

Abstract
Chlamydomonas reinhardtii is one of the most important microalgae model organisms and has been widely studied toward the understanding of chloroplast functions and various cellular processes. Further exploitation of C. reinhardtii as a model system to elucidate various molecular mechanisms and pathways requires systematic study of gene regulation. However, there is a general lack of genome-scale gene regulation study, such as global cis-regulatory element (CRE) identification, in C. reinhardtii. Recently, large-scale genomic data in microalgae species have become available, which enable the development of efficient computational methods to systematically identify CREs and characterize their roles in microalgae gene regulation. Here, we performed in silico CRE identification at the whole genome level in C. reinhardtii using a comparative genomics-based method. We predicted a large number of CREs in C. reinhardtii that are consistent with experimentally verified CREs. We also discovered that a large percentage of these CREs form combinations and have the potential to work together for coordinated gene regulation in C. reinhardtii. Multiple lines of evidence from literature, gene transcriptional profiles, and gene annotation resources support our prediction. The predicted CREs will serve, to our knowledge, as the first large-scale collection of CREs in C. reinhardtii to facilitate further experimental study of microalgae gene regulation. The accompanying software tool and the predictions in C. reinhardtii are also made available through a Web-accessible database (http://hulab.ucf.edu/research/projects/Microalgae/sdcre/motifcomb.html).
Chlamydomonas reinhardtii is a member of single-celled green algae that diverged from the streptophytes approximately one billion years ago. Being composed of multiple mitochondria, two anterior flagella, and a chloroplast, C. reinhardtii serves as an outstanding microalgae model organism, especially for analyzing eukaryotic chloroplast biology, action of flagella and basal bodies, and many biological pathways such as circadian rhythms, cell cycle control, and plant respiration (Rochaix, 2004; Wemmer and Marshall, 2004; Bisova et al., 2005; Cardol et al., 2005; Mittag et al., 2005). As a photosynthetic microalgae species, C. reinhardtii has also shown its potential in biofuel generation (Grossman et al., 2003; Beckmann et al., 2009; Langner et al., 2009; Nguyen et al., 2009). However, since cellular processes in general are coordinated by transcriptional regulation of functionally related genes, further exploitation of C. reinhardtii as a model system to elucidate various molecular mechanisms requires the systematic study of gene regulation (Bohne and Linden, 2002; Li et al., 2010; Sun et al., 2010). Genome-scale study of gene regulation in C. reinhardtii is currently in the very early stages. For example, cis-regulatory elements (CREs) are genomic DNA segments that play important roles in gene regulation by modulating gene activities through their interaction with RNAs or regulatory proteins called transcription factors (TFs). The discovery and functional annotation of CREs is thus one of the immediate next steps toward global understanding of gene regulatory mechanisms in C. reinhardtii (Wang et al., 2009). However, there are less than one dozen CREs annotated in C. reinhardtii to date, even though many TFs have been collected and deposited in a number of databases (Wingender et al., 1996; Higo et al., 1999; Rombauts et al., 1999; Portales-Casamar et al., 2010). The correspondences between CREs and regulatory proteins remain largely unknown.
Unprecedented large-scale microalgae genomic data have now become available, which makes it possible to perform the genome-wide identification of CREs in C. reinhardtii. For example, the complete nuclear genome sequence of C. reinhardtii was published in 2007, and around 15,000 protein-coding genes were predicted (Merchant et al., 2007). Since then, several updates of the C. reinhardtii genome and the genome annotation have become available, including versions 3.0 (15,256 genes), 4.0 (16,709 genes), and 4.3 (17,114 genes). Additionally, another green algal species, Volvox carteri, that is close to C. reinhardtii in evolution has been recently sequenced, which provides a great opportunity to study the C. reinhardtii genome by comparative genomics (Prochnik et al., 2010). In addition to the genomic sequence data, there is a large supply of expression data available as complementary DNA libraries, ESTs, microarray expression measurements, and RNA sequencing reads (Eberhard et al., 2006; Miller et al., 2010; Castruita et al., 2011; Fischer et al., 2012; Urzica et al., 2012). Integrative analysis of DNA sequence data and mRNA expression measurements have been shown to be successful in gene regulatory network studies at the whole-genome level (Segal et al., 2003; Wang and Stormo, 2003).
Existing CRE motif-finding methods often assume that a bona fide CRE motif must be overrepresented in the input sequences (i.e. the overrepresentation property of motifs; Stormo and Hartzell, 1989; Lawrence et al., 1993; Bailey and Elkan, 1994). However, because of the degenerative nature of motifs, overrepresentation alone is often not enough to distinguish true motifs from random patterns formed by DNA segments (Blanchette and Tompa, 2002; Wang and Stormo, 2003). To improve the sensitivity and specificity of CRE motif prediction, dozens of motif-finding methods have been developed to exploit the co-occurrence property of motifs (i.e. multiple CREs are often co-occurring in short regions; Frith et al., 2001; Zhou and Wong, 2004; Gupta and Liu, 2005; Hu et al., 2008). Alternative methods require evolutionary conservation of motifs to further filter false-positive discoveries (Loots et al., 2000; Blanchette and Tompa, 2002; Wang and Stormo, 2003; Liu et al., 2004; Sinha et al., 2004; Elemento and Tavazoie, 2005; Li and Wong, 2005; Li et al., 2005). The rationale is that functional motifs should be evolutionarily conserved across multiple species (Loots et al., 2000), Considering evolutionary conservation as an additional criterion indeed has been shown effective in identifying bona fide CRE motifs, whereas how to better quantify the evolutionary conservation for CRE motif finding is worth further investigation. For example, the current common practice to quantify the evolutionary conservation of a potential CRE motif is to score the multiple sequence alignment (MSA) of its corresponding DNA segments in orthologous sequences. For this quantification strategy, there can be at least two issues. One is that short DNA segments are not always well aligned with their corresponding segments in MSA, and consequently, a plethora of not-well-aligned CREs can be missed (Li and Wong, 2005). The other issue is that even for DNA segments that can be aligned well with their corresponding segments, current strategies to score conservation are often debatable. For example, one commonly used strategy is to compare a CRE candidate with its corresponding DNA segments in different orthologous sequences, and if the number of mismatches is smaller than a specified cutoff, then the CRE candidate is defined as conserved. Since species divergence time is not considered, strategies like this may result in inaccurate assessment for conservation (Li et al., 2005).
In this paper, we present, to our knowledge, the first genome-wide computational identification of CREs in C. reinhardtii using a comparative genomics-based approach named MERCED, short for “modeling evolution rate across species for cis-regulatory element discovery.” MERCED searches for CREs through uncovering CRE motifs (i.e. common patterns of CREs that can be bound by the same TFs). By simultaneously considering multiple properties of motifs, including overrepresentation, co-occurrence, and evolutionary conservation, MERCED is able to reduce false-positive predictions significantly. Most importantly, considering species divergence time when evaluating evolutionary conservation leads to better incorporation of the evolutionary information of individual DNA segments. By comparatively integrating V. carteri and C. reinhardtii genome information, MERCED predicted 66,530 CREs, corresponding to 317 CRE motifs. A total of 164 (51.7%) of these CRE motifs tend to frequently co-occur in regulatory sequences flanking the transcription start sites. The existence of many such frequently co-occurring motifs, termed motif combinations, indicates the potential of these motifs to coordinately regulate target genes. Many of these identified motifs and motif combinations are consistent with experimentally verified motifs from public databases. Further integration of gene transcriptional profiles and gene annotation data resources also provide multiple functional lines of evidence supporting the predictions. The motif predictions generated from this study and the accompanying software tool MERCED have been deposited into our Web-accessible database, which will be useful to experimental biologists interested in gene regulation in algae species.
RESULTS
MERCED: Genome-Scale Prediction of CRE Motifs in C. reinhardtii
We developed the MERCED algorithm to predict conserved CREs between microalgae species C. reinhardtii and V. carteri. Different from available methods, MERCED defines conserved DNA segments by carefully modeling the species divergence time. The algorithm consists of the following steps (for details, see “Materials and Methods”). (1) Define orthologous gene pairs in C. reinhardtii and V. carteri as reciprocal best hits obtained by applying PSI-BLAST (for Position-Specific Iterative Basic Local Alignment Search Tool) to the protein sequences in the two species (Altschul et al., 1997). (2) Construct a nucleotide substitution matrix to model the neutral evolution rate of nucleotide substitution between C. reinhardtii and V. carteri based on 4-fold degenerate sites in proteins of orthologous genes (Li et al., 1985). (3) Define conserved k-mers in regulatory sequences of orthologous genes based on their statistical significance estimated from the nucleotide substitution matrix constructed in step 2. A k-mer is a k-bp-long DNA segment. Generating functions are applied here to improve the efficiency for significance calculation (Huang et al., 2004). (4) Group conserved k-mers using hierarchical clustering with average linkage (Sokal and Michener, 1985; Fig. 1). In brief, we calculate the similarity between every pair of conserved k-mers. The similarity of two k-mers is estimated as 1 − d/k, where d is the number of mismatches between the two k-mers. We then apply the hierarchical clustering with the average linkage to cluster-conserved k-mers based on the pairwise similarities. We require any pair of conserved k-mers in a cluster have a similarity score greater than 0.6 at this step (Fig. 1). (5) Define CRE motifs based on patterns of k-mers in the same clusters obtained in step 4, and define k-mers underlying these patterns as the CRE instances of the corresponding motif. During this step, we require a cluster to contain a significant number of k-mers to take the overrepresentation property of motifs into account.
Figure 1.

Identified CRE motifs from conserved k-mers. [See online article for color version of this figure.]
The MERCED algorithm has several advantages over MSA-based methods. One of them is that MERCED is more practical for conservation estimation of short DNA segments compared with MSA-based methods. This is because the latter will not work for the not-well-aligned DNA segments, whereas DNA segments, especially the short ones, are often difficult to align well using existing MSA algorithms. Additionally, in contrast to MSA-based methods, MERCED considers species divergence time rather than merely the number of mismatched nucleotides between DNA segments in aligned orthologous sequences. As a result, we can obtain very accurate estimation of evolutionary conservation even for very short DNA segments. This is because the conservation between two DNA segments from two orthologous sequences depends on not only the number of mismatches but also the evolutionary distance between them, whereas the absolute value of mismatch number is not always proportional to the evolutionary distance (Frazer et al., 2001). In fact, highly similar sequences between closely related species (e.g. human [Homo sapiens] and mouse [Mus musculus]) can be present either as a result of active conservation due to functional constraints or as a result of shared ancestry due to insufficient divergence time (Frazer et al., 2001). For example, as illustrated in Figure 2B, one 8-mer in a C. reinhardtii sequence with a zero mismatch compared with a similar 8-mer in the orthologous V. carteri sequence may have a P value of 0.011 according to our model and thus will not be considered as a conserved 8-mer pair. On the contrary, another 8-mer with two mismatches compared with its similar 8-mer in the same orthologous sequences will be considered as conserved because of the much smaller P value, 2.84E-05. One additional beneficial side effect of the MERCED is that when comparing potential instances of a CRE motif in different species for CRE conservation quantification, MERCED avoids setting an arbitrary cutoff for the number of mismatches.
Figure 2.

A, The substitution matrix that models the neutral evolution rate of nucleotides. The (i,j) entry, Sij in S, represents the probability that the i-th type of nucleotides in C. reinhardtii corresponds to the j-th type of nucleotides in V. carteri, and i and j are one of the four types of nucleotides and are in the order A, C, G, and T. B, Three 8-mer pairs as examples to illustrate that smaller conservation P values are not always corresponding to smaller mismatch numbers. [See online article for color version of this figure.]
The Predicted Motifs Are Consistent with Experimentally Verified CRE Motifs
Applying MERCED to the upstream regulatory sequences of 8,741 groups of orthologous genes in C. reinhardtii and V. carteri, we predicted 317 conserved CRE motifs (false discovery rate [FDR] < 0.05 based on permutation) and 66,530 corresponding CREs (k = 8; Supplemental Table S1). We compared the predicted motifs with the known motifs in the PLACE database (http://www.dna.affrc.go.jp/PLACE/; Higo et al., 1999) based on the STAMP software tool (Mahony and Benos, 2007). PLACE has so far compiled 469 experimentally verified motifs in plants from literature (Higo et al., 1999), and the STAMP tool is commonly used to assess motif similarities with statistical significance estimation. We found that 195 (62.5%) of the 317 predicted motifs are similar to the PLACE motifs with the STAMP E-value cutoff as 1E-5, which was used in previous studies to define similar motifs (Fauteux et al., 2008; Reed et al., 2008; Supplemental Table S1). Take the predicted motif M75, for example; the consensus of its reverse complement is GNCGGCCA, which is similar to the PLACE motif GRAZMRAB17-GGGCGGCCAGTG (E-value = 4.18E-11). As another example, the predicted motif M78 is similar to the known motif LRENPCABE-ACGTGGCA, with the consensus of its reverse complement as CNTGGCA (E-value = 3.5E-11). We also found that 129 (27.5%) PLACE motifs are similar to the predicted 317 motifs, and multiple predicted motifs may be similar to the same PLACE motif. For instance, the predicted motifs M4 and M5 have the motif consensus GCAGCTGC and YCAGCAGC, respectively, which are similar to the same known PLACE motif ANAERO2CONSENSUS, with the consensus AGCAGC (Supplemental Table S1). In the mean time, many of these experimentally verified motifs in PLACE database are not similar to the predicted motifs. One possible explanation is that, rather than from the algae, the majority of the PLACE motifs are from land plant species, especially Arabidopsis (Arabidopsis thaliana; Higo et al., 1999). Therefore, the difference between the PLACE motifs and the predicted motifs might suggest the evolution of gene regulatory mechanism over the long evolutionary distance between the algae and the land plants.
In addition to the comparisons with experimentally verified motifs, we also found literature support for the predicted motifs. For instance, TGACGCCA is an experimentally verified CRE in the C. reinhardtii gene GPX5, which has been shown to be relevant to the oxidative stress response in C. reinhardtii (Fischer et al., 2009). This CRE is similar to the consensus of the predicted motif M44, T[CG]C[ACGT]GCCA, where nucleotides included in brackets are those frequently occurring at the specified positions. Moreover, we found that the target genes of the motif M44 significantly share the following functions based on Gene Ontology (GO) enrichment analysis (Boyle et al., 2004): response to stress (GO:0006950; corrected P value of 7.34E-08), which is consistent with the function of GPX5. We have also found the predicted motif M44 to be similar to PLACE motif GRAZMRAB28 (E-value = 1.78E-08). GRAZMRAB28 is found in the promoters of abscisic acid (ABA)-responsive genes (Busk and Pagès, 1998), which also supports the functionality of M44.
The Predicted Motif Combinations Are Consistent with Experimentally Verified Ones
In high eukaryotes, multiple TFs often coordinately regulate their target genes by binding to their respective CREs that co-occur in the short regions of a few hundred base pairs (Yuh et al., 1998; Blanchette et al., 2006; Ding et al., 2012). Many co-occurring CREs in plants have also been identified by experimental studies. These CREs can be associated with a pair of interacting TFs or individual TFs with multiple DNA-binding domains (Singh, 1998). To see whether CREs of the predicted motifs significantly co-occur in the regulatory sequences of C. reinhardtii, we applied our previously developed method (Cai et al., 2010) to identify motif combinations containing multiple motifs whose instances frequently co-occur in the regulatory sequences. In total, we predicted 92,694 motif combinations based on the identified CRE motifs (k = 8), each of which consists of two to six motifs. The percentage of motif combinations composed of two to six motifs is 0.08%, 40.40%, 47.44%, 11.75%, and 0.33%, respectively (Supplemental Table S2). As a way to evaluate our prediction, we compared the predicted motif combinations with seven well-known co-occurring motif pairs (Table I; Singh, 1998; Steffens et al., 2005). We found that five out of the seven known motif combinations were subsets of our predicted combinations (Supplemental Table S3). In general, a known combination can be a subset of multiple predicted combinations. For instance, one of the known motif combinations is composed of two motifs, CAACA and CACCTG, that can be bound by the APETALA2/Ethylene-Responsive element binding factor (AP2/ERF) and B3 domain-containing TF RAV1 at the APETALA2-like N-terminal domain and the B3-like C-terminal domain, respectively. A total of 378 predicted motif combinations have been found to include these two motifs. With different additional motifs, these different motif combinations containing the same known combinations can regulate different sets of target genes. For instance, two predicted motif combinations, MOD3428 and MOD3431, both include the above RAV1-binding motif combination of M164 (GTCNCGGC) and M27 (CAGGYGC; Table I; Supplemental Tables S2 and S3). MOD3428 includes one additional motif, M11 (CGYCGCCA), and MOD3431 includes one additional motif, M21 (CCCNCGCC). It turns out that MOD3428 regulates 204 predicted target genes and MOD3431 regulates 230 predicted target genes, and only 104 of these target genes are shared by both motif combinations, suggesting that the additional motifs M11 and M21 play extra roles in these two motif combinations.
Table I. Comparison of predicted motif combinations with seven known combinations.
bZIP, Basic Leucine Zipper Domain; Dof, DNA-binding with one finger; hox3, homeobox3; HD, homeodomain.
| Known Combinations | Recognition Motifs of Known Combinations | No. of Predicted Similar Motif Combinations |
|---|---|---|
| bHLH+MYB | bHLH-CANNTG | 1,299 |
| MYB-[AT]AACCA or [CT]AAC[GT]G | ||
| bZIP+Dof | bZIP-ACGT | 24 |
| Dof-AAAG | ||
| MADS+MADS | MADS-CC[AT][AT][AT][AT][AT][AT]GG | 0 |
| bZIP+bZIP | bZIP-ACGT | 0 |
| RAV1 | RAV1 AP2-like-CAACA | 378 |
| RAV1 B3-like-CACCTG | ||
| bZIP+MYB | bZIP-ACGT | 1 |
| MYB-CANNTG | ||
| hox3 | hox3-HD1-TCCT | 3,233 |
| hox3-HD2-GATC |
Further investigation of the motifs co-occurring with known motif combinations in a predicted motif combination showed that the functions of additional motifs relate to those of known motifs in the same combinations. For instance, MOD5159 is a combination of three motifs, M13, M167, and M0. The first two motifs comprise the known combination basic helix-loop-helix (bHLH)-MYB and correspond to the binding sites of TFs with bHLH and MYB protein domains, respectively. Both bHLH- and MYB-containing TFs have been shown to be involved in ABA signaling (Abe et al., 2003). The additional motif, M0, is also implicated in ABA signaling because of its similarity to the known PLACE motif AGCBOXNPGLB, which is known to be the binding sequence of the stress signal-response factors ERFs (Fujimoto et al., 2000). Therefore, the additional motif M0 is also related to ABA signaling, and its function is consistent with that of the known combination in terms of ABA signaling. In addition, we found that the target genes of this motif combination MOD5159 significantly share the following functional annotation term: response to stress (GO:0006950; corrected P value of 1E-4), which further supports the coherent function of the three motifs and that of the motif combination.
Functional Enrichment of Predicted Motif Combinations
We also performed functional analysis of the predicted motif combinations by investigating the overlap between their target genes and sets of genes with the same GO annotations (see “Materials and Methods”). Significant overlapping with function-annotated gene sets indicates the function coherence of a motif combination in terms of gene coregulation (Blanchette et al., 2006). In other words, motifs contained in such motif combinations are likely to work together to regulate their target genes. We found that target genes of 87.5% of the predicted motif combinations in C. reinhardtii significantly share the same functions (FDR < 0.05; see “Materials and Methods”). The following two examples illustrate the functional relevance of predicted motif combinations.
Example 1.
The motif combination MOD22676 contains four predicted motifs, M39, M4, M25, and M15. All of these four motifs are similar to known motifs that have been shown to be relevant to plant stress response. For example, M4 is similar to the PLACE motif AGCBOXNPGLB-AGCCGCC, which is the binding sequence of stress signal-response factors ERFs (Fujimoto et al., 2000). M39 is similar to the PLACE motif GRAZMRAB28-CATGCCGCC, which was found in the promoter of an ABA-responsive gene that also plays a critical role in response of environmental stress (Busk and Pagès, 1997). M15 is similar to the PLACE motif ABREMOTIFIIIOSRAB16B-GCCGCGTGGC, which is known to be required for ABA responsiveness as well (Ono et al., 1996). Finally, M25 is similar to ABRE3OSRAB1616-GTACGTGGCGC, which is the known CRE motif called ABA-responsive element found in rice (Oryza sativa) and related to biotic and abiotic stresses (Skriver et al., 1991). In addition, we also found that the target genes of MOD22676 significantly share the following GO annotation terms: response to stress (GO:0006950; corrected P value of 4.65E-3), response to oxidative stress (GO:0006979; corrected P value of 4.85E-3), response to stimulus (GO:0050896; corrected P value of 0.015), and response to chemical stimulus (GO:0042221; corrected P value of 0.033). These GO functional annotations agree well with the functions of the predicted motifs in this motif combination, supporting the functionality of this motif combination MOD22676.
Example 2.
The motif combination MOD9430 is composed of three predicted motifs, M60, M63, and M22. All three motifs have similar known motifs that have been reported to be involved in photoregulation. For example, M60 is similar to the PLACE motif BOXBPSAS1 that is found in the light-regulated Asymmetric Leaves1 gene (Fujimoto et al., 2000), M63 is similar to the PLACE motif PE3ASPHYA3 with a role in photoregulation (Bruce and Quail, 1990), and M22 is similar to the PLACE motif PREMOTIFNPCABE that is also related to photoregulation (Castresana et al., 1988). Additionally, we found that the target genes of MOD9430 significantly share the following GO term: photosynthesis (GO:0015979; corrected P value of 0.01). This GO term is consistent with the functions of the predicted motifs in this motif combination and thus supports the functionality of the predicted motif and motif combination.
In addition to the performed functional study using GO-annotated gene sets, we compared target genes of the 92,694 motif combinations with genes that are coexpressed in C. reinhardtii. Since coexpressed target genes are often coregulated (Allocco et al., 2004), the significant overlap of target genes of a motif combination with coexpressed gene sets supports the functionality of the predicted motifs and motif combinations as well (see “Materials and Methods”). We found that target genes of 314 predicted motif combinations significantly overlap with at least one coexpressed gene set obtained from four microarray data sets (FDR < 0.05; Supplemental Table S4). Consistently, for 289 of the 314 (92.0%) motif combinations, their target genes significantly share the same functions, based on the above GO analysis. In addition, we found that the functions of the predicted motifs are likely to be associated with the microarray experimental conditions. For instance, target genes of the motif combination MOD39131 are significantly coexpressed in the gene expression data set GSE30648, measuring genetic response of C. reinhardtii under different oxidative and electrophilic stress conditions. Accordingly, a motif in this combination, M21, is similar to the known motif REGION1OSOSEM, which has been reported to be an ABA-responsive element (Hattori et al., 1995) that plays a critical role in environmental stress response (Busk and Pagès, 1997). Besides, we also found that the target genes of MOD39131 significantly share the GO term response to stress (GO:0006950; corrected P value = 0.016).
Comparison with Other Methods
There is no computational study for genome-wide CRE prediction in C. reinhardtii. The MERCED presented here integrates multiple properties of potential CREs as motif-finding criteria. Different from existing motif-prediction methods applied in other species, the MERCED takes species divergence time into account when incorporating sequence conservation properties to define motifs. To see whether the consideration of species divergence time helps the CRE motif prediction in C. reinhardtii, we compared the MERCED with three alternative approaches using different strategies for sequence conservation evaluation.
The first alternative approach we compared with uses a mismatch number cutoff α to define conserved segments. We implemented this strategy by applying the same procedure of motif finding as in our method, except that we defined conserved k-mers using different mismatch number cutoff α here rather than the generating function-based statistical significance P value. In other words, for every pair of promoter sequences corresponding to the 8,741 orthologous genes, we defined a k-mer in C. reinhardtii as conserved if there exists at least one k-mer in its V. carteri orthologous sequence that has at most α mismatches with it. We chose the top x motifs based on the statistical significance, where x is equal to the number of predicted motifs using MERCED (Liu et al., 2002). We then compared the predicted motifs with known motifs in the PLACE database using the STAMP motif comparison tool (Mahony and Benos, 2007) with various E-value cutoffs. We found that under each of the STAMP E-value cutoffs, more motifs similar to known motifs are predicted by our method than those predicted by the method based on various mismatch number cutoff α (Table II).
Table II. Comparison of predicted motifs with known plant motifs.
The comparisons are based on 8-mers. The comparison becomes unnecessary when the mismatch number α is larger than 1, since 84.5% of 8-mer segments in C. reinhardtii will be defined as conserved 8-mers by the mismatch number-counting method when α = 2.
| Method | E Value Cutoff |
||
|---|---|---|---|
| 1E-5 | 1E-6 | 1E-7 | |
| MERCED | 195 (62%) | 112 (35%) | 72 (23%) |
| α = 0 | 171 (54%) | 103 (32.5%) | 52 (16.4%) |
| α = 1 | 179 (56.5%) | 100 (31.5%) | 57 (18.0%) |
| FastCompare | 169 (53.3%) | 97 (30.6%) | 50 (15.8%) |
| PhyloNet | 188 (59.3%) | 104 (32.8%) | 33 (10.4%) |
We next compared the MERCED with FastCompare, a more sophisticated approach (Elemento and Tavazoie, 2007). FastCompare determines conserved segments directly from sequence comparison, which essentially enumerates all k-mer segments in two species and assesses their significance based on the difference between their co-occurrence in orthologous sequences of the two species and their occurrence in individual species. We applied FastCompare to our regulatory sequences using the default parameters and only kept the same number of top predicted motifs as predicted using the MERCED for comparison. The results showed that our method predicted slightly more motifs that are similar to known plant motifs than FastCompare for various STAMP E-value cutoffs. For instance, for the STAMP E-value cutoff 1E-5, we predicted 195 8-mer motifs that are similar to known motifs in the PLACE database, while FastCompare predicted 169 motifs that are similar to known PLACE motifs. This result indicates the benefit of incorporating species divergence time to determine sequence conservation for CRE motif finding in C. reinhardtii.
We also compared the MERCED with another alternative strategy used in the PhyloNet (Wang and Stormo, 2005). Like FastCompare, PhyloNet is one of the only a few methods that can de novo identify motifs on the genome scale. Briefly, PhyloNet BLASTs conserved segments in a group of orthologous sequences against conserved segments in all other groups of orthologous sequences to identify motifs. The conserved segments in PhyloNet are defined by the wconsensus algorithm (Hertz and Stormo, 1999), which uses the information content to measure the similarity of segments and motifs. After applying PhyloNet to our data and comparing the same number of top-ranked motifs predicted by PhyloNet and the MERCED, we found that, again, the MERCED predicted more motifs that are similar to known plant motifs than PhyloNet for various STAMP E-value cutoffs (Table II). Not considering the species divergence time in PhyloNet can be the partial cause, since we observed that many highly similar segments in orthologous sequences that are defined as conserved in PhyloNet may not be conserved if we take species divergence time into account. For instance, GAGAAGAA is exactly shared by the regulatory sequences of two orthologous genes, Cre07.g327250 in C. reinhardtii and Vocar2001129m in V. carteri. However, it only has a relatively large conservation P value of 0.057 according to our model. It is thus not considered conserved with the FDR cutoff as 0.05. The overall comparisons shown in Table II demonstrated the necessity of considering species divergence time for CRE motif prediction.
A Public Database for cis-Regulatory Information in C. reinhardtii
Based on the results obtained in C. reinhardtii, we further developed a Web-accessible database (http://hulab.ucf.edu/research/projects/Microalgae/sdcre/motifcomb.html) where researchers can download the MERCED software tool and all the currently predicted CRE motifs, CREs, and motif combinations in C. reinhardtii. The database supports a variety of queries for specific predicted motifs, motifs similar to PLACE or TRANSFAC motifs, predicted and/or known motif combinations, target genes of specific motifs and/or motif combinations, and so on. A query-based search is able to output the position weight matrix-represented motifs and their target genes labeled with known expression profiles and GO annotation. The user can choose to view the query results in various formats.
DISCUSSION
Genome-wide identification of CREs in the C. reinhardtii genome is critical to further study gene regulation and molecular functions. We thus performed, to our knowledge, the first large-scale CRE prediction in C. reinhardtii. Different from available CRE prediction methods, our approach considers the species divergence time and the nucleotide content rather than depending on MSA and mismatch number counting to determine whether DNA segments are conserved CREs and motifs. According to sequence permutation test, the developed method has a very low FDR. Compared with alignment and mismatch number counting-based approaches, the developed method is more efficient in filtering divergent segments while keeping conserved segments that have quite a few mismatches compared with their counterpart segments. In total, we have predicted 66,530 CREs corresponding to 317 CRE motifs in C. reinhardtii that are conserved in the green alga V. carteri. We found that 62.5% of the predicted 317 motifs are similar to known PLACE motifs, and 27.5% of known PLACE motifs are included in our prediction. In addition, we discovered that the predicted motifs form 92,694 statistically significant motif combinations in C. reinhardtii. These statistically significant motif combinations are evaluated and supported by known motif combinations, GO enrichment analysis, and gene expression analysis. The large number of CRE motifs and combinations predicted in this study will further facilitate algae research for various applications.
In addition to the genome-wide CRE prediction in C. reinhardtii, further analysis of the predicted CREs in C. reinhardtii implies that the transcriptional regulation mechanisms may be significantly different between the green algae and land plant species. We found that two out of the seven well-known motif combinations shared by land plant species are not included in our predicted motif combinations. The missing well-known motif combinations in land plant species suggest that the green algae may have their specific regulatory mechanisms. In fact, whereas genes encoding MADS domains are widespread in land plant genomes (Riechmann and Meyerowitz, 1997; de Folter et al., 2005), there are only two predicted TFs with MADS domains in C. reinhardtii (Pérez-Rodríguez et al., 2010). This can also be a possible explanation for the lack of motif combinations that contain the well-known MADS-MADS motif combinations. Interestingly, the comparisons of the predicted motifs with experimentally verified known motifs also suggest that C. reinhardtii may have both plant-like and animal-like motifs. For instance, the predicted motif M3 is similar to several known motifs related to photosynthesis, such as PE3ASPHYA3 (Bruce et al., 1991). In the mean time, we found that some predicted motifs tend to have animal-like functions related to flagellum. For instance, the predicted motif M10, not similar to any known PLACE motif, was found in 76 out of 263 (29%) motif combinations whose target genes contain IFT88 (Cre.07.gee5750), which is known to be related to flagellum function (Lucker et al., 2010). This indicates that M10 may be a novel motif related to flagellum function.
A few caveats exist for applying our methods. First, the identification of input regulatory sequences is a complicated issue, since the CREs can be anywhere in the upstream sequences. In this study, the regulatory sequences were restricted to the upstream 1-kb-long sequences (Vandepoele et al., 2006; Ma and Bohnert, 2007). We compared the results with those obtained from shortened upstream sequences (800 bp) and those from extended upstream sequences (1,200 bp). We found that more than 83.5% of predicted motifs are shared by upstream regions defined in the three different ways. Second, the length of potential motifs to be identified needs to be determined. We used k-mer motifs to illustrate the MERCED algorithm for k = 8, while k could be any number between 6 and 14. We used 8-mers, because the most dominant length of motifs in the TRANSFAC database is eight (Wingender et al., 1996), and many previous studies have successfully identified meaningful motifs in plants and other species using 8-mers (Mariño-Ramírez et al., 2004; Yamamoto et al., 2011). We also predicted 7-mer and 9-mer motifs. We found that more than 88% of predicted 7-mer and 9-mer motifs are similar to the predicted 8-mer motifs (STAMP E-value < 1E-5 [Mahony and Benos, 2007]). In the MERCED software, a user can set different lengths for the regulatory sequences and different parameter k for k-mer CRE prediction. Third, one needs to pay attention to the criteria used to evaluate the similarity between motifs. We measured the similarity of predicted motifs and known motifs in the PLACE database by using the STAMP software. Despite the high significance of the similarity, the functional consequences of the dissimilar positions between the predicted motifs and the known motifs need to be carefully assessed by experiments.
Finally, although we have shown various sources of evidence that support our predictions, these predictions need to be experimentally validated. Currently, the number of genes with annotated functions in the two algae species is small and experimentally verified CREs are rare. With more and more genomic data available, the prediction accuracy can be further improved and better evaluated. For example, recent sequencing technology has generated more large-scale measurement of gene expression and TF binding and produced ChIP-seq (for chromatin immunoprecipitation followed by massively parallel DNA sequencing) and RNA-seq (for high throughout sequencing of RNA) data sets in many species (Johnson et al., 2007; Robertson et al., 2007). There are already four RNA-seq data sets available in the Gene Expression Omnibus (GEO) and 15 samples available in the Sequence Read Archive in C. reinhardtii (Miller et al., 2010; Castruita et al., 2011; Fischer et al., 2012; Urzica et al., 2012). Although the number is still small and the data-processing strategies (e.g. data normalization and isoform identification) are still under development, we can foresee in the near future that a large number of RNA-seq and ChIP-seq data sets can be integrated for CRE prediction and evaluation under different experimental conditions. Additionally, with more genomes sequenced, we will be able to integrate more species into our comparative genomics study, for which species divergence time incorporation is expected to produce more accurate conservation estimation.
MATERIALS AND METHODS
Protein, DNA Sequences, and Orthologous Gene Pairs
We defined the promoter sequence of a gene as the upstream 1,000-bp sequence relative to the translation start site of this gene. We used upstream sequences relative to the translation start sites instead of transcription start sites, because CREs have been found in sequences downstream of the transcription start sites and upstream of the translation start sites (Feldbrügge et al., 1994; Ovadia et al., 2010). In addition, translation start sites can be more reliably obtained than transcription start sites. All genes and their promoter sequences in Chlamydomonas reinhardtii and Volvox carteri were downloaded from the Phytozome database (http://www.phytozome.net/). The Joint Genome Institute (JGI) version 4.3 and the JGI version 1.0 releases were used for the two species, respectively. In total, we obtained 17,116 genes in C. reinhardtii and 14,546 genes in V. carteri. Repeats in the upstream sequences were then masked using RepeatMasker (http://www.repeatmasker.org/). We also obtained all protein sequences in the two species from the Phytozome database. The PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) program (Altschul et al., 1997) was then applied to these sequences to find the reciprocal best hits between proteins in the two species. In total, we defined 8,742 orthologous pairs based on reciprocal best hits (E-value ≤ 1E-10) in C. reinhardtii and V. carteri (Supplemental Table S5).
Gene Expression Data and Coexpressed Clusters
We selected four gene expression data sets in C. reinhardtii from the GEO database, requiring that each of them contains at least 15 samples (Edgar et al., 2002). The data sets we used were GSE20860, GSE20861, GSE30646, and GSE30648. We then BLASTed the probe sequence in each data set against the genes in the C. reinhardtii genome to identify genes corresponding to each probe sequence. If a BLAST hit (a partial gene sequence) matched more than 95% of a probe sequence with more than 95% identity, we assigned the corresponding gene to the probe from the GEO data set. We then calculated the pairwise Pearson’s correlation for every pair of genes in each data set. These gene correlations were then used to obtain coexpressed gene sets by applying a hierarchical clustering algorithm with the average linkage (Sokal and Michener, 1985). In total, we obtained 437 gene sets, each of which contains more than three genes and has gene correlation no less than 0.6 (Supplemental Table S6).
Model of the Neutral Evolution Rate of Nucleotide Substitution
We constructed a substitution matrix to describe the neutral evolution of nucleotides between C. reinhardtii and V. carteri. The construction is based on the 4-fold degenerate sites in orthologous proteins in the two algae species. We first aligned each pair of orthologous proteins by using the widely used protein-alignment software MUSCLE (Edgar, 2004). We then obtained all aligned 4-fold degenerate sites with the same amino acid in the alignments. A 4-fold degenerate site is a position in a codon where all nucleotide substitutions at this site are synonymous. For each of the positions containing 4-fold degenerate sites, we obtained their corresponding nucleotides and counted how many times a given type of nucleotide in C. reinhardtii corresponds to another given type of nucleotide in V. carteri. In the end, we obtained a four-by-four substitution matrix (S) involving the four types of nucleotides, A, C, G, and T, as illustrated in Figure 2A.
Identification of Conserved k-mers in C. reinhardtii
To identify k-mers in C. reinhardtii that are conserved between C. reinhardtii and V. carteri, we first defined conserved-to-be (CTB) k-mer pairs. A CTB
k-mer pair contains two k-mers, one from C. reinhardtii and the other from V. carteri, which are likely to be conserved between these two species. To find all CTB
k-mer pairs, we obtained the expected number of mismatches m between any k-mer in a promoter sequence μ in C. reinhardtii and any k-mer in μ’s orthologous sequence in V. carteri. Here, m is calculated as 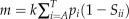 , where pi is the probability that the i-th type of nucleotides in C. reinhardtii and Sii is the probability that the i-th type of nucleotides in C. reinhardtii correspond to the i-th type of nucleotides in V. carteri. Note that m is the same for all k-mers. We defined two k-mers as a CTB
k-mer pair if the observed mismatch between them was smaller than m. With all the CTB
k-mer pairs in the two species defined, we then selected k-mers from these CTB
k-mer pairs as conserved k-mers by their statistical significance (P value) in terms of evolutionary conservation. To calculate the conservation P value of a CTB
k-mer pair that contains a specific k-mer, one needs to compare all the 4k different k-mers in V. carteri with this k-mer in C. reinhardtii. However, with the large number of k-mers in C. reinhardtii, directly enumerating all the 4k different k-mers in V. carteri is time consuming. Therefore, we exploited the mathematics concept of generating function to calculate the P value. The generating function is a representation of the probability mass function of a random variable. For a discrete random variable X, its generating function is defined as
, where pi is the probability that the i-th type of nucleotides in C. reinhardtii and Sii is the probability that the i-th type of nucleotides in C. reinhardtii correspond to the i-th type of nucleotides in V. carteri. Note that m is the same for all k-mers. We defined two k-mers as a CTB
k-mer pair if the observed mismatch between them was smaller than m. With all the CTB
k-mer pairs in the two species defined, we then selected k-mers from these CTB
k-mer pairs as conserved k-mers by their statistical significance (P value) in terms of evolutionary conservation. To calculate the conservation P value of a CTB
k-mer pair that contains a specific k-mer, one needs to compare all the 4k different k-mers in V. carteri with this k-mer in C. reinhardtii. However, with the large number of k-mers in C. reinhardtii, directly enumerating all the 4k different k-mers in V. carteri is time consuming. Therefore, we exploited the mathematics concept of generating function to calculate the P value. The generating function is a representation of the probability mass function of a random variable. For a discrete random variable X, its generating function is defined as 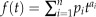 , where
, where 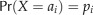 and n is the number of different values X can take. For every k-mer μ in C. reinhardtii that has similar k-mers in μ’s corresponding orthologous sequence in V. carteri, say a1a2…ak, we can define a generating function of the k-mer in C. reinhardtii as
and n is the number of different values X can take. For every k-mer μ in C. reinhardtii that has similar k-mers in μ’s corresponding orthologous sequence in V. carteri, say a1a2…ak, we can define a generating function of the k-mer in C. reinhardtii as 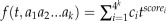 , where ci is the probability that the i-th k-mer occurs in the upstream 1,000-bp sequences in V. carteri and scorei is the substitution score of the k-mer
a1a2…ak in C. reinhardtii by the i-th k-mer in V. carteri. The generating function can be efficiently computed as in previous studies (Staden, 1989; Huang et al., 2004). The statistical significance for the evolutionary conservation between a1a2…ak and one of its similar k-mers in V. carteri can then be calculated as
, where ci is the probability that the i-th k-mer occurs in the upstream 1,000-bp sequences in V. carteri and scorei is the substitution score of the k-mer
a1a2…ak in C. reinhardtii by the i-th k-mer in V. carteri. The generating function can be efficiently computed as in previous studies (Staden, 1989; Huang et al., 2004). The statistical significance for the evolutionary conservation between a1a2…ak and one of its similar k-mers in V. carteri can then be calculated as 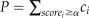 , where α is the substitution score of this k-mer pair. We claimed a k-mer pair as conserved if its Bonferroni corrected P ≤ 0.05.
, where α is the substitution score of this k-mer pair. We claimed a k-mer pair as conserved if its Bonferroni corrected P ≤ 0.05.
Prediction of Motifs and Motif Combinations
To predict CRE motifs from the conserved k-mers identified in C. reinhardtii, we first applied the hierarchical clustering algorithm (average linkage; Sokal and Michener, 1985) to cluster the conserved k-mers. Since conserved k-mers forming a cluster are likely to have the same underlying pattern and thus correspond to the same motif, we predicted a motif for each cluster. Each predicted motif is represented by a position weight matrix (Stormo and Hartzell, 1989). In the end, we obtained 66,530 conserved CREs and 317 underlying 8-mer motifs. With the predicted motifs, we further identified motif combinations by finding motifs frequently co-occurring in a large number of regulatory sequences in C. reinhardtii using our previously developed method (Cai et al., 2010). The statistical significance of each group of frequently co-occurring motifs was determined by Poisson clumping heuristic (Cai et al., 2010). The groups of motifs with sufficient significance (FDR < 0.05) were predicted as motif combinations.
Functional Analysis of the Predicted Motif Combinations
To see whether motif combinations are associated with specific functions, we investigated the overlap between the target genes of co-occurring motifs and functional gene sets. The functional gene sets are defined as sets of genes with specific GO function annotation (Ashburner et al., 2000) or sets of genes with correlated transcription expression, since correlated expression often implicates similar function (Altman and Raychaudhuri, 2001; Ideker et al., 2001). The GO function annotation was downloaded from the JGI Web site (http://genome.jgi-psf.org/cgi-bin/ToGo?species=Chlre4), which is the version 4.0 C. reinhardtii annotation. The statistical significance of any overlap between target genes of a motif combination and a given functional gene set is calculated as follows: let S be the set of all the N genes in a genome, S1 be a predicted target gene set of a motif combination, and S2 be a given functional gene set, and assume the number of genes in the intersection of the three sets S, S1, and S2 is n, M, and m, respectively. Then, the P value of the overlap of the set S1 and the set S2 can be estimated based on the hypergeometric test 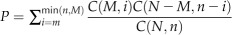 , where C(x,y) is the combinatorial number of choosing y items out of x items. From such obtained P values, we then calculate q values based on the Q-Value software to estimate the statistical significance of the overlap (Storey and Tibshirani, 2003). All the motif combinations with sufficient statistical significance (FDR < 0.05) in terms of overlapping with known functional gene sets are reported as functional motif combinations.
, where C(x,y) is the combinatorial number of choosing y items out of x items. From such obtained P values, we then calculate q values based on the Q-Value software to estimate the statistical significance of the overlap (Storey and Tibshirani, 2003). All the motif combinations with sufficient statistical significance (FDR < 0.05) in terms of overlapping with known functional gene sets are reported as functional motif combinations.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Table S1. Predicted motifs.
Supplemental Table S2. Predicted motif combinations.
Supplemental Table S3. Motif combinations consistent with known TF combinations.
Supplemental Table S4. Motif combinations with significantly coexpressed target genes.
Supplemental Table S5. Orthologous gene pairs in C. reinhardtii and V. carteri.
Supplemental Table S6. Coexpressed gene clusters.
Glossary
- CRE
cis-regulatory element
- TF
transcription factor
- MSA
multiple sequence alignment
- MERCED
modeling evolution rate across species for cis-regulatory element discovery
- k-mer
k-bp-long DNA segment
- FDR
false discovery rate
- ABA
abscisic acid
- GO
Gene Ontology
- GEO
Gene Expression Omnibus
- JGI
Joint Genome Institute
- CTB
conserved-to-be
References
- Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K. (2003) Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15: 63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocco DJ, Kohane IS, Butte AJ. (2004) Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 5: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman RB, Raychaudhuri S. (2001) Whole-genome expression analysis: challenges beyond clustering. Curr Opin Struct Biol 11: 340–347 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Beckmann J, Lehr F, Finazzi G, Hankamer B, Posten C, Wobbe L, Kruse O. (2009) Improvement of light to biomass conversion by de-regulation of light-harvesting protein translation in Chlamydomonas reinhardtii. J Biotechnol 142: 70–77 [DOI] [PubMed] [Google Scholar]
- Bisova K, Krylov DM, Umen JG. (2005) Genome-wide annotation and expression profiling of cell cycle regulatory genes in Chlamydomonas reinhardtii. Plant Physiol 137: 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Bataille AR, Chen X, Poitras C, Laganière J, Lefèbvre C, Deblois G, Giguère V, Ferretti V, Bergeron D, et al. (2006) Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res 16: 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Tompa M. (2002) Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res 12: 739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne F, Linden H. (2002) Regulation of carotenoid biosynthesis genes in response to light in Chlamydomonas reinhardtii. Biochim Biophys Acta 1579: 26–34 [DOI] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. (2004) GO:TermFinder: open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20: 3710–3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce WB, Deng XW, Quail PH. (1991) A negatively acting DNA sequence element mediates phytochrome-directed repression of phyA gene transcription. EMBO J 10: 3015–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce WB, Quail PH. (1990) Cis-acting elements involved in photoregulation of an oat phytochrome promoter in rice. Plant Cell 2: 1081–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk PK, Pagès M. (1997) Protein binding to the abscisic acid-responsive element is independent of VIVIPAROUS1 in vivo. Plant Cell 9: 2261–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk PK, Pagès M. (1998) Regulation of abscisic acid-induced transcription. Plant Mol Biol 37: 425–435 [DOI] [PubMed] [Google Scholar]
- Cai X, Hou L, Su N, Hu H, Deng M, Li X. (2010) Systematic identification of conserved motif modules in the human genome. BMC Genomics 11: 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardol P, González-Halphen D, Reyes-Prieto A, Baurain D, Matagne RF, Remacle C. (2005) The mitochondrial oxidative phosphorylation proteome of Chlamydomonas reinhardtii deduced from the Genome Sequencing Project. Plant Physiol 137: 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana C, Garcia-Luque I, Alonso E, Malik VS, Cashmore AR. (1988) Both positive and negative regulatory elements mediate expression of a photoregulated CAB gene from Nicotiana plumbaginifolia. EMBO J 7: 1929–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castruita M, Casero D, Karpowicz SJ, Kropat J, Vieler A, Hsieh SI, Yan W, Cokus S, Loo JA, Benning C, et al. (2011) Systems biology approach in Chlamydomonas reveals connections between copper nutrition and multiple metabolic steps. Plant Cell 23: 1273–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Folter S, Immink RG, Kieffer M, Parenicova L, Henz SR, Weigel D, Busscher M, Kooiker M, Colombo L, Kater MM, et al. (2005) Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 17: 1424–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Hu H, Li X. (2012) Thousands of cis-regulatory sequence combinations are shared by Arabidopsis and poplar. Plant Physiol 158: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard S, Jain M, Im CS, Pollock S, Shrager J, Lin Y, Peek AS, Grossman AR. (2006) Generation of an oligonucleotide array for analysis of gene expression in Chlamydomonas reinhardtii. Curr Genet 49: 106–124 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O, Tavazoie S. (2005) Fast and systematic genome-wide discovery of conserved regulatory elements using a non-alignment based approach. Genome Biol 6: R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O, Tavazoie S. (2007) FastCompare: a nonalignment approach for genome-scale discovery of DNA and mRNA regulatory elements using network-level conservation. Methods Mol Biol 395: 349–366 [PubMed] [Google Scholar]
- Fauteux F, Blanchette M, Strömvik MV. (2008) Seeder: discriminative seeding DNA motif discovery. Bioinformatics 24: 2303–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldbrügge M, Sprenger M, Dinkelbach M, Yazaki K, Harter K, Weisshaar B. (1994) Functional analysis of a light-responsive plant bZIP transcriptional regulator. Plant Cell 6: 1607–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer BB, Dayer R, Schwarzenbach Y, Lemaire SD, Behra R, Liedtke A, Eggen RI. (2009) Function and regulation of the glutathione peroxidase homologous gene GPXH/GPX5 in Chlamydomonas reinhardtii. Plant Mol Biol 71: 569–583 [DOI] [PubMed] [Google Scholar]
- Fischer BB, Ledford HK, Wakao S, Huang SG, Casero D, Pellegrini M, Merchant SS, Koller A, Eggen RI, Niyogi KK. (2012) SINGLET OXYGEN RESISTANT 1 links reactive electrophile signaling to singlet oxygen acclimation in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 109: E1302–E1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Sheehan JB, Stokowski RP, Chen X, Hosseini R, Cheng JF, Fodor SP, Cox DR, Patil N. (2001) Evolutionarily conserved sequences on human chromosome 21. Genome Res 11: 1651–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Hansen U, Weng Z. (2001) Detection of cis-element clusters in higher eukaryotic DNA. Bioinformatics 17: 878–889 [DOI] [PubMed] [Google Scholar]
- Fujimoto SY, Ohta M, Usui A, Shinshi H, Ohme-Takagi M. (2000) Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 12: 393–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AR, Harris EE, Hauser C, Lefebvre PA, Martinez D, Rokhsar D, Shrager J, Silflow CD, Stern D, Vallon O, et al. (2003) Chlamydomonas reinhardtii at the crossroads of genomics. Eukaryot Cell 2: 1137–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Liu JS. (2005) De novo cis-regulatory module elicitation for eukaryotic genomes. Proc Natl Acad Sci USA 102: 7079–7084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori T, Terada T, Hamasuna S. (1995) Regulation of the Osem gene by abscisic acid and the transcriptional activator VP1: analysis of cis-acting promoter elements required for regulation by abscisic acid and VP1. Plant J 7: 913–925 [DOI] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. (1999) Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577 [DOI] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. (1999) Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res 27: 297–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Hu H, Li X. (2008) MOPAT: a graph-based method to predict recurrent cis-regulatory modules from known motifs. Nucleic Acids Res 36: 4488–4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Kao MC, Zhou X, Liu JS, Wong WH. (2004) Determination of local statistical significance of patterns in Markov sequences with application to promoter element identification. J Comput Biol 11: 1–14 [DOI] [PubMed] [Google Scholar]
- Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, Bumgarner R, Goodlett DR, Aebersold R, Hood L. (2001) Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science 292: 929–934 [DOI] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Langner U, Jakob T, Stehfest K, Wilhelm C. (2009) An energy balance from absorbed photons to new biomass for Chlamydomonas reinhardtii and Chlamydomonas acidophila under neutral and extremely acidic growth conditions. Plant Cell Environ 32: 250–258 [DOI] [PubMed] [Google Scholar]
- Lawrence CE, Altschul SF, Boguski MS, Liu JS, Neuwald AF, Wootton JC. (1993) Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science 262: 208–214 [DOI] [PubMed] [Google Scholar]
- Li WH, Wu CI, Luo CC. (1985) A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol 2: 150–174 [DOI] [PubMed] [Google Scholar]
- Li X, Wong WH. (2005) Sampling motifs on phylogenetic trees. Proc Natl Acad Sci USA 102: 9481–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhong S, Wong WH. (2005) Reliable prediction of transcription factor binding sites by phylogenetic verification. Proc Natl Acad Sci USA 102: 16945–16950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Han D, Hu G, Sommerfeld M, Hu Q. (2010) Inhibition of starch synthesis results in overproduction of lipids in Chlamydomonas reinhardtii. Biotechnol Bioeng 107: 258–268 [DOI] [PubMed] [Google Scholar]
- Liu XS, Brutlag DL, Liu JS. (2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat Biotechnol 20: 835–839 [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu XS, Wei L, Altman RB, Batzoglou S. (2004) Eukaryotic regulatory element conservation analysis and identification using comparative genomics. Genome Res 14: 451–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA. (2000) Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136–140 [DOI] [PubMed] [Google Scholar]
- Lucker BF, Miller MS, Dziedzic SA, Blackmarr PT, Cole DG. (2010) Direct interactions of intraflagellar transport complex B proteins IFT88, IFT52, and IFT46. J Biol Chem 285: 21508–21518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Bohnert HJ. (2007) Integration of Arabidopsis thaliana stress-related transcript profiles, promoter structures, and cell-specific expression. Genome Biol 8: R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahony S, Benos PV. (2007) STAMP: a Web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35: W253–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariño-Ramírez L, Spouge JL, Kanga GC, Landsman D. (2004) Statistical analysis of over-represented words in human promoter sequences. Nucleic Acids Res 32: 949–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L, et al. (2007) The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R, Wu G, Deshpande RR, Vieler A, Gärtner K, Li X, Moellering ER, Zäuner S, Cornish AJ, Liu B, et al. (2010) Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol 154: 1737–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag M, Kiaulehn S, Johnson CH. (2005) The circadian clock in Chlamydomonas reinhardtii. What is it for? What is it similar to? Plant Physiol 137: 399–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Choi SP, Lee J, Lee JH, Sim SJ. (2009) Hydrothermal acid pretreatment of Chlamydomonas reinhardtii biomass for ethanol production. J Microbiol Biotechnol 19: 161–166 [DOI] [PubMed] [Google Scholar]
- Ono A, Izawa T, Chua NH, Shimamoto K. (1996) The rab16B promoter of rice contains two distinct abscisic acid-responsive elements. Plant Physiol 112: 483–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovadia A, Tabibian-Keissar H, Cohen Y, Kenigsbuch D. (2010) The 5′UTR of CCA1 includes an autoregulatory cis element that segregates between light and circadian regulation of CCA1 and LHY. Plant Mol Biol 72: 659–671 [DOI] [PubMed] [Google Scholar]
- Pérez-Rodríguez P, Riaño-Pachón DM, Corrêa LG, Rensing SA, Kersten B, Mueller-Roeber B. (2010) PlnTFDB: updated content and new features of the plant transcription factor database. Nucleic Acids Res 38: D822–D827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B, Wasserman WW, Sandelin A. (2010) JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38: D105–D110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnik SE, Umen J, Nedelcu AM, Hallmann A, Miller SM, Nishii I, Ferris P, Kuo A, Mitros T, Fritz-Laylin LK, et al. (2010) Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 329: 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed BD, Charos AE, Szekely AM, Weissman SM, Snyder M. (2008) Genome-wide occupancy of SREBP1 and its partners NFY and SP1 reveals novel functional roles and combinatorial regulation of distinct classes of genes. PLoS Genet 4: e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL, Meyerowitz EM. (1997) MADS domain proteins in plant development. Biol Chem 378: 1079–1101 [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4: 651–657 [DOI] [PubMed] [Google Scholar]
- Rochaix JD. (2004) Genetics of the biogenesis and dynamics of the photosynthetic machinery in eukaryotes. Plant Cell 16: 1650–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombauts S, Déhais P, Van Montagu M, Rouzé P. (1999) PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res 27: 295–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, Friedman N. (2003) Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 34: 166–176 [DOI] [PubMed] [Google Scholar]
- Singh KB. (1998) Transcriptional regulation in plants: the importance of combinatorial control. Plant Physiol 118: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Blanchette M, Tompa M. (2004) PhyME: a probabilistic algorithm for finding motifs in sets of orthologous sequences. BMC Bioinformatics 5: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skriver K, Olsen FL, Rogers JC, Mundy J. (1991) Cis-acting DNA elements responsive to gibberellin and its antagonist abscisic acid. Proc Natl Acad Sci USA 88: 7266–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal R, Michener C. (1985) A statistical method for evaluating systematic relationships. Univ Kans Sci Bull 38: 1409–1438 [Google Scholar]
- Staden R. (1989) Methods for calculating the probabilities of finding patterns in sequences. Comput Appl Biosci 5: 89–96 [DOI] [PubMed] [Google Scholar]
- Steffens NO, Galuschka C, Schindler M, Bülow L, Hehl R. (2005) AthaMap Web tools for database-assisted identification of combinatorial cis-regulatory elements and the display of highly conserved transcription factor binding sites in Arabidopsis thaliana. Nucleic Acids Res 33: W397–W402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD, Hartzell GW., III (1989) Identifying protein-binding sites from unaligned DNA fragments. Proc Natl Acad Sci USA 86: 1183–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun TH, Liu CQ, Hui YY, Wu WK, Zhou ZG, Lu S. (2010) Coordinated regulation of gene expression for carotenoid metabolism in Chlamydomonas reinhardtii. J Integr Plant Biol 52: 868–878 [DOI] [PubMed] [Google Scholar]
- Urzica EI, Adler LN, Page MD, Linster CL, Arbing MA, Casero D, Pellegrini M, Merchant SS, Clarke SG. (2012) Impact of oxidative stress on ascorbate biosynthesis in Chlamydomonas via regulation of the VTC2 gene encoding a GDP-L-galactose phosphorylase. J Biol Chem 287: 14234–14245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, Casneuf T, Van de Peer Y. (2006) Identification of novel regulatory modules in dicotyledonous plants using expression data and comparative genomics. Genome Biol 7: R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Stormo GD. (2003) Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 19: 2369–2380 [DOI] [PubMed] [Google Scholar]
- Wang T, Stormo GD. (2005) Identifying the conserved network of cis-regulatory sites of a eukaryotic genome. Proc Natl Acad Sci USA 102: 17400–17405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Haberer G, Mayer KF. (2009) Discovery of cis-elements between sorghum and rice using co-expression and evolutionary conservation. BMC Genomics 10: 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemmer KA, Marshall WF. (2004) Flagellar motility: all pull together. Curr Biol 14: R992–R993 [DOI] [PubMed] [Google Scholar]
- Wingender E, Dietze P, Karas H, Knüppel R. (1996) TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res 24: 238–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto YY, Yoshioka Y, Hyakumachi M, Maruyama K, Yamaguchi-Shinozaki K, Tokizawa M, Koyama H. (2011) Prediction of transcriptional regulatory elements for plant hormone responses based on microarray data. BMC Plant Biol 11: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh CH, Bolouri H, Davidson EH. (1998) Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science 279: 1896–1902 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wong WH. (2004) CisModule: de novo discovery of cis-regulatory modules by hierarchical mixture modeling. Proc Natl Acad Sci USA 101: 12114–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]