plump nuclei are prominent in the dermis (arrow) in contrast to normal skin(F). (G) Muscle is replaced by fibrous and adipose tissue with occasional residual muscle fibers (arrow) (scale bar 80 μm). (H) Normal muscle. Imaging of patient N45 showing (I)&(K) fibroadipose overgrowth of the legs including subcutaneous and muscular tissue,(J) fibroadipose overgrowth of the pelvis, and (L) &(M) plain radiographs showing distorting overgrowth of the knees . Plain radiograph of the hand of patient N68 showing distorting overgrowth of the second and third ray of the right hand (N). Plain radiograph (O) and photograph (P) of the left foot of patient N110 show overgrowth limited to the first and second rays of the left foot.
Eight of the 10 patients have undergone multiple surgical debulking or orthopedic procedures for overgrowth. Skeletal overgrowth was variable in character. Four had skeletal overgrowth with preserved architecture while others had distorting overgrowth (Figure 2). Patient N45 underwent orthopedic surgery for a 5 cm leg length discrepancy. Bone ages, when assessed, were normal, and no dental anomalies were reported. Several patients had enlarged peripheral nerves, cutaneous vascular malformations, and testicular or epididymal cysts and hydroceles. Four patients had strikingly lipoatrophy in areas not affected by overgrowth, but no evidence of either insulin resistance or hypoglycemia was reported. There have been no malignancies, although patient N108 did have benign nephrogenic rests.
All patients had been suggested to have Proteus syndrome, however on our evaluation none met the clinical criteria for this condition6 (Table 1), and 7 of the 10 had congenital segmental overgrowth, which is rare in Proteus syndrome. None had a cerebriform connective tissue nevus. Another diagnostic consideration is Klippel-Trenaunay syndrome (KTS). However, the vascular anomalies in the present patients comprised only capillary vascular malformations and none had the lateral venous anomaly or port wine stains typical of KTS7. Furthermore, the overgrowth in the present patients was not coincident with the vascular anomalies, which is characteristic of KTS. All patients had apparently normal intellectual and pubertal development and one patient has two unaffected children.
Table 1.
Summary of Clinical Features of Patients
| Patient Designation | C1 | C2 | N7 | N45 | N68 | N99 | N104 | N108 | N109 | N110 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age at time of manuscript preparation (years) | 37 | 31 | 17 | 31 | 49 | 1 | 11 | 6 | 4 | 1.5 |
| Sporadic occurrence* | + | + | + | + | + | + | + | + | + | + |
| Mosaic distribution* | + | + | + | + | + | + | + | + | + | + |
| Progressive course* | + | + | + | + | + | +/− | + | + | + | +/− |
| Linear epidermal nevus* | − | − | − | +/− | − | − | − | +/− | − | − |
| Asymm., disproportionate limb overgrowth* | + | + | + | + | + | + | + | + | + | + |
| Fibroadipose overgrowth* | − | − | + | + | + | + | + | + | + | |
| Regional lipohypoplasia* | + | − | + | + | − | − | + | − | − | |
| Vascular malformations: one or more* | − | − | − | + | + | − | − | − | − | − |
| Polydactyly | − | − | + | − | − | − | − | − | − | − |
| Non-spleen/thymus visceral overgrowth | − | − | − | + | − | − | − | − | ||
| Lipomatous infiltration of muscles | + | + | + | + | + | + | ||||
| Testicular/epididymal abnormalities | − | − | + | + | + | |||||
| Natural History Features | ||||||||||
| Congenital overgrowth | + | + | + | + | + | + | + | − | ||
| Progression of adipose dysregulation | + | + | + | + | + | + |
Abbreviations: +: feature present, −: feature evaluated and absent, +/−: some evidence to support presence of feature, blank cell: Not assessed. An asterisk indicates a diagnostic feature of Proteus syndrome. Other specific criteria for Proteus syndrome that were either absent or not assessed in all patients are not enumerated.
Exome sequencing of dermal fibroblast DNA from the unaffected arm and affected leg of patient C1 identified 45 non-synonymous genetic variants unique to leg-derived fibroblasts and absent from dbSNP and 894 controls. Sanger sequencing of DNA from a freshly cultured dermal fibroblasts from a second skin biopsy confirmed only 2 of these leg-specific genetic variants: the heterozygous PIK3CA mutations c.3140A>T (rs121913279, which predicts p.His1047Leu) and USHBP1 c.280C>G (which predicts p.Glu94Gln) (Figure 3, Supplementary Figure 1). USHBP1 encodes a putative Usher syndrome-related USH1C binding protein, highly expressed in the heart, and no link of USHBP1 to the phenotype was apparent from existing literature. In contrast, PIK3CA encodes the p110α catalytic subunit of the growth factor signal-transducing PI3K, and p.His1047Leu has been reported in >130 cancers by COSMIC, while the p.His1047Arg mutation, the commonest cancer-associated PIK3CA mutation8, stimulates both PI3K signaling and growth. This mutation was selected for further study.
Figure 3. Identification of PIK3CA mutations in affected cells and tissues.

A. PIK3CA c.3140A>T (p.His1047Leu) identified in cultured dermal fibroblasts from the affected left leg but not the unaffected right arm of patient C1 (left) and present at varying levels in left leg tissues (right) B. Location of His1047 near the carboxy-terminus of the kinase domain of the p110α catalytic subunit of type 1A PI3K. ABD = adaptor binding domain; RBD = ras binding domain C. PIK3CA c.3140A>G (p.His1047Arg) identified in cells derived from a variety of tissues of patients N7, N99, and N45.
Sequencing of DNA from 5 tissues obtained from the amputated left leg of patient C1 confirmed the PIK3CA mutation at all sites, with mutation burdens of 8% to 39% (Figure 3 and Supplementary Table 1) using a custom restriction enzyme-based assay (Supplementary Figure 2 and Supplementary Table 2). To determine whether mutations at codon 1047 might underlie similar forms of overgrowth we undertook targeted sequencing in 10 other patients with overgrowth syndromes with overlapping clinical features to those of patient C1. In 9 of 10 patients mosaicism for mutations at PIK3CA codon 1047 were identified. The cancer-associated PIK3CA variant, p.His1047Arg, was identified in 7 of the 9 patients, with mutation burdens of <1% to 35% in affected tissues and fibroblast cultures (Supplementary Table 1)). In 2 out of the 9 patients, the p.His1047Leu mutation was identified, with mutation burdens from 4% to 49%. The mutations were absent from blood and unaffected tissues from 9 of the reported patients, from both parents of 6 patients, and from 51 cell or tissue control samples.
These PIK3CA mutations were absent in >5,000 samples in the NHLBI Exome Sequencing Project. In the ClinSeq™ dataset (712 exomes) there were 21 variant sequence reads at this position among >92,000 reads, although no sample had >1 variant read. In the 1000 genomes data9 (933 low coverage genomes), there were >5,000 wild type reads and one variant read.
Both the p.His1047Arg and p.His1047Leu PIK3CA variants have increased kinase activity due to enhanced lipid binding10-12. We thus assessed PI3K activity in dermal fibroblasts from 3 patients by applying a mass spectroscopic assay for PIP3 (ref. 10) before and after stimulation of cells with EGF. PIP3 levels were increased 2-4X in affected cells at baseline and in response to EGF stimulation (Figure 4), and basal PIP3 levels in affected cells were indistinguishable from those in controls after stimulation. Basal hyperphosphorylation of downstream AKT and p70 S6 kinase signaling was detected in mutant PIK3CA cells. No amplification of stimulated phosphorylation was observed in affected cells, reflecting the maximum stimulation capacity of the signaling cascade. There was no increased signaling through the MEK/ERK pathway (Figure 4). We conclude that these patients harbor somatically mutated cells with enhanced basal activity of the PI3K/AKT pathway.
Figure 4. Hyperactivity of phosphatidylinositol-3-kinase in cells harboring PIK3CA mutations.
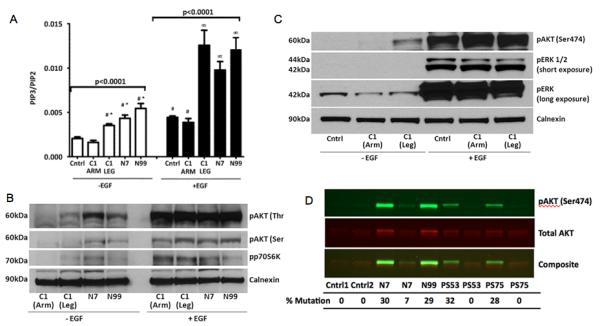
A. PIP3 levels determined by mass spectroscopy are elevated in fibroblasts harboring an activating PIK3CA mutation both in the basal state and after stimulation with EGF compared to unaffected cells. Cells from a healthy control (Cntrl), Patient C1 (unaffected arm and affected leg) and from patients N7 and N99 (both affected) are shown. * = Significantly higher than unstimulated unaffected cells (p<0.01); ∞ = significantly higher than EGF-stimulated unaffected cells (p<0.01); # = not significantly different; error bars are + 2 SEM. (B) & (C). Representative immunoblot showing abnormal basal phosphorylation of AKT and p70 S6 kinase (p70S6K) in affected dermal fibroblasts (B) but no increase in basal ERK phosphorylation (C). (D). Infrared detection of phosphorylated and total AKT in patients with PIK3CA or AKT1 mutations. Fibroblasts were serum starved for 24 hours before solubilization. Patients N7 and N99 were positive for the p.His1047Arg mutation in PIK3CA, PS53 and PS75 were positive for the p.Glu17Lys mutation in AKT1 (ref. 3), Cntrl1 and Cntrl2 were from unaffected individuals and negative for both mutations. Antibodies used in these experiments are described in Supplementary Table 3).
Our description of 10 patients with a previously uncharacterized severe segmental overgrowth syndrome due to somatic occurrence of activating mutations in the p110α catalytic subunit of PI3 kinase consolidates the paradigm of non-Mendelian, non-malignant growth disorders caused by isolated cancer-associated mutations in the PI3K/AKT signaling pathway. Moreover, the finding of distinct and homogeneous underlying genetic defects in stringently-defined Proteus syndrome, which is caused by somatic activation of AKT1, and in the currently delineated syndrome offers a mechanism-based validation of existing diagnostic criteria for Proteus syndrome. The syndrome we delineate does not fulfill the specific diagnostic criteria for PS13, instead falling within the ill-defined category of segmental overgrowth14. Attempts have been made to subclassify this group, and the patients reported here may be within category III of a recently proposed system15. However, we believe that future refinement of understanding of the phenotypic spectrum associated with PIK3CA mutations affords the prospect of further rationalization of diagnostic schemes.
PI3K signaling activates the serine/threonine kinases AKT1, AKT2 and AKT3. AKT1 is most widely expressed, and is associated with growth16, consistent with the Proteus phenotype, while AKT2 is highly expressed in insulin-responsive tissues including skeletal muscle, liver, and fat, and is more closely implicated in the metabolic actions of insulin17. AKT3 is most highly expressed in brain and heart, with lower expression in the tissues affected in the current patients. Recently somatic occurrence of both AKT2 and AKT3 p.Glu17Lys mutants, paralogous to the Proteus-associated AKT1 mutation, have been described. The AKT2 mutation causes severe insulin-independent hypoglycemia, mild asymmetric overgrowth, and progressive obesity18, while the AKT3 mutation was associated with brain overgrowth19.
The role of AKT2 in inhibiting adipose lipolysis and stimulating glucose uptake may explain the aggressive adipose expansion seen with PIK3CA mutations compared to AKT1 mutation alone. We speculate that the adipose tissue paucity in the non-overgrown areas of the patients is caused by chronic negative energy balance of those adipose depots consequent upon the demands of the pathologically growing and energy-sequestering adipose tissue in affected regions. The patients we describe do not exhibit a composite of the phenotypes associated with pathological AKT1, AKT2 and AKT3 activation, lacking key features of each condition such as hypoglycemia, cerebriform connective tissue nevi, and brain overgrowth. Some differences may be attributable to the timing and location of the founder mutation during embryogenesis. Thus, the lack of hypoglycemia and brain overgrowth likely reflect lack of PIK3CA activation in the liver and brain respectively in the current patients. Quantitative differences in the degree of activation of common downstream signaling pathways by the different mutants may also play a role, as may PI3K stimulation of non-AKT-dependent responses including rac-mediated cytoskeletal reorganization20. Some of the explanation for the phenotypic discrepancies of PIK3CA and AKT activation may also lie in the complexity of the PI3K enzyme: p110α is only one of three catalytic subunits coupled to receptor tyrosine kinase activation, each of which dimerizes with one of five regulatory subunits, lending significant combinatorial complexity to PI3K function. Specificity of coupling of different PI3K heterodimers to downstream pathways is not fully understood.
Given the prevalence of PIK3CA codon 1047 cancer mutations, a critical consideration is whether patients with these mutations are at increased risk of malignancy. Several observations indicate that such mutations can initiate cancer. Circumstantial evidence comes from the observation of PIK3CA mutations in the early stages of some human cancers21,22, while more direct evidence comes from murine studies. Transgenic expression of the Pik3ca p.His1047Arg mutation in lung23, or breast epithelium24-26 of mice has been shown to produce malignant tumors. However, in these studies the Pik3ca mutant was overexpressed, potentially exaggerating its oncogenicity. Expression of Pik3ca p.His1047Arg at endogenous levels in mouse ovaries did not produce tumors after one year27. It is thus possible that expression at endogenous levels in the cellular context of human mesodermal lineages has more benign consequences than implied by the mouse models overexpressing mutant Pik3ca. It is of note that codon 1047 oncogenic PIK3CA mutations have been reported at high prevalence in benign seborrheic keratoses and epidermal nevi in humans28, demonstrating that there is no obligate association of these mutations to malignancy. However, longitudinal studies are needed to properly assess this potential risk and to formulate surveillance recommendations, should such a risk be identified.
Treatment of segmental overgrowth disorders has relied upon surgical debulking6 and orthopedic procedures to limit growth29. The identification of activated PI3K/AKT signaling suggests potential utility of targeted therapy, either through inhibition of PI3K, of AKT, or of downstream pathways such as mTORC1, using clinically available drugs including rapamycin, which has been reported to produce major benefits in a child with somatic PTEN deficiency30. Intensive efforts are underway to develop novel inhibitors for use in cancer. When agents emerge with side effect profiles that are tolerable for long-term use in patients with non-malignant disease, their use in patients with severe progressive segmental overgrowth syndromes offers the prospect of rational mechanism-based therapy.
ONLINE METHODS
Patients
This study was approved by the UK National Research Ethics Committee and by the NHGRI Institutional Review Board. Written informed consent was obtained from all participants or their parents.
Body composition analysis and imaging
Body composition was measured by Lunar Prodigy dual-energy x-ray absorptiometry (GE Lunar).
Study Procedures
Using standard techniques, we isolated DNA from tissues and cell lines obtained either perioperatively, or using punch skin biopsy of areas with overgrowth (“affected”) or no overgrowth and distant from affected areas (“unaffected”). For exome sequencing DNA libraries were prepared from affected and unaffected cells from patient C1 as described previously32. Genotypes were called and filtered using a Phred-like genotype quality score of ≥50 as a cut-off, which we have found to provide a pragmatic balance between sensitivity and accuracy. In brief, sequence data were filtered to leave variants that were: 1) Predicted to change protein sequence and 2) Absent from the Single Nucleotide Polymorphism Database (dbSNP), 894 control genomes and 234 control exomes. Rare functional variants called in affected leg cells but not unaffected arm cells were verified by Sanger sequencing first in DNA used for exome sequencing, and then in DNA extracted from dermal fibroblasts established from a fresh skin biopsy. Remaining candidate pathogenic mutations were then sequenced in a panel of DNA derived from tissue samples obtained from the right leg of patient C1 after amputation. Interrogation of the codon affected by the putative pathogenic mutation was then undertaken in DNA from cells and tissues of 10 additional patients with overgrowth using Sanger sequencing and custom restriction enzyme digestion (primer sequences available on request).
Exome Sequencing
Genomic DNA was extracted from fibroblasts. For exome sequencing, 3 μg of DNA was sheared, end-repaired and A-tailed before ligation of Illumina sequencing adapters. 2 μg of the library was captured with an Agilent SureSelect Human All Exon 50 Mb bait designed to capture all exons from the NCBI consensus CDS (CCDS) database and coding exons from the GENCODE project (Agilent Technologies, Santa Clara, USA). The captured sample was eluted, amplified and sequenced on the Illumina Hi-Seq platform (Illumina, Little Chesterford, UK) as 75-bp paired-end reads. BWA software package 0.5.8c, SAMtools v0.1.7, Dindel v1.01 and Genome Analysis Toolkit (GATK) v1.0.4269M were used to map sequence reads to the human genome reference sequence (GRCh37) and the NCBI dbSNP database build ID 132 to calculate read quality, and to call single nucleotide variants and insertion/deletions. A total of 10.98 and 10.45 Gb of sequence was produced with a mean depth of 115x and 110x from arm and leg respectively. In the arm coverage was 96.7%, 87.8% and 85.4% at a depth of 4x, 20x and 25x, respectively, with 88.2% coverage at a depth of 10x, using high mapping quality alignments only (i.e., mapping quality ≥ 30 for reads mapped with BWA). Equivalent figures for coverage in the leg were 96.6%, 87.4%, 84.8% and 88.0%.
Variant Filtering
SNVs labeled as present within the NCBI dbSNP database Build ID132 were filtered and then perl scripts were used to further filter against SNVs in 894 individuals included in the 1000 Genomes Project phase 1 data release and exome-wide sequencing data from 236 control individuals from the CoLaus cohort33. Variants were annotated against Ensembl version 62 and were defined as coding if they were non-synonymous, resulted in loss or gain of a stop codon, or occurred within essential splice sites. Finally, all variants seen in both arm and leg, or in arm only, were excluded.
PIK3CA Codon 1047 Genotyping
Exon 21 of PIK3CA was amplified by PCR of genomic DNA and sequenced by conventional Sanger sequencing using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA). Primer sequences are available on request. Quantification of mutation burden was performed using a custom PCR restriction enzyme assay as described3 with the following modifications. Genomic DNA was amplified and digested with the primer and restriction enzyme combinationsogb specified in Supplementary Table 2. A sample was deemed positive if the mutant peak height was at least 100 relative fluorescence units. If in order to reach this threshold the wild type peak was off scale, the mutation percentage was assigned a value of <1%. When using the BsaBI assay, wild type DNA with and without enzyme was used in each case to confirm complete digestion of wild type alleles.
Phosphatidylinositol 3,4,5-trisphosphate (PIP3) quantification
PI3K/AKT signaling was assessed both at baseline and after stimulation with epidermal growth factor (EGF) in affected and unaffected dermal fibroblasts, by determining PIP3 and PIP2 using a recently developed mass spectrometry assay10. In brief, dermal fibroblasts were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1,000 U/l penicillin, 0.1 g/l streptomycin, and 2 mmol/l L-glutamine and were grown to 70% confluence in six-well culture plates. Cells were serum starved in serum-free medium supplemented with 1% bovine serum albumin (BSA), 1,000 U/l penicillin, 0.1 g/l streptomycin, and 2 mmol/l L-glutamine for 2 hours and then stimulated with either serum-free medium containing 10 nmol/l epidermal growth factor (EGF) or serum-free medium alone for 5 minutes. The medium was aspirated and 340 μl 1 mM hydrochloric acid (HCl) was added at 4 °C. Lysates were harvested and 10 μl of PIP3 internal standard (provided by the Babraham Institute) was added. 750 μl of kill mixture (484 ml Methanol (MeOH), 242 ml Chloroform (CHCl3) and 23.55 ml 1 M HCl) was added to each sample prior to centrifugation (15,000g, 5 min, 4 °C). 725 μl of CHCl3 was added prior to further centrifugation. The lower phase was collected and mixed with 708 μl of the upper phase of pre-derivitization wash mixture (240 ml CHCl3, 120 ml MeOH and 90 ml 0.01 M HCl) and samples were again centrifuged. The resultant lower phase was collected. 50 μl of 2 M TMS-diazomethane (Sigma) in hexane was added in a fume hood for 10 minutes at room temperature. The reaction was quenched with 6 μl glacial acetic acid. 700 μl of the upper phase of post-derivitization wash (240 ml CHCl3, 120 ml MeOH, 90 ml H2O) was added before mixing, centrifugation and collection of the lower phase. Samples were dried at room temperature and dissolved in 80 μl MeOH and 20 μl H2O. Analysis was undertaken using mass spectroscopy on a MDSCI EX 400Q Trap mass spectrometer (Applied Biosystems), connected to a Prominence HPLC system using a 1.0 × 50 mm column (Waters). The peak values of C38:4 species were expressed as a ratio (PIP3/PIP2). Statistical analysis was performed using one-way ANOVA and Bonferroni post-hoc analyses.
Cell Signaling Studies
Finally, phosphoblotting was used to assess phosphorylation of signaling intermediates of the PI3K/AKT pathway. Dermal fibroblasts were maintained, serum starved for 16 hours and EGF stimulated as described above. Following stimulation, cells were washed with ice cold PBS and solubilized in 80 μl lysis buffer (50 mmol/l HEPES, pH 7.5, 150 mmol/l NaCl, 30 mmol/l NaF, 10 mmol/l Na4P2O7, 1 mmol/l Na2VO4, 1% (v/v), Triton X-100, 10 mmol/l EDTA and 1 tablet of protease inhibitor mini complete cocktail (Roche, Mannheim, Germany)) per 7 ml of buffer. Protein concentration was quantified using the Bio-Rad DC protein assay (BioRad Laboratories, Hercules, CA, USA). Proteins were denatured at 95 °C then resolved by SDS-PAGE and transferred to polyvinylidene fluoride membranes using the iBlot system (Invitrogen). The membranes were blocked in buffer containing 50 mM TrisHCl, pH 7.6, 150 mM NaCl, 0.1% Tween-20 and 3% BSA for 1 hour at room temperature and probed for 16 hours at 4 °C with the relevant primary antibody (Supplementary Table 2). After washing the membranes, blots were incubated with horseradish peroxidase-linked secondary antibodies. Proteins were visualized by chemiluminescence (Enhanced Chemiluminescent kit, Amersham). Quantification was undertaken using Adobe PhotoShop CS3. Background subtraction was performed and bands quantified using control samples as base-line across six separate western blots. The mean relative integrated density in leg cells of patient C1 was +147% under basal conditions and +709% following stimulation with 10 nmol/L EGF. Statistical analysis was performed with one-way ANOVA and Bonferroni post hoc analyses.
Fibroblasts used for infrared imaging were serum starved for 24 hours before solubilization. Proteins were resolved on 10% Tris-glycine gels and transferred to nitrocellulose membranes using the iBlot system. Membranes were blocked in Odyssey blocking buffer (LiCor Biosciences) for 1 hour at room temperature. Primary antibody hybridization was performed overnight at 4 °C as before. After washing membranes, blots were incubated for 1 hour at room temperature in buffer containing 1:20,000 dilutions of anti-rabbit IRDye 800CW and anti-mouse IRDye 680LT secondary antibodies and 0.2% Tween. After washing, blots were scanned using the Odyssey infrared imaging system (LiCor Biosciences). Proteins were detected in the 700 nm channel using a scanning intensity of 5 and in the 800 nm channel using an intensity of 8.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank L. Ivey, Dr. J.J. Johnston, Dr. V. Tasic, Dr. M. Walters and E. Choolun for support and advice. The authors are especially grateful to the patients who participated in this research study and to the Proteus Syndrome Foundations of the United States and United Kingdom, who have supported and encouraged the patients and our research efforts. V.E.R.P., S.O.R., D.B.S., I.B., and R.K.S. were supported by the Wellcome Trust (grants 097721/Z/11/Z, 80952/Z/06/Z, 078986/Z/06/Z, 098051/Z/05/Z, and 091551/Z/10/Z), the Medical Research Council Centre for Obesity and Related Disorders, and the United Kingdom National Institute for Health Research (NIHR) Cambridge Biomedical Research Centre. L.G.B., M.J.L., J.C.S., A.M.W. were supported by the Intramural Research Program of the National Human Genome Research Institute. S.R., Q.F., and M.J.O.W. were supported by the Biotechnology and Biological Sciences Research Council (BBSRC). We are grateful for access to exome sequence data from the CoLaus cohort, which was sequenced as part of a partnership between the Wellcome Trust Sanger Institute, the CoLaus principal investigators and the Quantitative Sciences dept. of GlaxoSmithKline.
Footnotes
AUTHOR CONTRIBUTIONS M.J.L., V.E.R.P.,I.B., L.G.B. and R.K.S. designed the study and drafted the manuscript, M.J.L. and V.E.R.P. performed the research and analyzed the data, F.P., C.E.S., A.D. and I.B. performed analysis of WES data and wrote the corresponding methods in the manuscript, J.S. provided clinical data and wrote the corresponding clinical descriptions, A.M.W. performed mutation assays and assisted with immunoblotting experiments, S.H., L.T., M.L.C., T.N.D., J.G., Z.G., V.R.S., C.T. provided clinical data and/or fibroblast samples from affected patients, S.R., Q.Z. and M.J.O.W. performed PIP3 quantification and data analysis, J.H. assisted with collection of clinical data, M.P.G. assisted with immunoblotting experiments, T.H. performed histological analysis, A.K.D interpreted imaging, S.O.R. and D.B.S. contributed to study design and reviewed the manuscript, L.G.B and R.K.S. supervised the overall study.
Competing financial interests: I.B. and her spouse own stock in GlaxoSmithKline and Incyte Corporation.
URLs COSMIC: http://www.sanger.ac.uk/perl/genetics/CGP/cosmic?action=bygene&ln=PIK3CA&start=&end=& coords=AA%3AAA
NHLBI Exome Sequencing Project: accessed February 2012: http://evs.gs.washington.edu/EVS/http://evs.gs.washington.edu/EVS/
NCBI dbSNP database build ID 132: ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606/database/b132_archive/organism_data
1000Genomes Project phase 1 data release (July 2010): ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/pilot_data/release/2010_07/README.2010_07_release
Accession Numbersogb PIK3CA: GenBank accession number NM_006218.2
Editorial Summary (AOP and Month, same) Leslie Biesecker and colleagues report an exome sequencing study identifying somatic activating mutations in PIK3CA as the cause of a new progressive segmental overgrowth disorder. They identify mutations in PIK3CA in an additional 10 patients with overlapping syndromes.
References
- 1.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Happle R. The group of epidermal nevus syndromes Part I. Well defined phenotypes. J Am Acad Dermatol. 2010;63:1–22. doi: 10.1016/j.jaad.2010.01.017. quiz 23-4. [DOI] [PubMed] [Google Scholar]
- 3.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. New Engl J Med. 2011;365:611–9. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpten JD, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–44. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 5.Happle R. Type 2 segmental Cowden disease vs. Proteus syndrome. Br J Dermatol. 2007;156:1089–90. doi: 10.1111/j.1365-2133.2007.07818.x. [DOI] [PubMed] [Google Scholar]
- 6.Biesecker L. The challenges of Proteus syndrome: Diagnosis and management. Eur J Hum Genet. 2006;14:1151–7. doi: 10.1038/sj.ejhg.5201638. [DOI] [PubMed] [Google Scholar]
- 7.Oduber CE, van der Horst CM, Hennekam RC. Klippel-Trenaunay syndrome: diagnostic criteria and hypothesis on etiology. Ann Plast Surg. 2008;60:217–23. doi: 10.1097/SAP.0b013e318062abc1. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 9.Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark J, et al. Quantification of PtdInsP3 molecular species in cells and tissues by mass spectrometry. Nat Methods. 2011;8:267–72. doi: 10.1038/nmeth.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kalpha and the membrane. Proc Natl Acad Sci U S A. 2009;106:16996–7001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hon WC, Berndt A, Williams RL. Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases. Oncogene. 2011 Nov 28; doi: 10.1038/onc.2011.532. doi: 10.1038/onc.2011.532. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biesecker LG, et al. Proteus syndrome: Diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389–95. doi: 10.1002/(sici)1096-8628(19990611)84:5<389::aid-ajmg1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 14.Carty MJ, Taghinia A, Upton J. Overgrowth conditions: a diagnostic and therapeutic conundrum. Hand Clin. 2009;25:229–45. doi: 10.1016/j.hcl.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 15.Oduber CE, et al. A proposal for classification of entities combining vascular malformations and deregulated growth. Eur J Med Genet. 2011;54:262–271. doi: 10.1016/j.ejmg.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 16.Chen WS, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Devel. 2001;15:2203–8. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–51. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 18.Hussain K, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poduri A, et al. Somatic Activation of AKT3 Causes Hemispheric Developmental Brain Malformations. Neuron. 2012;74:41–8. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hawkins PT, Anderson KE, Davidson K, Stephens LR. Signalling through Class I PI3Ks in mammalian cells. Biochem Soc Trans. 2006;34:647–62. doi: 10.1042/BST0340647. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto S, et al. PIK3CA mutation is an early event in the development of endometriosis-associated ovarian clear cell adenocarcinoma. J Pathol. 2011;225:189–94. doi: 10.1002/path.2940. [DOI] [PubMed] [Google Scholar]
- 22.Miron A, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010;70:5674–8. doi: 10.1158/0008-5472.CAN-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams JR, et al. Cooperation between Pik3ca and p53 mutations in mouse mammary tumor formation. Cancer Res. 2011;71:2706–17. doi: 10.1158/0008-5472.CAN-10-0738. [DOI] [PubMed] [Google Scholar]
- 25.Meyer DS, et al. Luminal expression of PIK3CA mutant H1047R in the mammary gland induces heterogeneous tumors. Cancer Res. 2011;71:4344–51. doi: 10.1158/0008-5472.CAN-10-3827. [DOI] [PubMed] [Google Scholar]
- 26.Liu P, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011;17:1116–20. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinross KM, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553–7. doi: 10.1172/JCI59309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hafner C, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci U S A. 2007;104:13450–4. doi: 10.1073/pnas.0705218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tosi LL, Sapp JC, Allen ES, O’Keefe RJ, Biesecker LG. Assessment and management of the orthopedic and other complications of Proteus syndrome. J Child Orthop. 2011;5:319–327. doi: 10.1007/s11832-011-0350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh DJ, et al. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5:357–61. doi: 10.1038/ncponc1112. [DOI] [PubMed] [Google Scholar]
- 31.Ellis H, Logan BM, Dixon AK. Human sectional anatomy: Atlas of body sections, CT and MRI images. xvii. Hodder-Arnold; London: 2007. p. 267. [Google Scholar]
- 32.Raffan E, Semple RK. Next generation sequencing--implications for clinical practice. Br Med Bull. 2011;99:53–71. doi: 10.1093/bmb/ldr029. [DOI] [PubMed] [Google Scholar]
- 33.Firmann M, et al. The CoLaus study: a population-based study to investigate the epidemiology and genetic determinants of cardiovascular risk factors and metabolic syndrome. BMC Cardiovasc Disord. 2008;8:6. doi: 10.1186/1471-2261-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.