

Introduction
The congenital long QT syndrome (LQTS) is a life-threatening cardiac arrhythmia syndrome which represents a leading cause of sudden death in the young. LQTS is typically characterized by a prolongation of the QT interval on the electrocardiogram (ECG) and by the occurrence of syncope or cardiac arrest, mainly precipitated by emotional or physical stress.
Since 1975 (1) two hereditary variants, the Romano-Ward (RW) syndrome (2,3) and the very severe Jervell and Lange-Nielsen (JLN) syndrome (4,5), which is associated with congenital deafness, have been included under the comprehensive name of LQTS, one of the best understood monogenic diseases. The usual mode of inheritance for RW is autosomal dominant, whereas JLN shows autosomal recessive inheritance or sporadic cases of compound heterozygosity.
Several reasons make LQTS an important disease. It can often be a lethal disorder and symptomatic patients left without therapy have a very high mortality rate, 21% within one year from the first syncope (6). However, with proper treatment, mortality is now around 1% over a 15 year follow-up (7). This makes inexcusable the existence of symptomatic but undiagnosed patients. LQTS is without doubt the cardiac disease in which molecular biology and genetics have made the greatest progress and unquestionably is the best example of genotype-phenotype correlation. In this regard it represents a paradigm for sudden cardiac death and its progressive unravelling helps to better understand the mechanisms underlying sudden death in more complex disorders such as ischemic heart disease and heart failure.
This review will outline the current knowledge about the genetics of LQTS and provide essential clinical data, while its primary focus will be on our approach to the clinical management of these patients.
Genetics of LQTS
The electrocardiographic QT interval represents the depolarization and the repolarization phases of the cardiac action potential. The interplay of several ion channels determines the action potential duration. Decreases in repolarizing outward K+ currents or increases in depolarizing inward sodium or calcium currents can lead to prolongation of the QT interval, thus representing a pathophysiological substrate for LQTS. Not surprisingly, since the dawn of molecular era in LQTS, genes encoding ion channels responsible for the timely execution of the cardiac action potential were considered plausible targets for investigation. Following the identification of the first three genes associated with the most frequent variants (8–10), 10 more genes involved in fine-tuning the cardiac action potential have been associated with LQTS (Table 1).
Table 1.
LQTS genes
| Gene | Syndrome | Frequency | Locus | Protein (Functional effect) |
|---|---|---|---|---|
| KCNQ1 (LQT1) | RWS, JLNS | 40–55 | 11p15.5 | Kv7.1 (↓) |
| KCNH2 (LQT2) | RWS | 30–45 | 7q35-36 | Kv11.1 (↓) |
| SCN5A (LQT3) | RWS | 5–10 | 3p21-p24 | NaV1.5 (↑) |
| ANKB (LQT4) | RWS | <1% | 4q25-q27 | Ankyrin B (↓) |
| KCNE1 (LQT5) | RWS, JLNS | <1% | 21q22.1 | MinK (↓) |
| KCNE2 (LQT6) | RWS | <1% | 21q22.1 | MiRP1 (↓) |
| KCNJ2 (LQT7) | AS | <1% | 17q23 | Kir2.1 (↓) |
| CACNA1C (LQT8) | TS | <1% | 12p13.3 | L-type calcium channel (↑) |
| CAV3 (LQT9) | RWS | <1% | 3p25 | Caveolin 3 (↓) |
| SCN4B (LQT10) | RWS | <1% | 11q23.3 | Sodium channel -β4 (↓) |
| AKAP9 (LQT11) | RWS | <1%% | 7q21-q22 | Yotiao (↓) |
| SNTA1 (LQT12) | RWS | <1% | 20q11.2 | Syntrophin-α1 (↓) |
| KCNJ5 (LQT13) | RWS | <1% | 11q24 | Kir3.4 (↓) |
RWS = Romano-Ward Syndrome
JLNS = Jervell-Lange-Nielsen Syndrome
AS = Andersen Syndrome
TS = Timothy Syndrome
Functional effect: (↓) loss-of-function or (↑) gain-of-function at the cellular in vitro level
Major LQTS genes
By far, KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3) are the most common LQTS genes, accounting for approximately 90% of all genotype-positive cases (11,12). The KCNQ1 gene encodes the α-subunit of the K+ channel Kv7.1, generating the depolarizing IKs current which, being physiologically increased by sympathetic activation, is essential for QT adaptation when heart rate increases. When IKs is defective, the QT interval fails to shorten appropriately during tachycardia thus creating a highly arrhythmogenic condition. Heterozygous KCNQ1 mutations cause the dominant Romano-Ward LQT1 syndrome and account for the majority of disease-causing variants. Homozygous mutations in KCNQ1, or compound heterozygous mutations, cause the recessive JLN variant, characterized by deafness due to the reduced IKs in the inner ear (4,5).
The mutations may produce different effects in this multimeric K+ channel. Defective and wild-type protein subunits may co-assemble and exert a dominant negative effect on the current. Alternatively, some mutant subunits may not co-assemble with wild-type peptides, resulting in a loss of function that reduces the IKs current by 50% or less (haploinsufficiency). The latter may also result as a consequence of mutations interfering with intracellular subunits trafficking, preventing the mutated peptides from reaching the cell membrane.
However, neither the localization of a mutation nor its cellular electrophysiological effect are sufficient to predict the impact on clinical manifestations (13). A good example is represented by a large cohort of LQT1 patients from all over the world, all carrying the A341V hot-spot mutation located in the transmembrane portion of the K+ channel with a very mild dominant-negative functional effect; in these patients we demonstrated a strikingly higher clinical severity among LQT1 carriers of A341V compared to LQT1 non-A341V patients, with mutations either localized to transmembrane domains or exhibiting a dominant-negative effect (13).
The second most common gene harbouring LQTS mutations is KCNH2, encoding the α-subunit of the K+ channel conducting the IKr current. The rapid IKr (KCNH2) and the slow IKs (KCNQ1) are two independent components of the delayed rectifier IK current, the major determinant of the phase 3 of the cardiac action potential. Mutations in KCNH2 cause a reduction of IKr current, through mechanisms similar to the effects exhibited by KCNQ1 mutations on IKs current (7). Up to 10% of genotyped cases may harbour compound heterozygous mutations on the same or on two of the main LQTS genes (14,15). Not surprisingly, a more severe cardiac phenotype accompanies compound mutations (14–16).
The third major LQTS gene, identified at the end of March 1995 (8), is SCN5A, encoding the α-subunit of the cardiac sodium channel and conducting the depolarizing sodium inward current (INa). A ground-breaking in-vitro expression study by Bennett et al in September 1995 showed that the SCN5A-ΔKPQ mutation produces the LQTS phenotype by increasing the delayed Na+ inward current and therefore prolonging the action potential duration (17). Within a few months, in December 1995 this was followed by our report that the genetic defects in LQTS may be linked to differential responses to heart rate changes and to Na+ channel blockers (18) and to the first evidence that mexiletine reduces the late Na+ current (19). This finding paved the way to the search for gene-specific therapies.
A number of genetically heterogeneous disorders are also associated with alterations in the sodium current including Brugada syndrome, atrial fibrillation, sick sinus node syndrome, and the Lev-Lenègre disease. As a further complexity some SCN5A mutations show a pleiotropic behavior and are associated with more than one phenotype, the so called “overlap syndrome” (20). When a single mutation can have opposite functional effects (i.e. increase and decrease of the Na+ current) what matters clinically is the phenotype.
Given the large and growing number of genetic variants identified so far, to distinguish pathogenic mutations from rare variants is critically important. Based on almost 400 definite cases and 1300 controls (21), the probability for a missense mutation to be pathogenic appears to depend largely on location. In general, genetic variants located in the pore and transmembrane regions are much more likely to be pathogenic. Whenever a functional study of the specific mutation has been performed, the results may help in assessing its clinical relevance. When these data are missing, as it is often the case, it is important to establish whether within the family the mutation co-segregates with either symptoms or QT prolongation. An important take-home message is that the laboratory finding of an aminoacidic substitution should not be automatically taken as indication of a disease-causing mutation.
Minor LQTS genes
Following the identification of the first three LQTS genes (8–10), several others were and are being identified; the list will continue to grow for a while.
KCNE1 and KCNE2 encode the minimal K+ ion channel (minK) and the minK related peptide 1 (MiRP1), which represent the main ancillary single-transmembrane β-subunits associated with the α-subunits of KCNQ1 and KCNH2. Mutations in the KCNE1 gene may cause either the dominant RW (LQT5) or, if present in homozygosity or compound heterozygosity, the recessive JLN (7). The cases of KCNE2 mutations associated with LQTS are few and some of them represent acquired LQTS associated with specific drugs, almost all IKr blockers (7).
Among the sodium channel interacting proteins the CAV3, SCN4B, and SNTA1 genes are regarded as additional LQTS genes (LQT9, LQT10 and LQT12) (22–24). The A-kinase anchor protein 9 gene (AKAP9) is involved in the phosphorylation of KCNQ1 and its mutations have been described in LQT11 (25). Two missense mutations in CACNA1C, encoding a voltage-gated calcium channel, are linked to Timothy syndrome (LQT8), a rare and extremely malignant variant (26). Finally, in a large Chinese family a heterozygous mutation was identified in the inwardly rectifying K+ channel subunit Kir3.4, encoded by the KCNJ5 gene. The variant was present in all the nine affected family members and was absent in more than 500 ethnically matched controls, suggesting a role in the aetiology of the novel LQT13 variant (27).
On the other hand, the ANKB and KCNJ2 genes, often referred to as LQT4 and LQT7, are associated with complex clinical disorders in which the prolongation of the QT interval is modest and, in our opinion, should not be strictly considered as part of LQTS (7).
Prevalence
Even though it is customary, when dealing with any cardiac disease of genetic origin, to provide its prevalence, almost always what is presented is largely an educated guess. LQTS represents an exception. For too long the prevalence of LQTS was assumed to be anywhere between 1/5,000 and 1/20,000, without any supporting data. The first data-driven indication of the prevalence of LQTS was published in 2009, based on the largest prospective study of neonatal electrocardiography ever performed (28). In 18 Italian maternity hospitals an ECG was performed in 44,596 infants 15 to 25 days old; in this cohort, 0.07% had a QTc greater than 470 ms and 0.47% had a QTc between 451 and 470 ms. Molecular screening allowed the identification of a disease-causing mutation in 43% of the neonates with a QTc above 470 ms, and in 29% of those screened with a QTc between 461 and 470 ms. In total, 17 of 43,080 white infants were affected by LQTS, demonstrating a prevalence of at least 1:2,534 apparently healthy live births (95% CI, 1:1583 to 1:4350) (28). Considerations based on the number of infants with a QTc > 450 ms who were not molecularly screened actually suggest that the prevalence of LQTS is close to 1:2,000. This prevalence concerns only infants with an abnormally long QTc and cannot estimate the additional incidence of “silent” mutations carriers (individuals who carry a disease-causing mutation but who have a normal QT interval).
Clinical Presentation
The clinical manifestations of LQTS have been described in detail too often to deserve additional repetitions here. The reader unfamiliar with LQTS can find these descriptions in previous publications (6,7,29). Here, we will mention only a few specific aspects which carry, in our opinion, special significance.
The ventricular tachyarrhythmia which underlies the cardiac events of LQTS is Torsadesde-Pointes (TdP), a curious type of ventricular tachycardia which most of the time is self-limiting and produces transient syncope but which can also degenerate into ventricular fibrillation and cause cardiac arrest or sudden death (7). It would be extremely important to know what causes TdP to stop after a limited number of seconds or to continue, with devastating consequences, but we do not.
The morphology of the T wave is often very useful for the diagnosis and the precordial leads are especially informative when they reveal biphasic or notched T waves (30). T wave alternans in polarity or amplitude (Figure 1), when observed, is diagnostic as we proposed almost 40 years ago (31). T wave alternans is a marker of major electrical instability and identifies patients at particularly high risk; its presence in a patient already on treatment should alert the physician to persistent high risk and warrants an immediate reassessment of therapy. Sinus pauses, unrelated to sinus arrhythmia, are an additional warning signal especially in patients with SCN5A mutations (7,32).
Figure 1.

Examples of T wave alternans from a 2-year old LQTS patient with multiple episodes of cardiac arrest. Tracings are from a 24-h Holter recording.
Diagnosis and Genetic Testing
Typical cases present no diagnostic difficulty for physicians aware of the disease. However, borderline cases are more complex and require the evaluation of multiple variables besides clinical history and ECG. The diagnostic criteria for LQTS proposed in 1985 (6) remain essentially valid for a quick assessment; however, a more quantitative approach to diagnosis became possible with the presentation in 1993 of a diagnostic score which became known as the “Schwartz score”, which was updated in 2006 (33,34). The last update has just been made on the basis of the report on the diagnostic role of QT prolongation in the recovery phase of an exercise stress test (35,36) (Table 2). The persistent use by some investigators of the old scoring system leads to an underestimation of the patients identified as “probably affected” and should be discontinued; a score of 3.5 points is sufficient for a high probability of LQTS.
Table 2.
1993–2011 LQTS DIAGNOSTIC CRITERIA
| POINTS | |||
|---|---|---|---|
| ELECTROCARDIOGRAPHIC FINDINGS | |||
| A | QTc^ | ≥ 480 ms | 3 |
| 460 – 479 ms | 2 | ||
| 450 – 459 (male) ms | 1 | ||
| B | QTc^ 4th minute of recovery from exercise stress test ≥ 480 ms | 1 | |
| C | TORSADE DE POINTES * | 2 | |
| D | T WAVE ALTERNANS | 1 | |
| E | NOTCHED T WAVE IN 3 LEADS | 1 | |
| F | LOW HEART RATE FOR AGE @ | 0.5 | |
| CLINICAL HISTORY | |||
| A | SYNCOPE * | WITH STRESS | 2 |
| WITHOUT STRESS | 1 | ||
| B | CONGENITAL DEAFNESS | 0.5 | |
| FAMILY HISTORY | |||
| A | FAMILY MEMBERS WITH DEFINITE LQTS $ | 1 | |
| B | UNEXPLAINED SUDDEN CARDIAC DEATH BELOW AGE 30 AMONG IMMEDIATE FAMILY MEMBERS $ | 0.5 | |
# In the absence of medications or disorders known to affect these electrocardiographic features
QTc calculated by Bazett's formula where QTc = QT/√RR
Mutually exclusive
Resting heart rate below the 2nd percentile for age
The same family member cannot be counted in A and B
SCORE: ≤ 1 point: low probability of LQTS
1.5 to 3 points: intermediate probability of LQTS
≥ 5 points high probability
(From ref. 36)
The importance of a correct diagnosis has assumed a new dimension in the molecular era. A new responsibility for the clinician lies in the identification of the most logical candidates for molecular screening, and relates to the availability and cost of genetic testing. The best example of this situation comes from a study by Taggart et al (37). In a group of 176 consecutive patients diagnosed as affected by LQTS and sent to the Mayo Clinic for management and genetic testing they regarded 41% of them as unaffected, 32% as probably affected and only 27% as definite cases of LQTS. Genetic testing confirmed the clinical assessment as disease-causing mutations were found in none of the “unaffected”, in 34% of the “probably affected”, and in 78% of the “definitely affected”. It follows that an exceedingly large number of patients incorrectly received the clinical diagnosis of LQTS by their own cardiologists.
It is indeed in the selection of patients with a suspicion of LQTS that the Schwartz score becomes especially useful. As the score gives importance to the degree of QT prolongation, it should be obvious that it cannot help in the identification of the silent mutation carriers. The smart approach consists in the use of the Schwartz score for the selection of those patients who should undergo molecular screening (everyone with a score ≥ 3.0) and in the use of “cascade screening” (38–39) for the identification of all affected family members including the silent mutation carriers.
Malignant Subtypes
Two well-defined LQTS variants carry an especially high risk and are very difficult to manage, the Jervell and Lange-Nielsen syndrome (4,5) and the Timothy syndrome (LQT8)(26).
The recessive JLN has the same cardiac phenotype observed in the RW type of LQTS, complicated by a more malignant course and by congenital deafness. The largest study on JLN, based on 187 patients, did show that almost 90% of the patients have cardiac events, that they become symptomatic much earlier than in the other major genetic subgroups of LQTS (Figure 2), and they do not respond as well to traditional therapy (5). Of interest, the patients whose homozygous mutations involve the KCNE1 gene instead of KCNQ1 are at lower risk (5).
Figure 2.
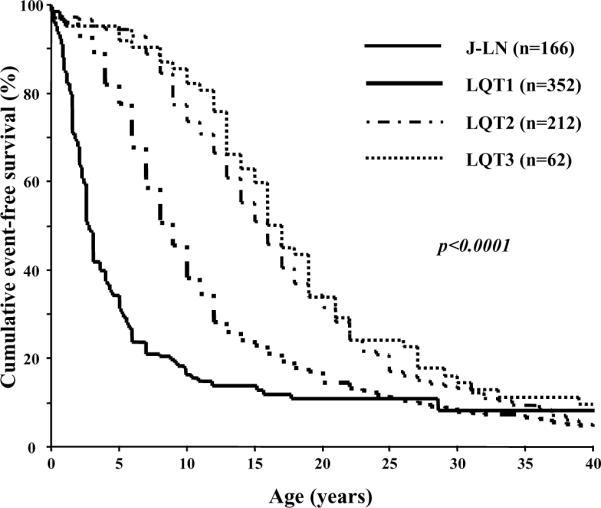
Kaplan-Meier curves of event-free survival comparing JLN patients vs LQT1, LQT2 and LQT3 symptomatic patients. (From ref. 5)
The Timothy syndrome is an extremely rare variant characterized by marked QT prolongation associated with syndactyly and often presenting with 2:1 functional AV block and macroscopic T wave alternans (26). Congenital heart diseases, intermittent hypoglycemia, cognitive abnormalities and autism can also be present. Of the 17 children reported by Splawski et al 10 (59%) died at a mean age of 2.5 years.
Genotype-Phenotype Correlation
The clinical manifestations of LQTS may vary according to the different genetic background. The disease-causing gene is the main determinant of the clinical phenotype, but also the position of the mutation in the protein and the specific disease-causing mutation can contribute to clinical severity.
Disease-causing gene and phenotype
In 2003 data on 647 patients of known genotype indicated that life-threatening events were lower among LQT1 patients, higher among LQT2 females than LQT2 males, and higher among LQT3 males than LQT3 females (40). This study also provided the rather unexpected and important information that the number of “silent mutation carriers”, i.e. individuals with a disease-causing mutation but with a normal QT interval, exceeds previous estimates and correlates with the specific genes. Indeed, silent mutation carriers represent 36% of LQT1, 19% of LQT2, and 10% of LQT3 patients.
In 2001 Schwartz et al examined the possible relationship between genotype and conditions (“triggers”) associated with the events in 670 symptomatic patients with LQTS and known genotype (41). As predicted by the impairment in IKs current (essential for QT shortening during increase in heart rate), most of the events in LQT1 patients occurred during exercise or stress (Figure 3). A highly specific trigger for LQT1 is represented by swimming. Many of the events in LQT2 patients occurred during arousal, especially from auditory stimuli such as sudden noises and telephone ringing, particularly when occurring at rest. Most of the events in LQT3 occurred while patients were asleep or at rest. LQT2 and LQT3 patients are at low risk for life-threatening arrhythmias during exercise because they have a well-preserved IKs current, allowing appropriate shortening of the QT interval whenever heart rate increases.
Figure 3.

Triggers for all lethal and non-lethal cardiac events in the three genotypes. (Modified from ref. 41)
In a study of a uniquely large South African LQT1 founder population we observed that faster basal heart rate and brisk autonomic responses are associated with a greater probability of being symptomatic, depending again on the gene-specific impairment of IKs (42,43). Relatively high values of baroreflex sensitivity imply an increased ability to change heart rate suddenly and this could be harmful especially in LQT1 patients: sudden heart rate increases with impaired QT shortening favor the R-on-T phenomenon and initiation of ventricular tachycardia-fibrillation; whereas, sudden pauses elicit early afterdepolarizations, which can trigger TdP (43).
Even in the postpartum period genotype is important because arrhythmic risk is higher for LQT2 than for LQT1 patients (44,45), probably due to sleep disruption and sudden awakening.
Genotype-phenotype correlation exists also in rare and malignant forms of LQTS as shown by the fact that among JLN patients those with KCNE1 mutations exhibit a markedly less severe clinical course than the majority of patients who have mutations on KCNQ1 (5).
Disease-causing mutations and phenotype
In 2002 Moss et al indicated that LQT2 patients with mutations in the pore region were at higher risk (46). In 2007 Moss et al demonstrated that in LQT1 patients both the transmembrane location of the mutation and their dominant-negative effect are independent risk factors for cardiac events (47). These studies were important because they called attention to the fact that not all mutations on the same gene produce a similar clinical phenotype and they were the beginning of a growing number of intriguing revelations on the complexity of the genotype-phenotype correlation.
Probably the most striking example of mutation-specific behavior comes from KCNQ1-A341V, a hot-spot mutation characterized by an unusual clinical severity demonstrated by 80% of the patients being symptomatic with over 30% experiencing cardiac arrest or sudden death (Figure 4) (13,42). What is puzzling is that A341V is only a mildly dominant mutation producing a relatively modest loss of IKs. The implication is that our current understanding of biophysical cellular studies is still incomplete and fails to allow a direct translation into the clinical reality.
Figure 4.

Unadjusted Kaplan-Meier estimate of the cumulative event-free survival (any first event) in the whole (non-SA– SA) A341V population plotted vs the LQT1 non-A341V group. Any cardiac event, whichever occurred first, was considered from birth through 40 years of age and before β-blocker therapy. Numbers at risk are indicated. (From ref. 13)
Another example of mutation-specific behavior is represented by the SCN5A-E1784K (48). This mutation is the most frequently described Brugada Syndrome mutation and it is also a relatively prevalent mutation among LQT3 patients. It can cause the Brugada pattern, long QT, sinus node dysfunction and life-threatening ventricular arrhythmias (48). The management of patients with this or other pleiotropic mutations (49) should be careful, taking into account that their genetic background could favor different clinical manifestations; their management should be based on the pattern manifested by multiple ECG recordings. However, even if the patient shows a pure LQT3 phenotype among sodium channel blockers it is wise to avoid flecainide, that may induce a Brugada pattern (50), whereas mexiletine is not contraindicated. In the study of family members carrying the same mutation physicians should be aware that they could show clinical signs of either LQTS or Brugada syndrome and should be treated accordingly.
Modifier Genes
The relationship between genotype and clinical phenotype is not necessarily linear in inherited arrhythmias. For example, in LQTS a genetic mutation may exhibit incomplete penetrance (51). Similarly, certain mutations may have variable expressivity, conferring different risk of disease expression in related individuals. Reasonably, both these aspects are linked to genetic, environmental or developmental factors that may modulate the disease onset or its clinical severity. The genetic factors involved in this modulation, referred to as “modifiers” or “modifier genes”, are distinct from the disease-causing mutation, and are the object of intense research activity.
Some common genetic variants of cardiac ion channel genes [single nucleotide polymorphisms (SNPs)] have detectable functional activity. These findings underlie the concept that SNPs may modulate the clinical severity of the primary mutation. By investigating a highly symptomatic LQTS proband and her relatives, Crotti et al provided the first evidence that the common polymorphism KCNH2-K897T (30% carrier frequency among whites) may modify the clinical expression of a latent LQT2 mutation (52).
The most powerful resource for studying modifier genes is represented by “founder populations”, in which a disease allele segregates in families descending from a common ancestor (53,54). In an unusually large South African LQT1 founder population (42), we demonstrated that two common variants in NOS1AP, encoding a nitric oxide synthase adaptor protein, were significantly associated with occurrence of symptoms, with clinical severity and with QT interval (55). Subsequently, the role of NOS1AP as a genetic modifier of LQTS has been confirmed in a large cohort of unrelated patients (56). This type of study exploiting the unique features of the founder populations are likely to provide the much needed information necessary to allow a more precise “custom-made” risk stratification for the individual carriers of LQTS-causing mutations.
Notably, the concept of “modifier genes” and of “modifier factors” illustrates the limitations of the cellular models currently used in the assessment of a functional defect. These models allow the biophysical investigation of putative mutations in single ion channels, potentially with the co-expression of specific, a priori selected variants (e.g. SNPs). However, these heterologous in-vitro systems may not completely reproduce all the possible channel interactions, such as the complex system of in vivo myocardial cell. Very recently, pluripotent stem cells were generated from dermal fibroblasts collected from LQTS patients (57–59). These cells were successfully differentiated into cardiac myocytes, exhibiting functional alterations typical of the disease. The use of induced pluripotent stem-cells may represent a novel and appropriate model to better elucidate the clinical heterogeneity in LQTS.
Current Management
The most significant information concerning therapy for LQTS still comes from a 1985 study (6), which included 233 symptomatic patients, and demonstrated the dramatic change in survival produced by pharmacologic or surgical antiadrenergic therapy when compared to any other therapy or no treatment. Such a large group of severely affected patients left without treatment is obviously no longer available.
β-adrenergic blockade
ß-adrenergic blocking agents represent the first choice therapy in symptomatic LQTS patients, barring specific contraindications. ß-blockers seldom result in excessive bradycardia, especially if the dosage is very gradually increased over several weeks.
Contrary to commonly held views, β-blockers are not all equally effective. Without question, the two most effective are propranolol and nadolol. Propranolol is still the most widely used drug, at 2–3 mg/kg/day; sometime the dosage is increased to 4 mg/kg and in very severe cases also higher doses are justified. Nadolol is also used very often as its longer half-life allows twice-a-day administration, usually at 1–1.5 mg/kg/day. Metoprolol is definitely less effective (60) and the switch from propranolol or nadolol to metoprolol has been associated to tragic recurrences. It is now clear that metoprolol should not be used in the management of LQTS. Also atenolol seems somewhat less effective but the data available are limited (61). Even though it is unclear whether or not differences in Na+ blocking activity (what in the old days was called the “membrane stabilizing effect”) play a role in the clearly different clinical efficacy of various β-blockers, it is interesting to note that this blocking effect is highest for propranolol, lower but present for nadolol, and completely absent for metoprolol (62).
In a study of 869 LQTS patients of unknown genotype, overall mortality on ß-blocker therapy was 2%, and it was 1.6% when limited to patients with syncope (no cardiac arrest) and without events in the first year of life (63). There is clear evidence that ß-blockers are extremely effective in LQT1 patients. Data from two large studies (64,65) indicate that mortality is 0.5% and sudden death combined with cardiac arrest reaches 1%. β-blocker noncompliance and use of QT-prolonging drug are responsible for almost all life-threatening “β-blocker failures” in LQT1 patients (65); conversely, compliance and the avoidance of QT-prolonging drugs is associated with 97% reduction in the risk for cardiac events. Compared to LQT1, LQT2 patients have more life-threatening events despite ß-blockers, but most of these are resuscitated cardiac arrests (6–7%) (64). Among LQT3 patients major events have been reported to occur more frequently (10–15%) despite ß-blockers (41,64) and have contributed to the incorrect notion that β-blockers are of limited or no value for LQT3 patients. This misconception is the consequence of including LQT3 patients who present with events in the first year of life with those who present with events at a later time (32). Indeed, the presence of a cardiac event in the first year of life is associated with an extremely poor prognosis independent of treatment. In patients presenting at an older age, mortality on β-blocker therapy is 2% and is highest in those with markedly prolonged QTc intervals, approaching 600 ms. This information comes from the largest study ever performed in LQT3, with data on 400 patients (Wilde A et al, - Unpublished data, 2011). Left cardiac sympathetic denervation appears to confer significant protection from life-threatening arrhythmias in the relatively small number of LQT3 patients who received this treatment (66). Patients with the severe JLN or TS often are not adequately protected by β-blockers and require additional protection (5,26).
Left Cardiac Sympathetic Denervation (LCSD)
LCSD, ideally performed by an extrapleural approach which makes thoracotomy unnecessary, requires removal of the first 3–4 thoracic ganglia. The cephalic portion of the left stellate ganglion is left intact to avoid the Horner's syndrome. An alternate surgical approach is represented by thoracoscopy (67). For small infants or whenever the local surgeons do not have adequate experience we recommend the traditional and easy approach represented by an opening in the third left intercostal space which allows a clear visualization of the stellate ganglion with the sympathetic chain. The technical details of our favored extrapleural approach have been recently described (68). The rationale for LCSD, largely based on its rather striking antifibrillatory effect (69), has also been recently reviewed (70).
The latest clinical data on LCSD were published in 2004 and included 147 LQTS patients who underwent sympathectomy during the last 35 years (66). They represented a group at very high risk [99% symptomatic, with an extremely long mean QTc (563±65 ms), previous cardiac arrest in 48%, a recurrent syncope despite full-dose ß-blockers in 75%]. During a mean follow-up of 8 years there was a 91% reduction in cardiac events. In 5 patients who underwent LCSD due to multiple ICD shocks and electrical storms, over a 4-year follow-up there was a 95% decrease in the number of shocks with a dramatic improvement in the quality of life of the patients and of their families. Interestingly, LCSD produced a mean QTc shortening of 39 ms, pointing to an action on the substrate as well as on the trigger. The unavoidable conclusion is that whenever syncopal episodes recur despite a full-dose ß-blocker therapy, LCSD should be considered and implemented without hesitation. Also, failure by the physician to provide the family with adequate information of the pro and cons of LCSD vs ICD implant may carry medico-legal consequences (71).
Implantable Cardioverter Defibrillator (ICD)
The decision to implant an ICD is relatively easy for the clinical cardiologist. In the case of appropriate shocks, he/she will have saved the life of the patient; in case of no shocks and possibly of complications, he/she will have done the best for the patient's protection. On the other hand, the decision to not implant an ICD could, in the case of a tragic outcome, lead to medico-legal consequences if such a decision was not supported by a valid rationale. Even though these considerations should play no or a minimal role in medical decisions, they actually do. In light of this, physicians should analyse the current state of knowledge.
There is an overall consensus for immediately implanting an ICD in cases where there has been a documented cardiac arrest, either on or off therapy. Some exceptions exist, such as in a clear case of a drug-induced event in an otherwise asymptomatic patient with modest QT prolongation. By contrast, opinions differ strongly regarding the use of ICDs in patients without cardiac arrest.
The current knowledge is essentially based on the largest ICD study ever published, which provided information on 233 LQTS patients (72). In this publication it is disquieting that the majority of ICDs were implanted in patients who had not had a previous cardiac arrest and many had not even failed ß-blocker therapy. Asymptomatic patients, almost absent among LQT1 and LQT2, reached the staggering number of 45% among LQT3 patients indicating that the mere presence of a SCN5A mutation, even in a totally asymptomatic individual, was deemed sufficient for ICD implant. During a mean follow-up of 4.6 years at least one appropriate shock was received by 28% of patients and adverse events occurred in 25% of the study population.
Given the practical importance to identify in advance those patients with the highest probability to receive appropriate shocks, which represents the justification for the ICD implant, we developed (72) a score (M-FACT) based on simple clinical variables available in a doctor's office during a first visit (Table 3). M-FACT considers QTc duration, age at implant, cardiac events despite therapy.
Table 3.
M-FACT * RISK SCORE
| − 1 point | 0 points | 1 point | 2 points | |
|---|---|---|---|---|
| Event free on therapy for > 10 yrs | Yes | |||
| QTc (ms) | ≤ 500 | > 500 – ≤ 550 | >550 | |
| Prior ACA | No | Yes | ||
| Events on therapy | No | Yes | ||
| Age at implant | > 20 yrs | ≤ 20 yrs |
Acronym derived from M (Minus 1 point for being free of cardiac events while on therapy for > 10 years); F (“Five Hundred” and “Five hundred and Fifty ms QTc); A for Age ≤ 20 years at implant; C (Cardiac arrest); T (events on Therapy); ACA = Aborted Cardiac Arrest
(From ref. 72)
Appropriate ICD therapies were predicted by age <20 years, a QTc above 500 ms, prior cardiac arrest and cardiac events despite therapy; within 7 years, appropriate shocks occurred in no patients with none of these factors and in 70% of those with all factors (Figure 5A and 5B).
Figure 5.
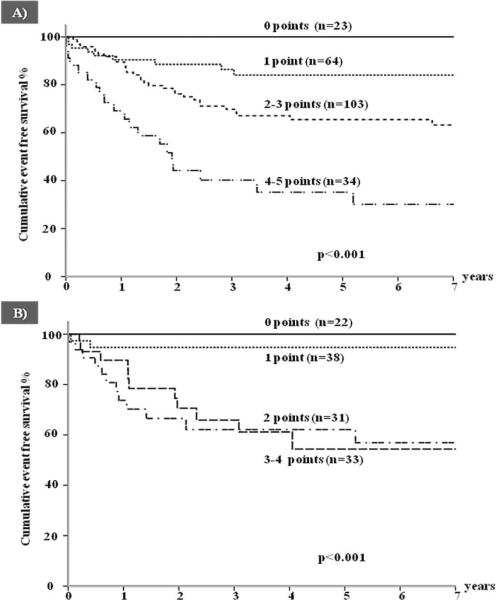
Cumulative event-free survival for a first appropriate ICD shock according to an increasing risk score (M-FACT) in (A) all patients and (B) in patients with no prior ACA. (From ref. 72)
Our current policy is to implant an ICD in: 1) all those who survived a cardiac arrest on therapy; 2) most of those who survived a cardiac arrest off therapy, except those with a reversible/preventable cause; 3) those with syncope despite full dose of β-blocker, whenever the option of LCSD either is not available or is discarded after discussion with the patients; 4) all patients with syncope despite full dose of β-blocker and LCSD; 5) exceptionally, the rare asymptomatic patients with a QTc above 550 ms who also manifests sign of high electric instability (e.g. T wave alternans) or other evidence of being at very high risk (e.g. very long sinus pauses followed by abnormal T wave morphologies) despite ß-blockade and LCSD.
For patients with J-LN or with Timothy syndrome who appear incompletely protected by antiadrenergic therapies we usually consider, in a case by case approach, the possibility of triple therapy, namely β-blockers plus LCSD plus ICD.
Gene-Specific Therapy and Management
There has been major progress in understanding the genotype-phenotype correlation and specifically several of the gene-specific triggers for life-threatening arrhythmias have been identified (41).This has made LQTS the first disease for which a gene-specific management has become possible and it is opening previously unforeseen preventive and therapeutic strategies.
LQT1 patients are at higher risk during sympathetic activation, such as during exercise and emotions (41). They should not participate in competitive sports. Swimming is particularly dangerous, as 99% of the arrhythmic episodes associated with swimming occur in LQT1 patients (41).
LQT2 patients are exquisitely sensitive to serum K+ levels, which should not be allowed to fall. When reasonable levels are not maintained by diet or by oral K+ supplements, a combination with K+ sparing agents should be considered. As these patients are at higher risk especially when aroused from sleep or rest by a sudden noise (7,41), we recommend that telephones and alarm clocks are removed from their bedrooms. Also when parents in the morning have to wake up their children, they should do it gently and without yelling. This combines good manners and gene-specific management.
The demonstration that LQT3-causing SCN5A mutations have a “gain-of-function” effect (17) led to test sodium channel blockers as possible adjuvants in the management of LQT3 patients (18). Among these drugs, flecainide is seldom used by our group (50). There is growing interest for ranolazine because of its specific effect on the delayed current but clinical data are still scanty, and most of the current clinical experience is with mexiletine. The effect of mexiletine is mutation-specific (73) and that is why we always test its effectiveness in all LQT3 patients under continuous ECG monitoring by the acute oral drug test technique - performed in hospital in an out-patient basis - using half of the daily dose,. Within 90 minutes the peak plasma concentration is reached and if the QTc is shortened by more than 40 ms then we add mexiletine to ß-blocker therapy. Even though there is no conclusive evidence for a beneficial effect and definite failures have occurred, there is also growing evidence of significant benefit in a number of individual cases. We observed highly malignant forms manifesting in infancy due to mutations causing extremely severe electrophysiological dysfunctions which were corrected by the combination of mexiletine and propranolol (74).
Independent of genotype, all LQTS patients should avoid any cardiac or non-cardiac drug that blocks the IKr current. A list of such drugs is available at www.torsades.org and should be given to every patient because their family physician may not be aware of these electrophysiological actions. This is a precise responsibility of the cardiologist who follows these patients.
Asymptomatic LQTS Patients and Patients with a normal QTc
ß-blocker treatment should be initiated in all patients including those still asymptomatic because in 10–12% of LQTS cases the first clinical manifestation is sudden death.. Among these, reasonable exceptions appear to be LQT1 males above age 40 because very seldom they have a first event above this age, and possibly individuals above age 50 with a QTc <480 ms. LQT2 women remain at risk throughout life and it is wise to always treat them, with few exceptions.
Patients with a normal QTc (<440 ms) constitute approximately 25% of the LQTS population and have a markedly lower risk for life-threatening events compared with phenotypically affected patients but are at higher risk compared with unaffected family-members (75). We usually do not treat them but an individual assessment, including age evaluation and family history, is appropriate. Additionally, follow-up visits are recommended to monitor the stability of their condition. Finally, among LQT1 and LQT3 patients with a normal QT interval, those with missense mutations in the transmembrane regions have a non-negligible risk of life-threatening arrhythmias (75); therefore, a mutation-specific evaluation should integrate the clinical evaluation whenever possible.
Acknowledgment
We are very grateful to Pinuccia De Tomasi for expert editorial support.
Funding Sources: NIH grant HL68880; Telethon Italy grant GGP09247
Footnotes
Conflict of Interest Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J. 1975;89:378–390. doi: 10.1016/0002-8703(75)90089-7. [DOI] [PubMed] [Google Scholar]
- 2.Romano C, Gemme G, Pongiglione R. Aritmie cardiache rare dell'età pediatrica. Clin Pediat. 1963;45:656–683. [PubMed] [Google Scholar]
- 3.Ward OC. A new familial cardiac syndrome in children. J Irish Med Ass. 1964;54:103–106. [PubMed] [Google Scholar]
- 4.Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am Heart J. 1957;54:59–68. doi: 10.1016/0002-8703(57)90079-0. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, Shkolnikova M, Berul CI, Bitner-Glindzicz M, Toivonen L, Horie M, Schulze-Bahr E, Denjoy I. The Jervell and Lange-Nielsen Syndrome. Natural history, molecular basis, and clinical outcome. Circulation. 2006;113:783–790. doi: 10.1161/CIRCULATIONAHA.105.592899. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J. 1985;109:399–411. doi: 10.1016/0002-8703(85)90626-x. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz PJ, Crotti L. Long QT and short QT syndromes. In: Zipes DP, Jalife J, editors. CARDIAC ELECTROPHYSIOLOGY: FROM CELL TO BEDSIDE. 5th Edition Elsevier/Saunders; Philadelphia: 2009. pp. 731–744. [Google Scholar]
- 8.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 9.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- 10.Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KvLQT1 mutations cause cardiac arrhythmias. Nature Genetics. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- 11.Kapplinger JD, Tester DJ, Salisbury BA, Carr JL, Harris-Kerr C, Pollevick GD, Wilde AA, Ackerman MJ. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. doi: 10.1016/j.hrthm.2009.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 13.Crotti L, Spazzolini C, Schwartz PJ, Shimizu W, Denjoy I, Schulze-Bahr E, Zaklyazminskaya EV, Swan H, Ackerman MJ, Moss AJ, Wilde AA, Horie M, Brink PA, Insolia R, De Ferrari GM, Crimi G. The common Long QT syndrome mutation KCNQ1/A341V causes unusually severe clinical manifestations in patients with different ethnic backgrounds: toward a mutation-specific risk stratification. Circulation. 2007;116:2366–2375. doi: 10.1161/CIRCULATIONAHA.107.726950. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz PJ, Priori SG, Napolitano C. How really rare are rare diseases? The intriguing case of independent compound mutations in the long QT syndrome. J Cardio siol. 2003;14:1120–1121. doi: 10.1046/j.1540-8167.2003.03339.x. [DOI] [PubMed] [Google Scholar]
- 15.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004;109:1834–1841. doi: 10.1161/01.CIR.0000125524.34234.13. [DOI] [PubMed] [Google Scholar]
- 16.Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, Makiyama T, Akao M, Ai T, Noda T, Miyazaki A, Miyamoto Y, Yamagishi M, Kamakura S, Horie M. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart Rhythm. 2010;7:1411–1418. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Bennett PB, Yazawa K, Makita N, George AL., Jr. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 19.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of Na+ channel-linked long-QT syndrome. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 20.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 21.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 23.Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, Valdivia C, Ueda K, Canizales-Quinteros S, Tusie-Luna MT, Makielski JC, Ackerman MJ. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–142. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci USA. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci USA. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. Cav1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Yang Y, Liang B, Liu J, Li J, Grunnet M, Olesen SP, Rasmussen HB, Ellinor PT, Gao L, Lin X, Li L, Wang L, Xiao J, Liu Y, Liu Y, Zhang S, Liang D, Peng L, Jespersen T, Chen YH. Identification of a Kir3.4 mutation in congenital Long QT Syndrome. Am J Hum Genet. 2010;86:872–880. doi: 10.1016/j.ajhg.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P, Spazzolini C. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3:18–33. doi: 10.1186/1750-1172-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malfatto G, Beria G, Sala S, Bonazzi O, Schwartz PJ. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol. 1994;23:296–301. doi: 10.1016/0735-1097(94)90410-3. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz PJ, Malliani A. Electrical alternation of the T wave. Clinical and experimental evidence of its relationship with the sympathetic nervous system and with the long QT syndrome. Am Heart J. 1975;89:45–50. doi: 10.1016/0002-8703(75)90008-3. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz PJ, Spazzolini C, Crotti L. All LQT3 patients need an ICD. True or false? Heart Rhythm. 2009;6:113–120. doi: 10.1016/j.hrthm.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–784. doi: 10.1161/01.cir.88.2.782. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz PJ. The congenital long QT syndromes from genotype to phenotype: clinical implications. J Intern Med. 2006;259:39–47. doi: 10.1111/j.1365-2796.2005.01583.x. [DOI] [PubMed] [Google Scholar]
- 35.Sy RW, Van der Werf C, Chatta I, Chockalingam P, Adler A, Healey JS, Perrin M, Gollob MH, Skanes AC, Yee R, Gula LJ, Leong –Sit P, Viskin S, Klein GJ, Wilde AA, Krahn AD. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation. doi: 10.1161/CIRCULATIONAHA.111.028258. In press. [DOI] [PubMed] [Google Scholar]
- 36.Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long QT syndrome. Circulation. doi: 10.1161/CIRCULATIONAHA.111.062182. In press. [DOI] [PubMed] [Google Scholar]
- 37.Taggart NW, Haglund CM, Tester DJ, Ackerman MJ. Diagnostic miscues in congenital long-QT syndrome. Circulation. 2007;115:2613–2620. doi: 10.1161/CIRCULATIONAHA.106.661082. [DOI] [PubMed] [Google Scholar]
- 38.Hofman N, Tan HL, Alders M, van Langen IM, Wilde AA. Active cascade screening in primary inherited arrhythmia syndromes: does it lead to prophylactic treatment? J Am Coll Cardiol. 2010;55:2570–2576. doi: 10.1016/j.jacc.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz PJ. Cascades or waterfalls, the cataracts of genetic screening are being opened on clinical cardiology. J Am Coll Cardiol. 2010;55:2577–2579. doi: 10.1016/j.jacc.2009.12.064. [DOI] [PubMed] [Google Scholar]
- 40.Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R, Cappelletti D. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147. [DOI] [PubMed] [Google Scholar]
- 41.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 42.Brink PA, Crotti L, Corfield V, Goosen A, Durrheim G, Hedley P, Heradien M, Geldenhuys G, Vanoli E, Bacchini S, Spazzolini C, Lundquist AL, Roden DM, George AL, Jr, Schwartz PJ. Phenotypic variability and unusual clinical severity of congenital Long QT Syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz PJ, Vanoli E, Crotti L, Spazzolini C, Ferrandi C, Goosen A, Hedley P, Heradien M, Bacchini S, Turco A, La Rovere MT, Bartoli A, George AL, Jr, Brink PA. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J Am Coll Cardiol. 2008;51:920–929. doi: 10.1016/j.jacc.2007.09.069. [DOI] [PubMed] [Google Scholar]
- 44.Khositseth A, Tester DJ, Will ML, Bell CM, Ackerman MJ. Identification of a common genetic substrate underlying postpartum cardiac events in congenital long QT syndrome. Heart Rhythm. 2004;1:60–64. doi: 10.1016/j.hrthm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 45.Heradien MJ, Goosen A, Crotti L, Durrheim G, Corfield V, Brink PA, Schwartz PJ. Does pregnancy increase cardiac risk for LQT1 patients? J Am Coll Cardiol. 2006;48:1410–1415. doi: 10.1016/j.jacc.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 46.Moss AJ, Zareba W, Kaufman ES, Gartman E, Peterson DR, Benhorin J, Towbin JA, Keating MT, Priori SG, Schwartz PJ, Vincent GM, Robinson JL, Andrews ML, Feng C, Hall WJ, Medina A, Zhang L, Wang Z. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation. 2002;105:794–799. doi: 10.1161/hc0702.105124. [DOI] [PubMed] [Google Scholar]
- 47.Moss AJ, Shimizu W, Wilde AA, Towbin JA, Zareba W, Robinson JL, Qi M, Vincent GM, Ackerman MJ, Kaufman ES, Hofman N, Seth R, Kamakura S, Miyamoto Y, Goldenberg I, Andrews ML, McNitt S. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation. 2007;115:2481–2489. doi: 10.1161/CIRCULATIONAHA.106.665406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makita N, Behr E, Shimizu W, Horie M, Sunami A, Crotti L, Schulze-Bahr E, Fukuhara S, Mochizuki N, Makiyama T, Itoh H, Christiansen M, McKeown P, Miyamoto K, Kamakura S, Tsutsui H, Schwartz PJ, George AL, Jr, Roden DM. The E1784K mutation in SCN5A gene is associated with mixed clinical phenotype of type 3 long QT syndrome. J Clin Invest. 2008;118:2219–2229. doi: 10.1172/JCI34057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crotti L. Pleiotropic mutations in ion channels: What lies behind them? Heart Rhythm Heart Rhythm. 2011;8:56–57. doi: 10.1016/j.hrthm.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Priori SG, Napolitano C, Schwartz PJ, Bloise R, Crotti L, Ronchetti E. The elusive link between LQT3 and Brugada syndrome. The role of flecainide challenge. Circulation. 2000;102:945–947. doi: 10.1161/01.cir.102.9.945. [DOI] [PubMed] [Google Scholar]
- 51.Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: clinical impact. Circulation. 1999;99:529–533. doi: 10.1161/01.cir.99.4.529. [DOI] [PubMed] [Google Scholar]
- 52.Crotti L, Lundquist AL, Insolia R, Pedrazzini M, Ferrandi C, De Ferrari GM, Vicentini A, Yang P, Roden DM, George AL, Jr, Schwartz PJ. KCNH2-K897T is a genetic modifier of latent congenital long QT syndrome. Circulation. 2005;112:1251–1258. doi: 10.1161/CIRCULATIONAHA.105.549071. [DOI] [PubMed] [Google Scholar]
- 53.Brink PA, Schwartz PJ. Of founder populations, long QT syndrome, and destiny. Heart Rhythm. 2009;6:S25–S33. doi: 10.1016/j.hrthm.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz PJ. Sudden cardiac death, founder populations and mushrooms. What is the link with gold mines and modifier genes? Heart Rhythm. 2011;8:548–550. doi: 10.1016/j.hrthm.2010.12.035. [DOI] [PubMed] [Google Scholar]
- 55.Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ, George AL., Jr. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomás M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, Spooner PM, Priori SG. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol. 2010;55:2745–2752. doi: 10.1016/j.jacc.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 57.Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A, Laugwitz KL. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363:1397–1409. doi: 10.1056/NEJMoa0908679. [DOI] [PubMed] [Google Scholar]
- 58.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471:225–229. doi: 10.1038/nature09747. [DOI] [PubMed] [Google Scholar]
- 59.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with Long-QT Syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011;109:841–847. doi: 10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- 60.Chockalingam P, Girardengo G, Crotti L, Johnson JN, Harris KM, van der Heijden JF, Hauer RNW, Beckmann BM, Rydberg A, Clur SAB, Fischer M, van den Heuvel F, Kääb S, Blom NA, Ackerman MJ, Schwartz PJ, Wilde AAM. β-blockers in the treatment of congenital long QT syndrome types 1 and 2, a comparison of propranolol, metoprolol and nadolol. Submitted for publication. [Google Scholar]
- 61.Chatrath R, Bell CM, Ackerman MJ. β-blocker therapy failures in symptomatic probands with genotyped long-QT syndrome. Pediatr Cardiol. 2004;25:459–465. doi: 10.1007/s00246-003-0567-3. [DOI] [PubMed] [Google Scholar]
- 62.Besana A, Wang DW, George AL, Jr, Schwartz PJ. Nadolol block of Nav1.5 does not explain its efficacy in the long QT syndrome. J Cardiovasc Pharmacol. 2011 Oct 25; doi: 10.1097/FJC.0b013e31823d2fd1. [Epub ahead of print] PMID: 22030895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J, Vincent GM, Locati EH, Priori SG, Napolitano C, Medina A, Zhang L, Robinson JL, Timothy K, Towbin JA, Andrews ML. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- 64.Priori SG, Napolitano C, Schwartz PJ, Grillo M, Bloise R, Ronchetti E, Moncalvo C, Tulipani C, Veia A, Bottelli G, Nastoli J. Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA. 2004;292:1341–1344. doi: 10.1001/jama.292.11.1341. [DOI] [PubMed] [Google Scholar]
- 65.Vincent GM, Schwartz PJ, Denjoy I, Swan H, Bithell C, Spazzolini C, Crotti L, Piippo K, Lupoglazoff JM, Villain E, Priori SG, Napolitano C, Zhang L. High efficacy of beta-blockers in long QT syndrome type 1: contribution of non-compliance and QT prolonging drugs to the occurrence of beta-blocker treatment “failures”. Circulation. 2009;119:215–221. doi: 10.1161/CIRCULATIONAHA.108.772533. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz PJ, Priori SG, Cerrone M, Spazzolini C, Odero A, Napolitano C, Bloise R, De Ferrari GM, Klersy C, Moss AJ, Zareba W, Robinson JL, Hall WJ, Brink PA, Toivonen L, Epstein AE, Li C, Hu D. Left cardiac sympathetic denervation in the management of high-risk patients affected by the long QT syndrome. Circulation. 2004;109:1826–1833. doi: 10.1161/01.CIR.0000125523.14403.1E. [DOI] [PubMed] [Google Scholar]
- 67.Collura CA, Johnson JN, Moir C, Ackerman MJ. Left cardiac sympathetic denervation for the treatment of long QT syndrome and catecholaminergic polymorphic ventricular tachycardia using video-assisted thoracic surgery. Heart Rhythm. 2009;6:752–759. doi: 10.1016/j.hrthm.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 68.Odero A, Bozzani A, De Ferrari GM, Schwartz PJ. Left cardiac sympathetic denervation for the prevention of life-threatening arrhythmias. the surgical supraclavicular approach to cervico thoracic sympathectomy. Heart Rhythm. 2010;7:1161–1165. doi: 10.1016/j.hrthm.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 69.Schwartz PJ, Snebold NG, Brown AM. Effects of unilateral cardiac sympathetic denervation on the ventricular fibrillation threshold. Am J Cardiol. 1976;37:1034–1040. doi: 10.1016/0002-9149(76)90420-3. [DOI] [PubMed] [Google Scholar]
- 70.Schwartz PJ. Cutting nerves and saving lives. Heart Rhythm. 2009;6:760–763. doi: 10.1016/j.hrthm.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 71.Schwartz PJ. Efficacy of left cardiac sympathetic denervation has an unforeseen side effect: medicolegal complications. Heart Rhythm. 2010;7:1330–1332. doi: 10.1016/j.hrthm.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 72.Schwartz PJ, Spazzolini C, Priori SG, Crotti L, Vicentini A, Landolina M, Gasparini M, Wilde AAM, Knops RE, Denjoy I, Toivonen L, Mönnig G, Al-Fayyadh M, Jordaens L, Borggrefe M, Holmgren C, Brugada P, De Roy L, Hohnloser SH, Brink PA. Who are the long-QT syndrome patients who receive an implantable cardioverter defibrillator and what happens to them? Data from the European long-QT syndrome implantable cardioverter-defibrillator (LQTS ICD) Registry. Circulation. 2010;122:1272–1282. doi: 10.1161/CIRCULATIONAHA.110.950147. [DOI] [PubMed] [Google Scholar]
- 73.Ruan Y, Liu N, Bloise R, Napolitano C, Priori SG. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation. 2007;116:1137–1144. doi: 10.1161/CIRCULATIONAHA.107.707877. [DOI] [PubMed] [Google Scholar]
- 74.Wang DW, Crotti L, Shimizu W, Pedrazzini M, Cantù F, De Filippo P, Kishiki K, Miyazaki A, Ikeda T, Schwartz PJ, George AL., Jr. Malignant perinatal variant of long QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circ Arrhythm Electrophysiol. 2008;1:370–378. doi: 10.1161/CIRCEP.108.788349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldenberg I, Horr S, Moss AJ, Lopes CM, Barsheshet A, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Platonov PJ, Priori SG, Qi M, Schwartz PJ, Shimizu W, Towbin JA, Vincent GM, Wilde AAM, Zhang L. Risk for life-threatening cardiac events in patients with genotype-confirmed Long-QT Syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–59. doi: 10.1016/j.jacc.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]