

Hep1 acts as a specialized chaperone to mediate the de novo folding of yeast mitochondrial Hsp70.
Abstract
Chaperones mediate protein folding and prevent deleterious protein aggregation in the cell. However, little is known about the biogenesis of chaperones themselves. In this study, we report on the biogenesis of the yeast mitochondrial Hsp70 (mtHsp70) chaperone, which is essential for the functionality of mitochondria. We show in vivo and in organello that mtHsp70 rapidly folds after its import into mitochondria, with its ATPase domain and peptide-binding domain (PBD) adopting their structures independently of each other. Importantly, folding of the ATPase domain but not of the PBD was severely affected in the absence of the Hsp70 escort protein, Hep1. We reconstituted the folding of mtHsp70, demonstrating that Hep1 and ATP/ADP were required and sufficient for its de novo folding. Our data show that Hep1 bound to a folding intermediate of mtHsp70. Binding of an adenine nucleotide triggered release of Hep1 and folding of the intermediate into native mtHsp70. Thus, Hep1 acts as a specialized chaperone mediating the de novo folding of an Hsp70 chaperone.
Introduction
Molecular chaperones help newly synthesized proteins to fold efficiently into their three-dimensional native structure in the cell (Frydman, 2001; Bukau et al., 2006; Hartl et al., 2011). A major class of molecular chaperones is comprised of the Hsp70 chaperones. In addition to their role in protein folding, Hsp70 chaperones have a variety of other crucial cellular functions, including prevention of protein aggregation, intracellular transport of proteins, assembly of oligomeric complexes, and regulation of protein degradation (Kang et al., 1990; Frydman, 2001; Bukau et al., 2006; Eisenberg and Greene, 2007; Nakatsukasa and Brodsky, 2008; Douglas and Cyr, 2010; Hartl et al., 2011). Because of their fundamental role in cellular proteostasis, Hsp70 chaperones are important for cell survival and present in almost all cellular compartments.
Mitochondria provide the cell with several essential metabolites and compounds required for the life of the cell but also with components triggering cell death. The homeostasis and function of mitochondria are crucially dependent on mitochondrial Hsp70 (mtHsp70) chaperones, the closest eukaryotic homologues of bacterial DnaK (Craig et al., 1987). The main mtHsp70 chaperone in Saccharomyces cerevisiae, the mtHsp70 protein, is also known in higher eukaryotes as mortalin because of its role in cell survival, cell death, and diseases (Wadhwa et al., 2002; Burbulla et al., 2010). It resides mainly in the mitochondrial matrix, where it promotes the folding of proteins (Kang et al., 1990). Another fraction of mtHsp70 is associated with the translocation channel of the TIM23 translocase on the matrix side of the inner membrane and functions as a central component of the import motor of the translocase (Endo and Yamano, 2009; Mokranjac and Neupert, 2010; van der Laan et al., 2010).
The structural organization and the molecular mechanism of Hsp70 chaperones are well conserved. The mtHsp70 chaperones, similar to other members of the Hsp70 chaperone family, consist of an N-terminal ATPase domain that is connected via a short hydrophobic interdomain linker to a C-terminal peptide-binding domain (PBD; Bukau and Horwich, 1998; Frydman, 2001; Hartl et al., 2011). The PBD interacts with the unfolded substrates. Its affinity to the substrates is regulated by the nucleotide state of the ATPase domain. In contrast, substrate binding to the PBD results in a conformational change of the ATPase domain stimulating its ATPase activity. The communication between the domains is mediated by the interdomain linker, which is therefore important for the function of Hsp70 proteins (Mayer et al., 1999; Han and Christen, 2001; Vogel et al., 2006; Jiang et al., 2007; Swain et al., 2007; Zhuravleva and Gierasch, 2011). Of similar importance is the interaction and cooperation of the Hsp70 chaperones with their cochaperones (Frydman, 2001; Bukau et al., 2006; Hartl et al., 2011). The J domain–containing cochaperones stimulate the ATPase activity of Hsp70, and the nucleotide exchange factors catalyze the exchange of ADP versus ATP, thereby accelerating the ATP-dependent reaction cycle of the Hsp70 chaperones. The mtHsp70 chaperone interacts with the nucleotide exchange factor Mge1 and employs as J cochaperone either Mdj1 in the process of protein folding or Tim14 in the process of protein translocation (Rowley et al., 1994; Miao et al., 1997; D’Silva et al., 2003; Mokranjac et al., 2003; Truscott et al., 2003; Voos, 2009).
mtHsp70 requires another crucial interaction partner, Hep1 (also known as Tim15 and Zim17), the Hsp70 escort protein which is highly conserved throughout the eukaryotic kingdom (Burri et al., 2004; Sanjuán Szklarz et al., 2005; Sichting et al., 2005; Yamamoto et al., 2005; Willmund et al., 2008; Zhai et al., 2008; Blamowska et al., 2010). Its chloroplast homologue HEP2 also interacts with plant Hsp70B and is important for the function of Hsp70B (Willmund et al., 2008). In the absence of Hep1, yeast cells have multiple defects such as reduced import of precursor proteins, altered mitochondrial morphology, reduced activity of Fe/S cluster–containing enzymes, and nuclear genome instability and are not viable at elevated temperature (Burri et al., 2004; Sanjuán Szklarz et al., 2005; Sichting et al., 2005; Yamamoto et al., 2005; Díaz de la Loza et al., 2011). All of the phenotypes described can be explained as secondary effects of reduced levels of functional mtHsp70 chaperones, although a role of Hep1 in the TIM23 translocase has also been suggested (Burri et al., 2004; Sanjuán Szklarz et al., 2005; Sichting et al., 2005; Yamamoto et al., 2005). We and others have previously shown that the mtHsp70 chaperone accumulates as insoluble aggregates in the absence of Hep1 in yeast cells (Sanjuán Szklarz et al., 2005; Sichting et al., 2005). It is the ATPase domain in the presence of the linker segment that makes mtHsp70 prone to aggregation (Zhai et al., 2008; Blamowska et al., 2010). Moreover, the ATPase domain plus linker is the minimal binding entity of mtHsp70 crucial for interaction with Hep1 (Zhai et al., 2008; Blamowska et al., 2010). Hep1 prevents self-aggregation of folded mtHsp70 in its nucleotide-free form and thereby maintains the function of the folded mtHsp70 chaperone (Sichting et al., 2005). This suggested that native mtHsp70 in an off-pathway of its reaction cycle forms an aggregation-prone conformer recognized by Hep1. Interestingly, the human mitochondrial HEP was reported to stimulate the ATPase activity of the cognate recombinant mtHsp70, conceivably representing an additional function as cochaperone similar to the one of J proteins (Zhai et al., 2008; Goswami et al., 2010). Although the importance of Hep proteins for mtHsp70 chaperones is well documented, the physiological function of their interaction has remained unclear.
Despite the fundamental cellular importance of Hsp70 chaperones, there is limited knowledge about the biogenesis of these chaperones. In particular, the de novo folding of mtHsp70 in mitochondria and a potential role of Hep1 in this process is an open question. We performed experiments in vivo and in organello that revealed that the folding of mtHsp70, in particular of its ATPase domain, is affected in the absence of Hep1. Moreover, we reconstituted the folding of mtHsp70 and the individual domains using the isolated proteins. In this reconstituted system, Hep1 and ATP/ADP were required and sufficient for the folding of mtHsp70 into its active conformation. In summary, our results demonstrate for the first time that the de novo folding of an Hsp70 chaperone in the cell, mtHsp70, depends on a specialized chaperone, the Hep1 chaperone.
Results
mtHsp70 and the individual domains of mtHsp70 fold rapidly upon import into mitochondria
To analyze the biogenesis of mtHsp70 in mitochondria, we established an assay monitoring the folding state of newly imported mtHsp70 in mitochondria. After import of the radiolabeled mtHsp70 precursor protein into isolated mitochondria and solubilization of mitochondria, the folding state of imported mtHsp70 was assessed by treatment with trypsin because the folded native mtHsp70 is trypsin resistant (Sichting et al., 2005). Indeed, almost all imported mtHsp70 was protected against trypsin, suggesting that the imported mtHsp70 folds rapidly in organello (Fig. 1 A).
Figure 1.

The chaperone mtHsp70 and its individual domains fold rapidly in organello. (A–E) Folding of mtHsp70 (A), PBD-DHFR (B), ATPase-DHFR (C), ATPaseLinker-DHFR (D), and ATPaseA4-DHFR (E) upon import into mitochondria. Radiolabeled precursor proteins were imported into wild-type mitochondria for the time periods indicated. Nonimported material was digested with trypsin. Mitochondria were reisolated and solubilized. Half of the sample was mock treated (−) to assess import, and the other half was treated with trypsin (+) to assess the folding state of the imported mtHsp70. Samples were analyzed by SDS-PAGE and autoradiography and, in the case of mtHsp70, quantification by densitometry (A, right). The amount of mtHsp70 imported at the latest time point was set to 100%. The graph represents the mean values ± standard deviation (n = 5). The asterisks indicate the folded DHFR domain with nondigested spacer.
We also tested the de novo folding of the individual mtHsp70 domains. To mimic the two-domain structure of full-length mtHsp70, the individual domains were analyzed in the context of dihydrofolate reductase (DHFR) fusion proteins, from now on referred to as PBD-DHFR and ATPase-DHFR, respectively, in which the PBD and the ATPase domain were each fused with an 80-aa-residue-long spacer to the mouse DHFR. Trypsin treatment of imported PBD-DHFR generated two protease-resistant fragments, corresponding to the PBD and the DHFR domain (Fig. 1 B). The kinetics of import of PBD-DHFR and of the formation of the protease-resistant fragments were very similar, indicating that the PBD-DHFR folds immediately after its import into the mitochondrial matrix. In the case of the ATPase-DHFR construct, the DHFR domain was protease resistant, but the ATPase domain was almost completely protease sensitive (Fig. 1 C). Thus, the ATPase domain does not fold properly in the context of the DHFR fusion protein. On the contrary, the PBD folds independently of the ATPase domain.
Because the interdomain linker between the ATPase domain and PBD is required for the communication between both domains and affects the native conformation of the ATPase domain, we asked whether the interdomain linker has an effect on the de novo folding of the ATPase domain. We followed the folding of imported ATPaseLinker-DHFR protein and tested in parallel, as a control, the folding of the ATPaseA4-DHFR variant, in which the linker amino acid residues 412–415 were replaced by four alanine residues. The ATPase domain in imported ATPaseLinker-DHFR but not in the ATPaseA4-DHFR variant folded into a trypsin-resistant conformation (Fig. 1, D and E). In both cases, the folded DHFR domain was protease resistant. We conclude that the folding of the ATPase domain depends on the presence of the interdomain linker. This is supported by experiments using a variant of full-length mtHsp70 protein, mtHsp70A4, in which the linker residues of mtHsp70 were likewise replaced by four alanine residues. This variant generated in the folding assay only a 35-kD stable fragment that corresponds in size to the PBD (Fig. S1). By immunoprecipitation experiments with antibodies recognizing specifically the ATPase domain or the C terminus of mtHsp70, we confirmed the fragment to be the PBD (Fig. S1). Thus, in contrast to the PBD, the ATPase domain in the mtHsp70A4 mutant was not able to fold into a protease-resistant form.
In summary, mtHsp70 folds rapidly after its import into mitochondria. Both domains are independent folding units. The ATPase domain folds in the context of mtHsp70 only in presence of the interdomain linker, whereas the PBD folds without the interdomain linker.
Hep1 is required for the folding of the ATPase domain of mtHsp70 in organello
We asked whether other components are involved in the folding process of mtHsp70 in the cell. Because Hep1 interacts with mtHsp70 and is crucial for maintaining native mtHsp70 in its functional state (Sichting et al., 2005), we analyzed whether Hep1 also acts in the folding of mtHsp70. To this end, we imported radiolabeled mtHsp70 into mitochondria lacking Hep1 and checked its folding state by treating mitochondrial extracts with trypsin. A 35-kD stable fragment of mtHsp70 was observed but not the full-length mtHsp70 (Fig. 2 A). Thus, in contrast to wild-type mitochondria, the folding of mtHsp70 was impaired in mitochondria lacking Hep1. The stable fragment could be immunoprecipitated with antibodies against the C terminus of mtHsp70, demonstrating that it corresponds to the PBD (Fig. S2). This shows that the PBD folds independently of Hep1, whereas the ATPase domain has a folding defect in the absence of Hep1.
Figure 2.
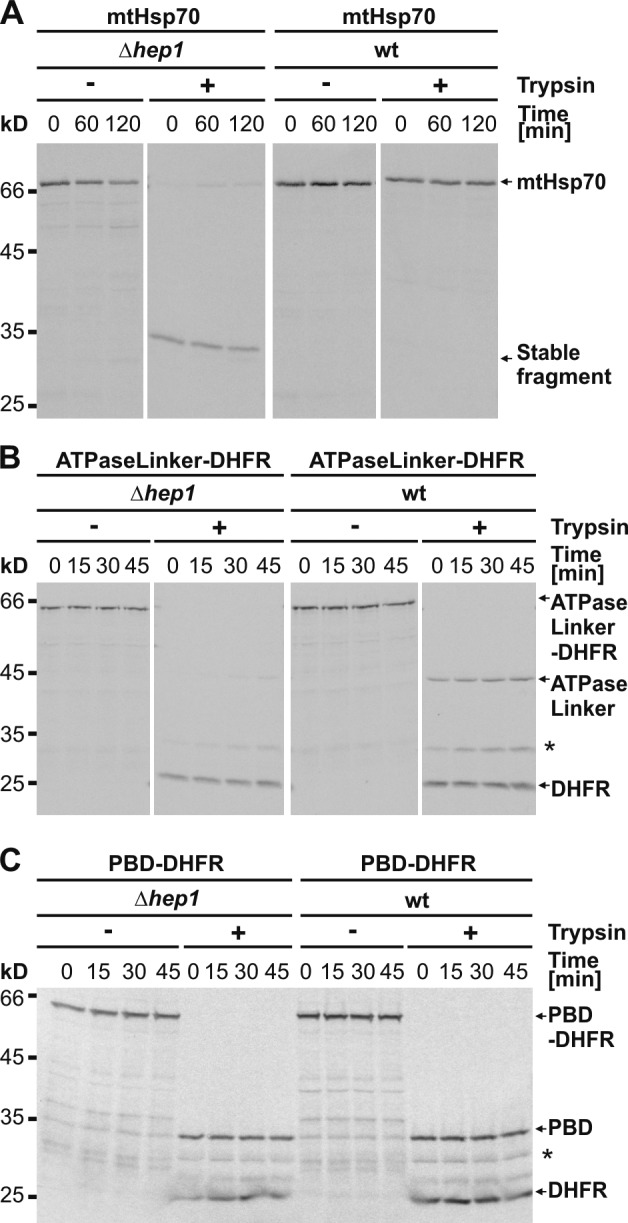
Folding of mtHsp70 in organello depends on the Hep1 chaperone. (A–C) Folding of mtHsp70 (A), ATPaseLinker-DHFR (B), and PBD-DHFR (C) upon import into mitochondria lacking Hep1. Radiolabeled precursor proteins were imported into Δhep1 and wild-type (wt) mitochondria. Import was stopped by addition of valinomycin, and samples were further incubated for the time periods indicated and treated as described in Fig. 1 A. Asterisks indicate folded DHFR domain with nondigested spacer.
To corroborate these findings, we assessed the folding of the two mtHsp70 constructs, ATPaseLinker-DHFR and PBD-DHFR, in Δhep1 mitochondria. The ATPaseLinker domain did not become folded, in contrast to the situation with wild-type mitochondria (Fig. 2 B). As expected, the PBD was able to fold also in the Hep1-deficient mitochondria (Fig. 2 C).
In conclusion, these results demonstrate that the chaperone Hep1 is required for de novo folding of mtHsp70 in organello. It mediates the correct folding of the ATPase domain plus linker of mtHsp70.
Folding of mtHsp70 is independent of Hsp60, Hsp78, and the mtHsp70 chaperone system
The mitochondrial matrix contains several chaperones promoting protein folding such as the Hsp60 chaperonin, the mtHsp70 chaperone system, and Hsp78, a member of the Clp/Hsp100 family (Voos, 2009). Do these mitochondrial chaperones assist in the de novo folding of mtHsp70? We addressed this question by using strains harboring deletions or temperature-sensitive mutants of these chaperones. Folding of mtHsp70 was not affected in the mif4 temperature-sensitive mutant (Fig. 3 A). In this mutant, Hsp60 does not assemble and therefore aggregates at nonpermissive temperature, as previously reported (Cheng et al., 1990). Moreover, imported mtHsp70 folded properly with comparable kinetics in the presence and absence of Hsp78 (Fig. 3 B). Thus, neither Hsp60 nor Hsp78 are crucial for folding of mtHsp70. We also tested the mtHsp70 system itself. Deletion of the J domain cochaperone Mdj1 did not affect the folding rates of imported mtHsp70 (Fig. 3 C), suggesting that the folding of mtHsp70 is independent of the mtHsp70 system.
Figure 3.
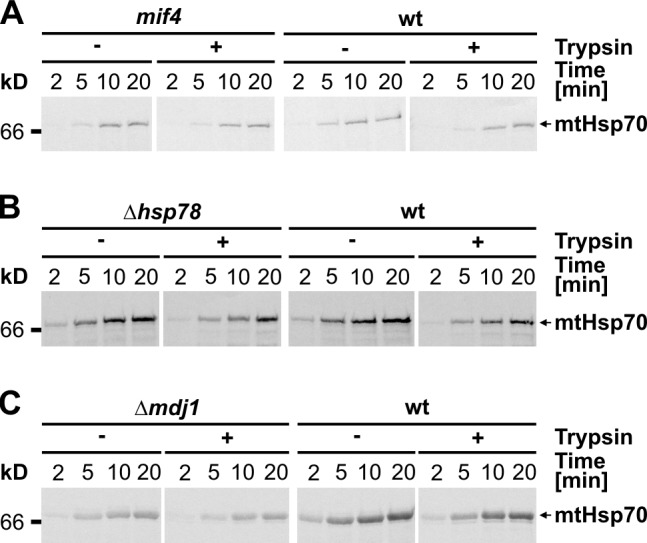
Defects in mitochondrial chaperone systems do not affect folding of mtHsp70. (A–C) Radiolabeled mtHsp70 precursor was imported into mitochondria isolated from the mif4 mutant cells (A), Δhsp78 cells (B), and Δmdj1 cells (C) and from the corresponding wild-type (wt) cells for the time periods indicated. Samples were treated and analyzed as described in Fig. 1 A.
Collectively, besides Hep1, none of the tested chaperones appear to be involved in the de novo folding of mtHsp70. This suggests Hep1 to be the major, if not the only, chaperone mediating the folding of mtHsp70 in mitochondria.
Hep1 is required for de novo folding of mtHsp70 in vivo
To confirm the role of Hep1 in the biogenesis of mtHsp70 in intact cells, we performed a labeling experiment with wild-type cells and with cells lacking Hep1. Cells were grown in the presence of radioactive methionine to synthesize radiolabeled proteins. This enabled us to follow their biogenesis. Mitochondria were isolated and mitochondrial extracts were prepared. Treatment of these extracts with trypsin followed by the immunoprecipitation with antibodies directed against mtHsp70 allowed the specific detection of the folding state of the newly synthetized and imported mtHsp70 protein. In mitochondrial extracts from wild-type cells, the majority of mtHsp70 was trypsin resistant, indicating its presence in the folded state. In contrast, in mitochondrial extracts from Δhep1 cells, only a minor fraction of mtHsp70 was trypsin resistant, and a stable fragment corresponding to the PBD was generated (Fig. 4). Apparently, de novo folding of the ATPase domain of mtHsp70 was impaired in cells lacking Hep1. Thus, consistent with the observations in organello, Hep1 plays an important role in the de novo folding of mtHsp70 in intact cells.
Figure 4.
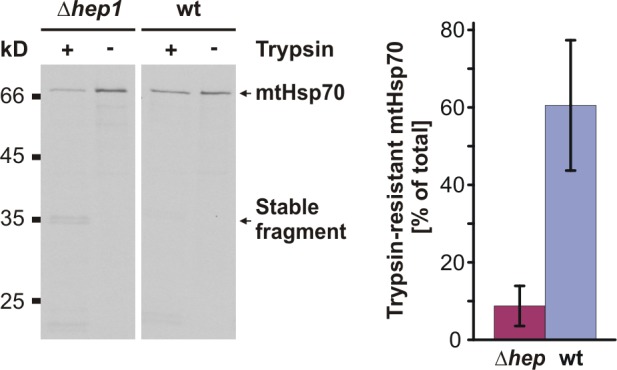
Folding of mtHsp70 in vivo requires Hep1. Δhep1 and wild-type (wt) cells were radiolabeled with [35S]methionine for 10 min. Then protein synthesis was stopped and mitochondria were isolated and solubilized. The lysate was incubated without and with trypsin to assess the folding of mtHsp70. mtHsp70 was immunoprecipitated with antibodies against mtHsp70. Samples were analyzed by SDS-PAGE and autoradiography (left) and quantified by densitometry (right). Total material (−Trypsin) was set to 100%, and trypsin-resistant material is shown as the percentage of total material. The diagram represents the mean values ± standard deviation (n = 3).
Hep1 is crucial for the folding of mtHsp70 in a reconstituted system
We reconstituted the folding process of mtHsp70 using purified components to elucidate the molecular mechanisms of how mtHsp70 folds in mitochondria. To this end, recombinant mtHsp70 was denatured by precipitation and resuspension in buffer containing urea. The sample was diluted in ATP-containing buffer lacking urea with and without addition of recombinant Hep1. Refolding of mtHsp70 was monitored by its trypsin resistance. In the presence of equimolar amounts of Hep1, mtHsp70 became resistant against trypsin in the timescale of minutes (Fig. 5 A, left). In contrast, virtually no trypsin-resistant full-length mtHsp70 was detected in absence of Hep1 (Fig. 5 A, right). Thus, Hep1 promotes refolding of denatured mtHsp70 in the presence of ATP in vitro. Interestingly, two fragments of mtHsp70 with apparent molecular masses of ∼45 and 35 kD were observed when samples were taken from the refolding mix containing Hep1 at early time points and treated with trypsin. These fragments represent folding intermediates of mtHsp70 that correspond, according to their molecular masses, to the ATPase domain and the PBD, respectively. Apparently, each of the domains of mtHsp70 adopts independently a trypsin-resistant conformation in the de novo folding of mtHsp70 before the full-length protein folds into its native, fully protease-protected state. Upon incubation in the absence of Hep1, only the 35-kD fragment was detected, indicating the presence of a folded PBD. This suggests that the PBD is able to fold without any chaperone, whereas the ATPase domain requires Hep1 for its folding in vitro.
Figure 5.

Isolated mtHsp70 folds in the presence of isolated Hep1 and adenine nucleotides into its active form. (A) Unfolded mtHsp70 was diluted into ATP-containing buffer with (left) or without (right) the addition of recombinant Hep1. At the time points indicated, samples were withdrawn and half of the samples were treated with trypsin. Samples were analyzed by SDS-PAGE and Coomassie staining. (B) Unfolded mtHsp70 was diluted into buffer containing ATP and recombinant Hep1 in molar ratios of 1:1, 0.5:1, or 0.25:1 of Hep1 to mtHsp70. Samples were treated and analyzed as described in A, and the signals of trypsin-protected full-length mtHsp70 were quantified by densitometry. Signals of mock-treated samples of the longest time period were set to 100%. The graph represents the mean values ± standard deviation (n = 5). (C) Native and unfolded mtHsp70 were incubated in ATP-containing buffer in the presence of recombinant Hep1 when indicated. Then the ATPase activities of the samples were measured. The ATPase activity of native mtHsp70 was set to 100%. Control indicates unfolded mtHsp70 incubated in absence of Hep1. The diagram represents the mean values of three independent measurements ± standard deviation. (D) Unfolded mtHsp70 was incubated in buffer containing recombinant Hep1 and the indicated nucleotides. After the indicated time periods, samples were withdrawn, treated with trypsin, and further analyzed as described in A. (E and F) Unfolded ATPaseLinker (E) or PBD (F) was diluted into the buffer containing ATP and/or recombinant Hep1. Samples were incubated and analyzed as described in D. Input indicates 100% of protein added in the refolding reactions. (G) Unfolded mtHsp70 or ATPaseLinker was diluted into buffer containing ATP and recombinant Mge1. As a control, recombinant Hep1 was used. Samples were treated as described in D. Input indicates 100% of protein added in the refolding reactions.
Is the rate of refolding of mtHsp70 dependent on the ratio of unfolded mtHsp70 to the added Hep1? The kinetics of refolding was measured at molar ratios of mtHsp70 to Hep1 of 1:1 to 1:0.25 over a period of 120 min. The initial rates of refolding increased with increasing amount of Hep1; the efficiency of refolding, however, was at the same level of ∼100% in the samples with a molar ratio of mtHsp70 to Hep1 of 1:0.5 and of 1:1 (Fig. 5 B). In samples with a lower Hep1 concentration and a molar ratio of 1:0.25, the efficiency of refolding was ∼85%. During incubation with Hep1, mtHsp70 may adopt a conformation that is refractory to refolding and/or Hep1 may lose functional activity. In summary, the data suggest that a Hep1 molecule can support folding of more than one mtHsp70 molecule.
To test whether folding of mtHsp70 in vitro leads to acquisition of catalytic activity, we performed an ATPase activity assay with purified native mtHsp70 and with fractions of mtHsp70 that were subjected to the refolding procedure in the presence and absence of Hep1. mtHsp70 refolded in the presence of Hep1 regained ∼70% of the activity of the native mtHsp70 protein (Fig. 5 C). In contrast, mtHsp70 remained inactive when subjected to the refolding procedure in the absence of Hep1 (Fig. 5 C). Thus, the trypsin-resistant conformation adopted by mtHsp70 upon refolding in the presence of Hep1 obviously represents the active form of the protein.
Are nucleotides involved in the folding process? To address this, we tested the effects of different nucleotides on the folding of mtHsp70 in the presence of Hep1. ATP and ADP promoted the folding of mtHsp70 to the protease-resistant, native form with similar efficiencies. In the presence of a nonhydrolyzable ATP analogue, ATPγS, mtHsp70 became trypsin resistant as well, albeit with slightly reduced efficiency. Trypsin-protected mtHsp70 was formed very inefficiently in the presence of AMP-PNP, another nonhydrolyzable ATP analogue (Fig. 5 D). Folded mtHsp70 was also not observed in the absence of any nucleotides. In conclusion, the de novo folding of mtHsp70 requires nucleotides in addition to Hep1. Because ADP and ATPγS act similarly to ATP, it is rather the binding of nucleotides than the hydrolysis of ATP that appears to be needed for folding. The different effects of the nucleotides suggest that a certain conformer of the ATPase domain is formed as an intermediate during the folding process, which is specifically recognized by nucleotides, thereby triggering further folding to the native state of mtHsp70.
Next, we analyzed the de novo folding of the individual domains of mtHsp70 in the reconstitution system. Denatured ATPaseLinker and PBD were diluted in buffer with and without Hep1 and ATP. The ATPaseLinker protein became trypsin resistant only in the presence of Hep1 and ATP (Fig. 5 E). In contrast, the PBD efficiently refolded even without the addition of Hep1 and ATP (Fig. 5 F). These findings are consistent with the results obtained from the analysis of the folding of the full-length protein in organello and in vitro. Apparently, it is the ATPase domain in full-length mtHsp70 that requires Hep1 and nucleotide for its de novo folding.
Can the function of Hep1 be taken over by Mge1, the nucleotide exchange factor of mtHsp70? It was previously reported that Mge1 is able to keep mtHsp70 soluble upon coexpression in Escherichia coli (Momose et al., 2007). Therefore, we tested the refolding of mtHsp70 in the presence of Mge1 in vitro. In contrast to Hep1, a fourfold molar excess of Mge1 compared with mtHsp70 did not result in folded mtHsp70 and ATPaseLinker (Fig. 5 G). Thus, interaction partners of mtHsp70 such as Mge1 appear not to promote de novo folding of mtHsp70. We conclude that the role of Hep1 in the folding of mtHsp70 reflects a specific chaperone function of Hep1.
Hep1 interacts with a folding intermediate of mtHsp70
A chaperone function of Hep1 in the de novo folding process of mtHsp70 implies a transient interaction of Hep1 with mtHsp70. We tested for such an interaction by chemical cross-linking with glutaraldehyde. To this end, we incubated the purified proteins in the absence or presence of ATP in the refolding buffer with or without cross-linking agent. Samples were analyzed by SDS-PAGE and Coomassie staining or immunodecoration with antibodies against Hep1 and mtHsp70. Upon incubation of mtHsp70 with Hep1, a cross-linked adduct containing Hep1 and mtHsp70 was generated (Fig. 6, A [lanes 8, 13, and 18] and B [lanes 5 and 11]). Importantly, much more adduct was formed in the absence than in the presence of ATP (Fig. 6 A, lanes 8, 9, 13, and 14). Because nucleotides such as ATP are needed to generate folded mtHsp70, a folding intermediate containing Hep1 was most likely trapped in the refolding process in the absence of ATP. In the presence of ATP, a large fraction of the intermediate was converted to fully folded mtHsp70, which was detected as a sharp band on the Coomassie blue–stained gel and in the immunodecoration with mtHsp70 (Fig. 6 A, lane 19). The weak and diffuse signal of mtHsp70 in the lanes lacking either Hep1 or ATP probably reflects the unfolded or partly folded nature of the protein, which allowed extensive modification and intramolecular cross-linking of the protein. In contrast to Hep1, the nucleotide exchange factor Mge1 did not form a cross-linked adduct with refolding mtHsp70 (Fig. 6 B, lanes 5 and 9). Apparently, not every interaction partner of the ATPase domain of native mtHsp70 has the ability to interact with refolding mtHsp70. This lack of interaction between Mge1 and refolding mtHsp70 is consistent with the observation that Mge1 cannot replace Hep1 in the folding of mtHsp70 (Fig. 5 G). In conclusion, binding of Hep1 to a folding intermediate of mtHsp70 is a crucial step of the de novo folding pathway of mtHsp70 that precedes the nucleotide-dependent step.
Figure 6.

A folding intermediate of mtHsp70 interacts with Hep1. (A and B) Unfolded mtHsp70 was diluted into the buffer containing Hep1 in twofold molar excess over mtHsp70 in the presence or absence of ATP, as indicated (A and B), or in a twofold molar excess of Mge1 (B). Samples were incubated at 30°C for 20 min and then treated with or without the cross-linking reagent (glutaraldehyde). Samples were analyzed by SDS-PAGE and Coomassie staining. Aliquots of the samples (15%) were analyzed by SDS-PAGE and immunodecoration with antibodies against Hep1, mtHsp70 (A and B), and Mge1 (B). (C) Unfolded mtHsp70 was incubated with a twofold molar excess of Hep1 for 10 min at 30°C (no chase). Samples were further incubated for 10 min in the absence (−) or presence of nucleotides, when indicated. All samples were treated with glutaraldehyde and analyzed as described in A.
We further analyzed the effect of nucleotides on the formation of the cross-linked adduct. Addition of ATP or ADP after formation of the mtHsp70–Hep1 complex led to release and addition of ATPγS to partial release of mtHsp70 from Hep1, whereas almost no mtHsp70 dissociated from Hep1 upon addition of AMP-PNP (Fig. 6 C). These observations are consistent with the results obtained for the folding of mtHsp70 in the presence of various nucleotides (Fig. 5 D).
In summary, our results confirm a transient interaction of Hep1 with mtHsp70 during de novo folding of mtHsp70. Binding of nucleotide triggers release of Hep1 and further folding of the intermediate to native mtHsp70. Thus, Hep1 fulfils a crucial chaperone function in the de novo folding pathway of mtHsp70.
Discussion
In this study, we report the first example that an Hsp70 chaperone requires a specialized chaperone for a specific step in its biogenesis, the attainment of its native structure. In intact cells and in isolated mitochondria, mtHsp70 folds rapidly de novo upon import into mitochondria. Under both conditions, folding of the ATPase domain of mtHsp70 is impaired in the absence of Hep1 but not folding of its PBD. In addition, isolated mtHsp70 that was unfolded by high concentrations of urea folds efficiently into its catalytically active form in the presence of Hep1 and adenine nucleotides. So far, it had been widely assumed that chaperones fold either spontaneously or with the help of the general chaperone machineries resembling the folding of other proteins. The folding and assembly of the mitochondrial Hsp60 chaperonin, for example, depend on assembled Hsp60 complexes (Cheng et al., 1990). However, as we show here, folding of mtHsp70 depends on Hep1 and cannot be mediated by the general chaperone systems.
Our results allow us to suggest a hypothetical model for the de novo folding of mtHsp70 (Fig. 7). The ATPase domain and the PBD start folding independently of each other. The PBD folds without the help of any additional factor. The ATPase domain forms together with the interdomain linker a folding unit in the context of mtHsp70. Both domains probably fold in parallel rather than sequentially. In the model, the PBD, nonetheless, is illustrated in a folded state because it folds on its own. The ATPase domain needs the chaperone Hep1 and adenine nucleotides, as well as the interdomain linker, to acquire its native structure. The formation of an early folding intermediate plays a central role in the folding of the ATPase domain plus linker. Hep1 recognizes this intermediate and binds to it. The folding intermediate of the ATPase domain in complex with Hep1 has the ability to form a nucleotide-binding site and, therefore, to bind ATP or ADP. Binding of the nucleotide then triggers the release of Hep1 and folding of the ATPase domain into a compact trypsin-resistant conformation. Folded ATPase domain and PBD cooperate together to form the native and catalytically active conformer of full-length mtHsp70. The model presented integrates previous findings that the nucleotide-free form of folded mtHsp70 has the propensity to adopt an aggregation-prone conformation (Sichting et al., 2005). By binding to the aggregation-prone conformer, Hep1 prevents aggregation of mtHsp70 and allows for the recovery of native mtHsp70. Interestingly, as observed for de novo folding, it is also the ATPase domain that requires the help of Hep1 (Zhai et al., 2008; Blamowska et al., 2010). Moreover, in both processes, the ATPase domain in combination with the interdomain linker is necessary for the interaction with Hep1 (Zhai et al., 2008; Blamowska et al., 2010). However, the exact nature of these interactions still has to be elucidated.
Figure 7.

Model of the folding of mtHsp70. The ATPase domain together with the interdomain linker and the PBD fold as individual units in the de novo folding pathway of mtHsp70. The PBD folds on its own; the ATPase domain does not. The ATPase domain in the context of the linker adopts a folding intermediate that is recognized by Hep1. Binding of Hep1 induces conformational changes that allow binding of adenine nucleotides (ATP/ADP). Hep1 is released. The folded ATPase domain and the PBD together form the native and catalytically active mtHsp70. Because the PBD folds independently of any factor, it is presented in a folded state in all folding intermediates, although PBD and ATPase domain probably fold in parallel. The model also depicts the transition of native mtHsp70 in its nucleotide-free state into an aggregation-prone conformer. Hep1 binds to the conformer, preventing aggregation and allowing mtHsp70 to regain its native structure.
The model implies that the folding pathway of mtHsp70 starts from completely unfolded mtHsp70. In the cell, newly synthesized mtHsp70 is imported into mitochondria as an unfolded precursor protein with an N-terminal presequence. Its import is driven by binding of native mtHsp70. In the matrix, newly imported mtHsp70 is released from native mtHsp70 and its presequence is cleaved off. It is unclear whether these two processes have been completed before the polypeptide chain of mtHsp70 starts folding. However, because all results obtained for the de novo folding in vivo and in organello are consistent with the results obtained in the reconstituted system, we conclude that the folding of imported mtHsp70 in mitochondria is reflected by the folding of urea-denatured mtHsp70 in vitro.
Based on the comparative analysis of protein sequences, homologues of Hep1 are found in mitochondria and chloroplasts throughout the eukaryotic kingdom. This raises the question of how Hsp70 chaperones present in other subcellular compartments fold into their native structure. These other Hsp70 chaperones might use functional homologues of Hep1, yet to be identified, that have no sequence homology with Hep1. Alternatively, they might not need an escort protein for their biogenesis because they have structural properties different from those of Hep-dependent Hsp70 chaperones. A Hep-specific folding pathway might have evolved with mitochondrial and chloroplast Hsp70s to assure that these chaperones do not fold in the cytosol and thus stay import competent. In contrast, the Hep-dependent Hsp70 chaperones might need a highly flexible and dynamic structure to interact with various partner proteins and to fulfill various functions in parallel such as protein folding and mitochondrial protein import. Such dynamic structure might come along with a particularly complex and delicate folding pathway requiring Hep1 as folding helper. The closest homologue of mtHsp70, bacterial DnaK, was observed to be present in a soluble form when targeted to the mitochondrial matrix of Δhep1 cells (Blamowska et al., 2010). This suggests that not all Hsp70 chaperones form aggregates in the absence of Hep1 after posttranslational import into mitochondria. DnaK has been reported to form oligomers in vitro and in intact cells (Schönfeld et al., 1995; Thompson et al., 2012). The process of oligomerization is reversible because ATP, excess of peptide substrates, and the cochaperones GrpE and the J domain of DnaJ support monomerization of DnaK (Schönfeld et al., 1995; Thompson et al., 2012). Most likely, the PBD mediates oligomerization of DnaK and cytosolic Hsp70 proteins (Benaroudj et al., 1996; Thompson et al., 2012). Thus, oligomerization of DnaK appears to differ from aggregation of mtHsp70. The ATPase domain of DnaK might be more compact and less susceptible to aggregation than that of mtHsp70. Therefore, DnaK might also need no help in its de novo folding by a Hep1-like protein. Indeed, purified unfolded DnaK can be refolded without any additional component (Clerico et al., 2010). Moreover, the unfolding of recombinant DnaK has been shown to be reversible (Palleros et al., 1992; Montgomery et al., 1993, 1999). Yet it remains to be analyzed whether the efficiency of DnaK folding is increased by proteinaceous factors and whether DnaK requires folding helpers in vivo.
The experiments presented here unambiguously demonstrate that Hep1 plays an important role in the biogenesis of mtHsp70. In vitro, no other chaperone than Hep1 is needed to refold denatured mtHsp70 in the presence of nucleotides. Moreover, other interaction partners of mtHsp70, the nucleotide exchange factor Mge1, as well as the J cochaperone Mdj1, were found not to be able to mediate the refolding of mtHsp70. Therefore, acting as a chaperone in the folding of mtHsp70 is a primary function of Hep1 and not only an effect of its interaction with mtHsp70. This is consistent with the observations that mitochondrial and chloroplast Hsp70s were only present in a soluble active form in E. coli, when they were coexpressed with their cognate Hep1 proteins (Sichting et al., 2005; Momose et al., 2007; Willmund et al., 2008; Zhai et al., 2008). Our results substantiate the suggestion that HEP2 may allow folding and activation of chloroplast Hsp70B after removal of the transit peptide from Hsp70B (Willmund et al., 2008). Hep1 has also been reported to prevent aggregation of native mtHsp70 chaperones (Sanjuán Szklarz et al., 2005; Sichting et al., 2005). Thus, Hep1 acts in protein folding as well as in prevention of protein aggregation, a property typical for many chaperones.
Hep1 is crucial for cell viability at higher temperatures (Sanjuán Szklarz et al., 2005; Sichting et al., 2005). Because Hep1 is required for folding of mtHsp70 also at lower temperatures in vitro (Fig. S3), other factors might help to fold mtHsp70 at lower temperatures in the absence of Hep1 in vivo. However, they probably do so less efficiently than Hep1 and are, therefore, not sufficient at higher temperatures. Potential candidates are the other mitochondrial chaperone systems and/or cochaperones that interact with mtHsp70. The nucleotide exchange factor Mge1, by binding to native mtHsp70, might shift the equilibrium of the partially unfolded to the native conformation of mtHsp70. However, our in vitro data show that Mge1 alone is not sufficient to promote de novo folding of mtHsp70, although Mge1, similar to Hep1, was reported to increase the solubility of mtHsp70 upon coexpression in E. coli (Momose et al., 2007). At present, it cannot be excluded that low amounts of mtHsp70 might fold in vivo without the help of any proteinaceous component or that because of effects such as molecular crowding, more factors are needed in vivo than in vitro.
In summary, our results indicate that Hep1 interacts with a folding intermediate of mtHsp70, thereby promoting the de novo folding of mtHsp70. Hep1 is the first chaperone known that selectively mediates the de novo folding of another chaperone. It will be interesting to find out whether the action of specialized chaperones is also important for the biogenesis of other chaperones in the cell.
Materials and methods
Plasmids
To express radiolabeled mtHsp70 precursor in vitro, the nucleotide sequence encoding full-length mtHsp70 (amino acid residues 1–654) was cloned into the pGEM4 vector. To generate radiolabeled DHFR fusion proteins, nucleotide fragments encoding PBD (residues 416–654), ATPase (residues 1–410), ATPaseLinker (residues 1–415), and ATPaseA4 (residues 1–411 plus four alanine residues) were cloned into the pGEM4 vector containing the fragment encoding a spacer of 80 residues of yeast cytochrome b2 (residues 87–167) and mouse DHFR. In the case of PBD-DHFR, the fragment encoding the presequence of subunit 9 of the yeast F1F0 ATPase was cloned in front of the PBD sequence. The mtHsp70A4 mutant, in which residues 412–415 were exchanged to alanine residues, was generated by overlap extension.
Strains and cell growth
A haploid HEP1 deletion strain (Δhep1) was generated in the FY16-79a background obtained from EUROSCARF (accession no. 10000R) by exchanging the ORF of HEP1 gene by the KanMX cassette (Wach et al., 1994). The HEP1 deletion strain and the corresponding wild-type strain were grown on lactate medium at 24°C. The temperature-sensitive mif4 strain (Cheng et al., 1989), the Δhsp78 strain (Schmitt et al., 1995), and the Δmdj1 strain (Rowley et al., 1994) and their corresponding wild-type strains were grown in YPG or YPGal medium at 24°C. The cultures of the mif4 strain and of the corresponding wild type were shifted to the nonpermissive temperature of 37°C for 2 h before isolation of mitochondria.
Protein expression and purification
Mature forms of mtHsp70 (residues 24–654) and ATPaseLinker (residues 24–415) were expressed from the plasmids pETDuet-his6Hep-mtHsp70 (Sichting et al., 2005) and pETDuet-his6Hep-ATPaseLinker (Blamowska et al., 2010), respectively, in cells of the BL21(DE3) ΔdnaK::52 strain (Mapa et al., 2010). Proteins were purified by affinity chromatography using a His6-Mge1-NiNTA affinity column as described before (Weiss et al., 2002). The His6-Mge1-NiNTA affinity column was prepared by binding of his-tagged Mge1 to a NiNTA agarose column. In brief, the bacterial cells expressing the mtHsp70 variants were resuspended in buffer A (20 mM Hepes-KOH, pH 7.4, 250 mM KCl, 20 mM imidazole, 2 mM β-mercaptoethanol, 10% glycerol [vol/vol], and 1 mM PMSF). 1 mg/ml lysozyme was added, and the suspension was incubated for 45 min at 4°C. Then cells were disrupted by sonication on ice (Branson Sonifier, 10 times for 12 s, 80% duty cycle). After a clarifying spin (27,200 g, 15 min, 4°C), the supernatant was loaded twice onto the His6-Mge1-NiNTA affinity column. The column was washed with 20 column volumes of buffer A, and bound proteins were eluted with elution buffer (20 mM Hepes-KOH, pH 7.4, 250 mM KCl, 2 mM β-mercaptoethanol, 10% glycerol [vol/vol], 1 mM PMSF, 10 mM MgCl2, and 5 mM ATP). The buffer was exchanged, and the protein obtained was kept at −80°C in storage buffer (20 mM Hepes-KOH, pH 7.4, 100 mM KCl, 5 mM MgCl2, and 5% glycerol [vol/vol]). The PBD (residues 410–654) was expressed in the BL21(DE3) ΔdnaK::52 strain from pETDuet-his6PBD as N-terminally his-tagged protein (Blamowska et al., 2010). The mature forms of Mge1 (residues 44–228) and Hep1 (residues 74–205) were expressed as N-terminally his-tagged proteins and purified on a NiNTA agarose column as described before (Horst et al., 1997; Sichting et al., 2005). In brief, cell lysate was prepared as described for the lysate of the cells expressing the mtHsp70 variants. Then the cell lysate was loaded onto a NiNTA agarose column. The column was washed with 20 column volumes of buffer A, and bound protein was then eluted with buffer A containing 300 mM imidazole. The proteins were stored at −80°C in storage buffer (20 mM Hepes-KOH, pH 7.4, 100 mM KCl, 5 mM MgCl2, and 5% glycerol [vol/vol]).
In organello folding assay
Radiolabeled precursor proteins were synthesized in vitro in a reticulocyte lysate system and imported into isolated mitochondria essentially as described before (Mokranjac et al., 2003). In brief, import was performed at 25°C in standard import buffer containing 1 mg/ml BSA, 5 mM NADH, 2.5 mM ATP, 10 mM creatine phosphate, and 100 µg/ml creatine kinase with the addition of 1–3% (vol/vol) of the lysate. After the indicated time periods, import was stopped by the addition of 2 µM valinomycin, and nonimported material was digested with 50 µg/ml trypsin at 4°C. Trypsin digestion was stopped with 20-fold excess of soybean trypsin inhibitor, and mitochondria were reisolated, washed, and solubilized with 1% Triton X-100 in 20 mM Tris-HCl, pH 7.5, and 80 mM KCl. Samples were halved, and one half was mock treated and the other half was treated with 50 µg/ml trypsin to analyze the folding state of the imported protein. After 10 min, proteolysis was stopped by TCA precipitation, and samples were analyzed by SDS-PAGE and autoradiography or dissolved in SDS-containing buffer (1% SDS [m/v] and 100 mM Tris, pH 7.4). The SDS-dissolved fraction was incubated for 5 min at 95°C, diluted 30-fold in immunoprecipitation buffer (20 mM Tris-HCl, pH 7.4, 300 mM NaCl, 0.2% [vol/vol] Triton X-100, 0.5 mM PMSF, and protease inhibitor mix [Roche]), and, after a clarifying spin, subjected to immunoprecipitation. The samples were incubated with the indicated antibodies prebound to protein A–Sepharose beads for 2 h at 4°C. Then beads were washed four times with immunoprecipitation buffer, and bound proteins were eluted with Laemmli buffer and analyzed by SDS-PAGE and autoradiography.
In vivo labeling
Yeast cells were grown in lactate medium to early logarithmic phase, harvested, resuspended in selective medium containing 2% (vol/vol) lactate, and grown further in the presence of [35S]methionine for 10 min at 25°C. 100 µg/ml cycloheximide and 10 mM cold methionine were added, and cells were harvested, resuspended in SH-KCl buffer (0.6 M sorbitol, 20 mM Hepes, pH 7.4, and 80 mM KCl) in the presence of 1 mM PMSF, and disrupted by shaking with glass beads at 4°C. Crude mitochondria were isolated by differential centrifugation and solubilized with 1% (vol/vol) Triton X-100 in 20 mM Tris-HCl, pH 7.5, and 80 mM KCl. Samples were halved; one half was mock treated, and the other half was digested with 2.5 µg/ml trypsin. Digestion was stopped by TCA precipitation, and samples were subjected to immunoprecipitation with antibodies directed against mtHsp70 and analyzed by SDS-PAGE, autoradiography, and quantification.
In vitro refolding assay
Recombinant proteins were precipitated with ammonium sulfate and resuspended in 8 M urea and 20 mM Tris-HCl, pH 7.4, to denature them. Samples were diluted 20-fold to a protein concentration of 0.3 mg/ml in refolding buffer (20 mM Hepes-KOH, pH 7.4, 100 mM KCl, 5 mM MgCl2, and 5% [vol/vol] glycerol) with the addition of 0.1 mg/ml cytochrome c. When indicated, Hep1 in an equimolar ratio to the unfolded protein (unless stated otherwise) or Mge1 in fourfold molar excess over unfolded protein and/or 2.5 mM of the indicated nucleotide was added. Samples were incubated at 25°C to allow protein refolding. Aliquots containing 6 µg of the refolding protein were withdrawn, diluted in one volume of ice-cold refolding buffer, and placed on ice, and 5 µg/ml trypsin was added. After 15 min at 4°C, trypsin was stopped by soybean trypsin inhibitor. After centrifugation for 10 min at 39,000 g, supernatants were analyzed by SDS-PAGE and Coomassie staining.
Cross-linking
3 µM of unfolded mtHsp70 was incubated for 20 min at 30°C in 50 mM Hepes-KOH, pH 7.4, 200 mM NaCl, 100 mM KCl, and 5 mM MgCl2. Where indicated, 6 µM Hep1, 6 µM Mge1, and/or 2.5 mM of the indicated nucleotide was added. Glutaraldehyde was added to a final concentration of 1 mM, and samples were further incubated for 30 min at 30°C. The cross-linking reaction was stopped by the addition of Laemmli buffer containing 200 mM glycin. Samples were analyzed by SDS-PAGE and Coomassie staining and co-immunodecoration with the indicated antibodies.
ATPase activity assay
To measure ATPase activity, a coupled ATP-regenerating enzyme system was used essentially as described previously (Nørby, 1988). 4.3 µM mtHsp70 was incubated for 20 min at 25°C in refolding buffer with 8.3 mM ATP and 0.3 mg/ml BSA and, when indicated, 8.6 µM Hep1. Samples were diluted 4.3-fold in refolding buffer containing 0.12 mM NADH, 3 mM phosphoenolpyruvate, 10 U lactate dehydrogenase, 15 U pyruvate kinase, 2 µM Mge1, and 2 µM DnaJ, and the change of absorbance at 360 nm at 25°C was monitored in time by spectrophotometry (JASCO).
Miscellaneous
Quantification was performed by densitometry using ImageMaster 1D Elite Software version 4.00 (GE Healthcare). On the gel images, nonrelevant gel lanes were excised digitally, as indicated by the white spacers. All the experiments presented were repeated at least three times.
Online supplemental material
Fig. S1 shows the analysis of an interdomain linker mutant demonstrating that the interdomain linker is needed for folding of the ATPase domain of mtHsp70. Fig. S2 confirms that the trypsin-resistant stable fragment of imported mtHsp70 corresponds to the PBD of mtHsp70. Fig. S3 shows that the folding of mtHsp70 in vitro at lower temperature is dependent on Hep1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201205012/DC1.
Acknowledgments
We are grateful to Alexandra Stiegler and Heiko Germeroth for excellent technical assistance. We thank Andreas Ladurner for his generous support. We thank Arthur Horwich for providing the mif4 mutant.
This work was supported by grants, SFB 594 (B13), from the Deutsche Forschungsgemeinschaft and by a predoctoral fellowship from the Elite Netzwerk Bayern to M. Blamowska.
Footnotes
Abbreviations used in this paper:
- DHFR
- dihydrofolate reductase
- mtHsp70
- mitochondrial Hsp70
- PBD
- peptide-binding domain
References
- Benaroudj N., Triniolles F., Ladjimi M.M. 1996. Effect of nucleotides, peptides, and unfolded proteins on the self-association of the molecular chaperone HSC70. J. Biol. Chem. 271:18471–18476 10.1074/jbc.271.31.18471 [DOI] [PubMed] [Google Scholar]
- Blamowska M., Sichting M., Mapa K., Mokranjac D., Neupert W., Hell K. 2010. ATPase domain and interdomain linker play a key role in aggregation of mitochondrial Hsp70 chaperone Ssc1. J. Biol. Chem. 285:4423–4431 10.1074/jbc.M109.061697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B., Horwich A.L. 1998. The Hsp70 and Hsp60 chaperone machines. Cell. 92:351–366 10.1016/S0092-8674(00)80928-9 [DOI] [PubMed] [Google Scholar]
- Bukau B., Weissman J., Horwich A. 2006. Molecular chaperones and protein quality control. Cell. 125:443–451 10.1016/j.cell.2006.04.014 [DOI] [PubMed] [Google Scholar]
- Burbulla L.F., Schelling C., Kato H., Rapaport D., Woitalla D., Schiesling C., Schulte C., Sharma M., Illig T., Bauer P., et al. 2010. Dissecting the role of the mitochondrial chaperone mortalin in Parkinson’s disease: functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 19:4437–4452 10.1093/hmg/ddq370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri L., Vascotto K., Fredersdorf S., Tiedt R., Hall M.N., Lithgow T. 2004. Zim17, a novel zinc finger protein essential for protein import into mitochondria. J. Biol. Chem. 279:50243–50249 10.1074/jbc.M409194200 [DOI] [PubMed] [Google Scholar]
- Cheng M.Y., Hartl F.-U., Horwich A.L. 1990. The mitochondrial chaperonin hsp60 is required for its own assembly. Nature. 348:455–458 10.1038/348455a0 [DOI] [PubMed] [Google Scholar]
- Cheng M.Y., Hartl F.U., Martin J., Pollock R.A., Kalousek F., Neupert W., Hallberg E.M., Hallberg R.L., Horwich A.L. 1989. Mitochondrial heat-shock protein hsp60 is essential for assembly of proteins imported into yeast mitochondria. Nature. 337:620–625 10.1038/337620a0 [DOI] [PubMed] [Google Scholar]
- Clerico E.M., Zhuravleva A., Smock R.G., Gierasch L.M. 2010. Segmental isotopic labeling of the Hsp70 molecular chaperone DnaK using expressed protein ligation. Biopolymers. 94:742–752 10.1002/bip.21426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig E.A., Kramer J., Kosic-Smithers J. 1987. SSC1, a member of the 70-kDa heat shock protein multigene family of Saccharomyces cerevisiae, is essential for growth. Proc. Natl. Acad. Sci. USA. 84:4156–4160 10.1073/pnas.84.12.4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz de la Loza Mdel.C., Gallardo M., García-Rubio M.L., Izquierdo A., Herrero E., Aguilera A., Wellinger R.E. 2011. Zim17/Tim15 links mitochondrial iron-sulfur cluster biosynthesis to nuclear genome stability. Nucleic Acids Res. 39:6002–6015 10.1093/nar/gkr193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Silva P.D., Schilke B., Walter W., Andrew A., Craig E.A. 2003. J protein cochaperone of the mitochondrial inner membrane required for protein import into the mitochondrial matrix. Proc. Natl. Acad. Sci. USA. 100:13839–13844 10.1073/pnas.1936150100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas P.M., Cyr D.M. 2010. Interplay between protein homeostasis networks in protein aggregation and proteotoxicity. Biopolymers. 93:229–236 10.1002/bip.21304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E., Greene L.E. 2007. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 8:640–646 10.1111/j.1600-0854.2007.00568.x [DOI] [PubMed] [Google Scholar]
- Endo T., Yamano K. 2009. Multiple pathways for mitochondrial protein traffic. Biol. Chem. 390:723–730 10.1515/BC.2009.087 [DOI] [PubMed] [Google Scholar]
- Frydman J. 2001. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu. Rev. Biochem. 70:603–647 10.1146/annurev.biochem.70.1.603 [DOI] [PubMed] [Google Scholar]
- Goswami A.V., Chittoor B., D’Silva P. 2010. Understanding the functional interplay between mammalian mitochondrial Hsp70 chaperone machine components. J. Biol. Chem. 285:19472–19482 10.1074/jbc.M110.105957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W., Christen P. 2001. Mutations in the interdomain linker region of DnaK abolish the chaperone action of the DnaK/DnaJ/GrpE system. FEBS Lett. 497:55–58 10.1016/S0014-5793(01)02435-8 [DOI] [PubMed] [Google Scholar]
- Hartl F.U., Bracher A., Hayer-Hartl M. 2011. Molecular chaperones in protein folding and proteostasis. Nature. 475:324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Horst M., Oppliger W., Rospert S., Schönfeld H.J., Schatz G., Azem A. 1997. Sequential action of two hsp70 complexes during protein import into mitochondria. EMBO J. 16:1842–1849 10.1093/emboj/16.8.1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J., Maes E.G., Taylor A.B., Wang L., Hinck A.P., Lafer E.M., Sousa R. 2007. Structural basis of J cochaperone binding and regulation of Hsp70. Mol. Cell. 28:422–433 10.1016/j.molcel.2007.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P.J., Ostermann J., Shilling J., Neupert W., Craig E.A., Pfanner N. 1990. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 348:137–143 10.1038/348137a0 [DOI] [PubMed] [Google Scholar]
- Mapa K., Sikor M., Kudryavtsev V., Waegemann K., Kalinin S., Seidel C.A., Neupert W., Lamb D.C., Mokranjac D. 2010. The conformational dynamics of the mitochondrial Hsp70 chaperone. Mol. Cell. 38:89–100 10.1016/j.molcel.2010.03.010 [DOI] [PubMed] [Google Scholar]
- Mayer M.P., Laufen T., Paal K., McCarty J.S., Bukau B. 1999. Investigation of the interaction between DnaK and DnaJ by surface plasmon resonance spectroscopy. J. Mol. Biol. 289:1131–1144 10.1006/jmbi.1999.2844 [DOI] [PubMed] [Google Scholar]
- Miao B., Davis J.E., Craig E.A. 1997. Mge1 functions as a nucleotide release factor for Ssc1, a mitochondrial Hsp70 of Saccharomyces cerevisiae. J. Mol. Biol. 265:541–552 10.1006/jmbi.1996.0762 [DOI] [PubMed] [Google Scholar]
- Mokranjac D., Neupert W. 2010. The many faces of the mitochondrial TIM23 complex. Biochim. Biophys. Acta. 1797:1045–1054 10.1016/j.bbabio.2010.01.026 [DOI] [PubMed] [Google Scholar]
- Mokranjac D., Sichting M., Neupert W., Hell K. 2003. Tim14, a novel key component of the import motor of the TIM23 protein translocase of mitochondria. EMBO J. 22:4945–4956 10.1093/emboj/cdg485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momose T., Ohshima C., Maeda M., Endo T. 2007. Structural basis of functional cooperation of Tim15/Zim17 with yeast mitochondrial Hsp70. EMBO Rep. 8:664–670 10.1038/sj.embor.7400990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery D., Jordan R., McMacken R., Freire E. 1993. Thermodynamic and structural analysis of the folding/unfolding transitions of the Escherichia coli molecular chaperone DnaK. J. Mol. Biol. 232:680–692 10.1006/jmbi.1993.1418 [DOI] [PubMed] [Google Scholar]
- Montgomery D.L., Morimoto R.I., Gierasch L.M. 1999. Mutations in the substrate binding domain of the Escherichia coli 70 kDa molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling. J. Mol. Biol. 286:915–932 10.1006/jmbi.1998.2514 [DOI] [PubMed] [Google Scholar]
- Nakatsukasa K., Brodsky J.L. 2008. The recognition and retrotranslocation of misfolded proteins from the endoplasmic reticulum. Traffic. 9:861–870 10.1111/j.1600-0854.2008.00729.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørby J.G. 1988. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 156:116–119 10.1016/0076-6879(88)56014-7 [DOI] [PubMed] [Google Scholar]
- Palleros D.R., Reid K.L., McCarty J.S., Walker G.C., Fink A.L. 1992. DnaK, hsp73, and their molten globules. Two different ways heat shock proteins respond to heat. J. Biol. Chem. 267:5279–5285 [PubMed] [Google Scholar]
- Rowley N., Prip-Buus C., Westermann B., Brown C., Schwarz E., Barrell B., Neupert W. 1994. Mdj1p, a novel chaperone of the DnaJ family, is involved in mitochondrial biogenesis and protein folding. Cell. 77:249–259 10.1016/0092-8674(94)90317-4 [DOI] [PubMed] [Google Scholar]
- Sanjuán Szklarz L.K., Guiard B., Rissler M., Wiedemann N., Kozjak V., van der Laan M., Lohaus C., Marcus K., Meyer H.E., Chacinska A., et al. 2005. Inactivation of the mitochondrial heat shock protein zim17 leads to aggregation of matrix hsp70s followed by pleiotropic effects on morphology and protein biogenesis. J. Mol. Biol. 351:206–218 10.1016/j.jmb.2005.05.068 [DOI] [PubMed] [Google Scholar]
- Schmitt M., Neupert W., Langer T. 1995. Hsp78, a Clp homologue within mitochondria, can substitute for chaperone functions of mt-hsp70. EMBO J. 14:3434–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld H.J., Schmidt D., Schröder H., Bukau B. 1995. The DnaK chaperone system of Escherichia coli: quaternary structures and interactions of the DnaK and GrpE components. J. Biol. Chem. 270:2183–2189 10.1074/jbc.270.5.2183 [DOI] [PubMed] [Google Scholar]
- Sichting M., Mokranjac D., Azem A., Neupert W., Hell K. 2005. Maintenance of structure and function of mitochondrial Hsp70 chaperones requires the chaperone Hep1. EMBO J. 24:1046–1056 10.1038/sj.emboj.7600580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain J.F., Dinler G., Sivendran R., Montgomery D.L., Stotz M., Gierasch L.M. 2007. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol. Cell. 26:27–39 10.1016/j.molcel.2007.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A.D., Bernard S.M., Skiniotis G., Gestwicki J.E. 2012. Visualization and functional analysis of the oligomeric states of Escherichia coli heat shock protein 70 (Hsp70/DnaK). Cell Stress Chaperones. 17:313–327 10.1007/s12192-011-0307-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscott K.N., Voos W., Frazier A.E., Lind M., Li Y., Geissler A., Dudek J., Müller H., Sickmann A., Meyer H.E., et al. 2003. A J-protein is an essential subunit of the presequence translocase-associated protein import motor of mitochondria. J. Cell Biol. 163:707–713 10.1083/jcb.200308004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan M., Hutu D.P., Rehling P. 2010. On the mechanism of preprotein import by the mitochondrial presequence translocase. Biochim. Biophys. Acta. 1803:732–739 10.1016/j.bbamcr.2010.01.013 [DOI] [PubMed] [Google Scholar]
- Vogel M., Mayer M.P., Bukau B. 2006. Allosteric regulation of Hsp70 chaperones involves a conserved interdomain linker. J. Biol. Chem. 281:38705–38711 10.1074/jbc.M609020200 [DOI] [PubMed] [Google Scholar]
- Voos W. 2009. Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res. Microbiol. 160:718–725 10.1016/j.resmic.2009.08.003 [DOI] [PubMed] [Google Scholar]
- Wach A., Brachat A., Pöhlmann R., Philippsen P. 1994. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 10:1793–1808 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- Wadhwa R., Taira K., Kaul S.C. 2002. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones. 7:309–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss C., Niv A., Azem A. 2002. Two-step purification of mitochondrial Hsp70, Ssc1p, using Mge1(His)(6) immobilized on Ni-agarose. Protein Expr. Purif. 24:268–273 10.1006/prep.2001.1563 [DOI] [PubMed] [Google Scholar]
- Willmund F., Hinnenberger M., Nick S., Schulz-Raffelt M., Mühlhaus T., Schroda M. 2008. Assistance for a chaperone: Chlamydomonas HEP2 activates plastidic HSP70B for cochaperone binding. J. Biol. Chem. 283:16363–16373 10.1074/jbc.M708431200 [DOI] [PubMed] [Google Scholar]
- Yamamoto H., Momose T., Yatsukawa Y., Ohshima C., Ishikawa D., Sato T., Tamura Y., Ohwa Y., Endo T. 2005. Identification of a novel member of yeast mitochondrial Hsp70-associated motor and chaperone proteins that facilitates protein translocation across the inner membrane. FEBS Lett. 579:507–511 10.1016/j.febslet.2004.12.018 [DOI] [PubMed] [Google Scholar]
- Zhai P., Stanworth C., Liu S., Silberg J.J. 2008. The human escort protein Hep binds to the ATPase domain of mitochondrial hsp70 and regulates ATP hydrolysis. J. Biol. Chem. 283:26098–26106 10.1074/jbc.M803475200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuravleva A., Gierasch L.M. 2011. Allosteric signal transmission in the nucleotide-binding domain of 70-kDa heat shock protein (Hsp70) molecular chaperones. Proc. Natl. Acad. Sci. USA. 108:6987–6992 10.1073/pnas.1014448108 [DOI] [PMC free article] [PubMed] [Google Scholar]