

Abstract
Previous studies in humans and animals have suggested a possible association between lead (Pb) exposure and the etiology of Alzheimer’s disease (AD). Animals acutely exposed to Pb display an over-expressed amyloid precursor protein (APP) and the ensuing accumulation of beta-amyloid (Aβ) in brain extracellular spaces. This study was designed to examine whether in vivo Pb exposure increased brain concentrations of Aβ, resulting in amyloid plaque deposition in brain tissues. Human Tg-SWDI APP transgenic mice, which genetically over-express amyloid plaques at age of 2-3 months, received oral gavages of 50 mg/kg Pb acetate once daily for 6 wk; a control group of the same mouse strain received the same molar concentration of Na acetate. ELISA results revealed a significant increase of Aβ in the CSF, brain cortex and hippocampus. Immunohistochemistry displayed a detectable increase of amyloid plaques in brains of Pb-exposed animals. Neurobehavioral test using Morris water maze showed an impaired spatial learning ability in Pb-treated mice, but not in C57BL/6 wild type mice with the same age. In vitro studies further uncovered that Pb facilitated Aβ fibril formation. Moreover, the synchrotron X-ray fluorescent studies demonstrated a high level of Pb present in amyloid plaques in mice exposed to Pb in vivo. Taken together, these data indicate that Pb exposure with ensuing elevated Aβ level in mouse brains appears to be associated with the amyloid plaques formation. Pb apparently facilitates Aβ fibril formation and participates in deposition of amyloid plaques.
Keywords: Lead or Pb, beta-amyloid or Aβ, amyloid plaques, Tg-SWDI mouse, fibril formation, X-ray fluorescence or XRF, hippocampus
1. Introduction
Alzheimer’s disease (AD) is one of the most common causes of dementia in the elderly. About 4.5 million people live in the United States are affected by this disorder (Hebert et al., 2003). AD is characterized by the progressive impairment of cognitive function and memory loss. Beta-amyloid (Aβ) accumulation in brain extracellular space is believed to be an initial feature of AD pathogenesis (Ogomori et al., 1989). The majority of AD cases (>95%) are in non-familial, late onset sporadic forms with age of >65 years that possess no clear genetic association (Migliore and Coppede, 2009), suggesting that the environmental factors such as exposure to toxic chemicals may play a significant role in AD pathogenesis.
Lead (Pb) is a well-known environmental neurotoxin, and chronic Pb exposure can produce detrimental effects for brain development and cognitive functions (Tong et al., 2000; Hu et al., 2006; Schnaas et al., 2006; Jiang et al., 2008). Although Pb has been removed from gasoline three decades ago in the United States, it still remains one of the most environmental hazards facing humans today (White et al., 2007). Pb exposure is associated with both peripheral and central neurological effects including memory deficits and AD-like pathology (Shih et al., 2007; Stewart et al., 2006). One case study also provides the clinical evidence of AD-type neurofibrillary tangles in hippocampus of an adult who suffered from childhood Pb encephalopathy (Niklowitz and Mandybur, 1975). The potential link between Pb exposure and increased risk of developing AD in rat and monkey is currently under active investigation (Basha et al., 2005a, b; Huang et al., 2011).
The accumulation of β-amyloid (Aβ), which is derived from the proteolytic cleavage of a larger 120 kDa transmembrane protein, the amyloid beta precursor protein (APP), within brain extracellular space is believed to be an initial feature of AD pathogenesis (Ogomori et al., 1989). Plaque formation that follows is brought about by the transformation of Aβ from a soluble form to neurotoxic aggregated, fibrillary structure. Some reports show that copper (Cu) and zinc (Zn), which are co-localized with the plaques, may play a role in this transformation (Miller et al., 2006; Lee et al., 1999). However, the question as to whether other metal ions such as Pb may participate in the plaque formation remains largely unanswered.
The absence of naturally occurring rodent forms of AD has imposed a research challenge for the use of animal models to study mechanistic alterations occurred in AD. We have successfully used an early-onset transgenic mouse model, known as Tg-SwDI which over-expresses human Swedish, Dutch, and Iowa triple-mutant APP in the brain in our previous studies (Gu et al., 2011). These transgenic mice develop amyloid plaques at the 2-3 months of age (Davis et al., 2004), serving as an ideal model to explore the effect of in vivo Pb exposure on the onset and severity of Aβ burden in the brain. In our acute Pb exposure studies, a significant accumulation of Aβ in the cerebrospinal fluid (CSF) and selected brain regions was observed (Gu et al., 2011). Nonetheless, a larger research puzzle, such as whether the increased Aβ would lead to an ultimate deposition of senile plaques in brain tissues as a result of Pb exposure, was unaddressed.
This study was designed to investigate the formation of plagues in Tg-SwDI mice following 4-6 wk Pb exposure. More specifically, we determined (1) whether Pb exposure altered Aβ levels in the CSF and brain tissues, (2) whether this led to the deposition of insoluble plagues in brain regions, (3) if the essential learning behavior may change as a result of the plaque formation, and (4) how the plaque formation may relate to Pb concentrations in the plaques. For the last study aim, we employed the state-of-the-art synchrotron-based X-ray fluorescence (XRF) technique to determine the Pb concentrations in amyloid plaque.
2. Material and Methods
2.1. Materials
Chemicals and assay kits were purchased from the following sources: Anti-mouse IgG conjugated with horse-radish peroxidase and β-actin were purchased from Sell Signaling (Danvers, MA), biotinylated anti-mouse IgG from Vector (Burlingame, CA), beta (β)-secretase fluorometric assay kit from BioVision (Mountain View, California), fluorogenic gamma (γ)-secretase substrate from Calbiochem (Merck KGaA, Darmstadt, Germany), Aβ1-40 from American Peptide Company (Sunnyvale, CA), 4-12% Bis-Tris gel and nitrocellulose membrane from Invitrogen (Carlsbad, California), and protease inhibitor cocktail from Roche (Indianapolis, IN). 3, 3’, 5, 5’-Tetramethylbenzidine (TMB), thioflavin S, thioflavin T, RIPA buffer and all other chemicals were obtained from Sigma Aldrich (St. Louis, MO). CytoFluor 2350, a fluorescence measurement system, was purchased from Millipore (Billerica, Massachusetts). The antibodies 2G3 and 21F12, the biotin labeled 3D6, and 8E5 were generous gifts from Eli Lilly. All reagents were of analytical grade, HPLC grade or the best available pharmaceutical grade.
2.2 Animals and treatments
Tg-SWDI homozygous mice and WT (wild type) mice (all with a C57BL/6 genetic background) were purchased from the Jackson Laboratory (Bar Harbor, Maine) and bred in the laboratory of Animal Center at Indiana University School of Medicine. Mice were housed 3-5 per cage, fed with food and water ad libitum, and maintained in a 12-h light/dark cycle facility. Tg-SWDI mice at the time of experimentation were 8 and 4 weeks old, and they were administered with 50 mg Pb-acetate/kg (i.e., 27 mg Pb/kg) by oral gavage or with an equivalent molar concentration of Na-acetate once daily for 6 or 4 weeks. Groups of WT mice were also treated with Pb or Na acetate in the same dose regimen by oral gavage. This dose regimen has been shown to produce a significant accumulation of Pb (60 μg/dL) in mice brain and similar to those found in children (Gu et al., 2011). All solutions were adjusted to pH7.2 prior to the oral gavage. At the end of dose administration, the mice were injected with ketamine/xylazine (75:10 mg/mL, 1 mL/kg body weight) prior to termination. Whole blood and CSF samples were collected from each animal. The brains were then perfused with ice-cold saline for 2 min by infusing saline via carotid artery, removed and flash–frozen for further experimentation. The blood was centrifuged for 10 min (2000g) to separate plasma and blood cells.
2.3. Quantification of Aβ by ELISA
Levels of Aβ were assayed by sandwich ELISA as described previously (Hyslop and Bender, 2002). Briefly, Hippocampus and cortex from one half of mouse brain were extracted with 5 M guanidine buffer. Hippocampus, cortex, CSF, and plasma were processed in ninety-six-well ELISA plates that had been coated with antibodies 2G3 and 21F12 to determine Aβ1-40 and Aβ1-42, respectively. After incubation of plates with casein buffer (0.25% casein and 0.05% sodium azide in PBS) for 2 h, samples were loaded overnight at 4°C. Biotin-3D6 was incubated for 1 h at room temperature, followed by horseradish peroxidase for 1 h. After incubation with TMB, the plates were read for absorbance at 450 nm. Total protein concentrations in the brain were determined using the Bradford protein assay, and the concentration of Aβ in the tissues was reported as ng/mg of total protein.
2.4. Histology and immunohistochemistry
The Tg-SWDI homozygous mice at age of 8 weeks old can develop Aβ immune reactivity in brain, indicating the formation of Aβ plaques. To determine how Pb exposure may affect amyloid plaque formation, one half of the brain from Tg-SWDI mice (see above) was rapidly frozen by plunging into liquid nitrogen and sectioned with a vibratome at 30 μm for plaques quantization. Plaques were stained with 3D6 (1:50), a monoclonal antibody against Aβ, and followed by a biotin anti-mouse secondary antibody (1:500). After applying DAB substrate solution, sections were visualized under the microscope. Positive plaques were counted in 9 sections (spaced 210 μm apart) from one hemibrain (Huang et al. 1999).
The total surface area of amyloid deposit was also measured and expressed as a percentage of the total surface of the hippocampus. Thioflavin S staining was used to measure the senile plaques. The frozen brain sections were thawed at room temperature, fixed with 75% ethanol for 1 min, stained with 1% thioflavin-S for 8 min, washed 2 times in 80% and 95% ethanol for 3 min, and then washed in distilled water for 3 times. Slides were observed using a fluorescence microscope.
2.5. Determination of brain soluble and insoluble Aβ levels and brain APP protein levels by Western blot
Mouse brain was homogenized in 1:20 (w/v) ice-cold carbonate buffer (50 mM NaCl and 100 mM Na2CO3, pH 11.5) with protease inhibitor cocktail. The extract was centrifuge at 14000 rpm for 15 min. Supernatant was neutralized with Tris-HCl (pH 6.8) and used to measure soluble Aβ. The pellet of the sample was treated with RIPA buffer to determine insoluble Aβ. Mouse brain was also directly homogenized in RIPA buffer with protease inhibitor cocktail for APP quantification. The protein concentration was determined by using the Bradford method. The protein extract (40 μg of protein) was loaded onto a 4-12% Bis-Tris gel, electrophoresed, and then transferred to a nitrocellulose membrane. The blots were probed with antibody 3D6 directly against Aβ (1:1000) and 8E4 directly against APP (1:2000), followed by a secondary antibody conjugated with horse-radish peroxidase (1:5000) and visualized by utilizing enhanced chemiluminescence. β-actin was also assayed as loading controls by using its antibody (1:1000). Band intensities were quantified and results were reported as a ratio of APP to β-actin (Tan et al., 2005).
2.6. Morris water maze test
Spatial learning ability was tested in the Morris water maze (Morris, 1984). The water maze was a 1.2-m diameter light yellow cylindrical pool filled with water at 26°C that contained a 20-cm diameter Plexiglas escape platform hidden 0.5 cm below the surface of the water. Pool water was made opaque by adding 150 ml of nontoxic white tempera paint. This was located in an experimental room rich in environmental cues, such as a window, a door, experimental equipment, and tables. After 4 wk-Pb exposure, visible (platform with a flag) and invisible (platform) trials were performed on Tg-SWDI and WT mice. The platform was placed in the second quadrant and mice were gently placed in the pool at the fourth quadrant facing the pool wall at the first trial. After first trial, mice were randomly placed into any of the three quadrants other than the platform quadrant. The time that it took for mouse to find the platform was recorded by an observer. Mice were given 18 training (6 time per day/3 days) trials during which their latency to find the platform was measured up to a maximum of 60 sec. If mice could not locate the platform in 60 seconds, it was manually placed on the platform (in which case 60 seconds was recorded as the time). After each time training, mice were allowed to rest on the platform for 30 seconds before the trial was repeated. The invisible platform test was performed immediately after the visible platform experiment. The flag was removed and the trials were repeated as described. After 3 days training, tests were performed on each of day 4, day 5 and day 6 (Wei et al., 2009).
2.7. β- and γ-Secretase activities assay
β- and γ-Secretase activities in brain were measured in 96-well transparent flat-bottom plates using commercial kits following the manufacturer’s instructions (β-Secretase Assay Protocol, Biovision). The signal was visually read in CytoFluor 2350 at excitation and emission wavelengths of 360 and 490 nm, respectively, followed by quantification and normalization. Each enzyme activity assay was repeated triple times (Yan et al., 2007).
2.8. Fluorometric experiments
Fluorometric experiment was performed as described previously (Naiki et al., 1989; Du et al., 2003). Synthetic Aβ1-40 was incubated with or without Pb in PBS buffer at 37°C overnight. The samples were added to 50 mM glycine pH 9.2, 2 mM thioflavin T (ThT) (Sigma) at a total volume of 2 ml. Fluorescence was measured spectrophotometrically in CytoFluor 2350 at excitation and emission wavelengths of 435 and 485 nm, respectively. Samples were run in triplicate.
2.9. Atomic absorption spectrophotometry (AAS) analysis
Pb concentrations in brain tissues were quantified by a Varian SpectroAA-20 Plus GTA-96 flameless graphite furnace AAS. The brain tissues were digested with 0.20 ml of concentrated HNO3 (Mallinckrodt, AR Select grade) in MARSXpress microwave-accelerated reaction system for 4 h. The standard curves were freshly prepared daily in a ranges of 0-5 μg/L. Samples were diluted directly with distilled, deionized water so as to keep the absorbance reading within the linear rages of the measurement. The detection limit was 1.35 ng Pb/ml of assay solution. The Intra-day and inter-day precisions of the method were 1.6 % and 2.8 %, respectively.
2.10. Preparation of brain sections for XRF
The mouse brain specimens were prepared as described previously (Leskovjan et al., 2009). Briefly, frozen samples were cut in half along the sagittal plane and one half mounted to a cryo-cassette for cutting. Sections were chosen such that the subiculum, known to contain amyloid plaques, was the largest (around Bregma -3.24 mm). One horizontal section of 30 μm was cut, thawed on 4 μm polypropylene film stretched on frames, frozen immediately, and stored at -80 °C until analysis. Three 30-μm sections were subsequently cut and mounted to glass slides for DAB staining to identify amyloid plaques. This process was repeated four times per brain (4 XRF samples and 12 samples for staining). No chemical fixation was used to avoid modifications of metal distribution and oxidation state. Care was also taken to use plastic whenever possible to avoid metal contamination.
2.11. Synchrotron based XRF
A description of the Biophysics Collaborative Access Team (BioCat) beamline 18 of the Advanced Photon Source can be found in previous publications (Barrea et al. 2010; Fischetti et al. 2004). In brief, X-rays produced by a 3.3-cm period undulator are monochromatized before being focused by KB mirrors onto the sample. Two 8-μm thick Al foils were placed between the sample and detector in order to attenuate the strong contributions to the spectrum from K and Ca (Kα attenuation lengths of 6.24 μm & 8.42 μm respectively). The detector position relative to the sample was adjusted to optimize the signal to noise ratio of the Pb Lα peak (unfitted). Imaging scans were performed at 13.3 keV with a flux of 1.4 × 1011 photons/s. A dwell time of 1.5 s/pixel was chosen as to have appropriate statistics for fitting the recorded pixel spectrum. Images were taken with 20 μm × 20 μm pixel size.
2.12. XRF data processing
The MAPS program (Vogt, 2003) was used to fit the spectrum of each pixel individually. Conversion of elemental fluorescence intensities to areal densities in μg/cm2 was performed by comparing the XRF intensities with those from thin film standards NBS-1832 and NBS-1833 (NIST, Gaithersburg, MD). Amyloid plaque areas were visually identified by comparing XRF images with DAB stained adjunct sections. Plaque areas were delineated and applied to XRF elemental maps to obtain the average pixel spectrum and the average metal concentrations.
2.13. Statistical analysis
Statistical analyses of the differences between groups were carried out by a one-way ANOVA with post hoc comparisons by the Dunnett’s test. All data are expressed as mean±SD. Differences between two means were considered significant when p was equal or less than 0.05.
3. Results
3.1. Increased brain levels of Aβ and amyloid plaques deposition in Pb-exposed mice
Acute Pb exposure has been shown to increase brain levels of Aβ in APP transgenic mice (Gu et al., 2011). To verify Pb effect on brain Aβ in a Pb exposure scenario, we treated 8-wk-old Tg-SWDI transgenic mice (n=7 per group) for 6 wk by oral gavage of 50 mg/kg of Pb acetate. Amounts of Pb in brain tissues and blood were analyzed by AAS. The Pb concentrations in control mice (n =4) were as follows, blood 1.83 ± 0.29 (SD) μg/dL, cortex 0.05 ± 0.01 μg/g, hippocampus 0.05 ± 0.03 μg/g, and cerebellum 0.09 ± 0.02 μg/g (n=4). In Pb-treated mice (n=4), the Pb level in blood was 29.5 ± 4.89 μg/dL, cortex 0.37 ± 0.01 μg/g, hippocampus 0.29 ± 0.03 μg/g, and cerebellum 0.39 ± 0.02 μg/g. These data indicated that brain Pb concentration and blood Pb concentration were about 6 and 16 times, respectively, higher in Pb-treated animals than in the controls (p<0.01).
ELISA analyses showed that the CSF concentrations of Aβ1-40 and Aβ1-42 were increased by 103% (p<0.05) and 108% (p<0.01), respectively, in Pb-treated mice as compared to controls (Fig. 1A). No statistical significant differences in plasma concentrations of Aβ were observed (Fig.1A). Pb exposure also significantly increased hippocampal levels of Aβ1-40 by 35.4% (p<0.05) and Aβ1-42 by 47.2% (p<0.05) in comparison to controls (Fig. 1B). Moreover, the cortical levels of Aβ1-40 and Aβ1-42 were higher in Pb-treated mice than the control mice (by 148% increase for Aβ1-40 and 240% for Aβ1-42, p<0.05) (Fig.1B). We further calculated the ratio of Aβ1-42/Aβ1-40. In the control group, the ratios of Aβ1-42/Aβ1-40 in CSF, plasma, cortex and hippocampus were 0.65±0.47, 0.50±0.13, 0.09±0.02, and 0.17±0.03, respectively. In the Pb group, the ratios were 0.62±0.27, 0.48±0.20, 0.11±0.03, and 0.20±0.07, respectively. No significant differences between control and Pb-treated groups were observed. The Western blot was also used to test whether Pb exposure may increase the level of soluble and/or insoluble Aβ (Fig. 1C). Compared to control mice, the levels of both soluble and insoluble Aβ were significantly increased after Pb exposure (p<0.05) (Fig. 1D).
Fig. 1.
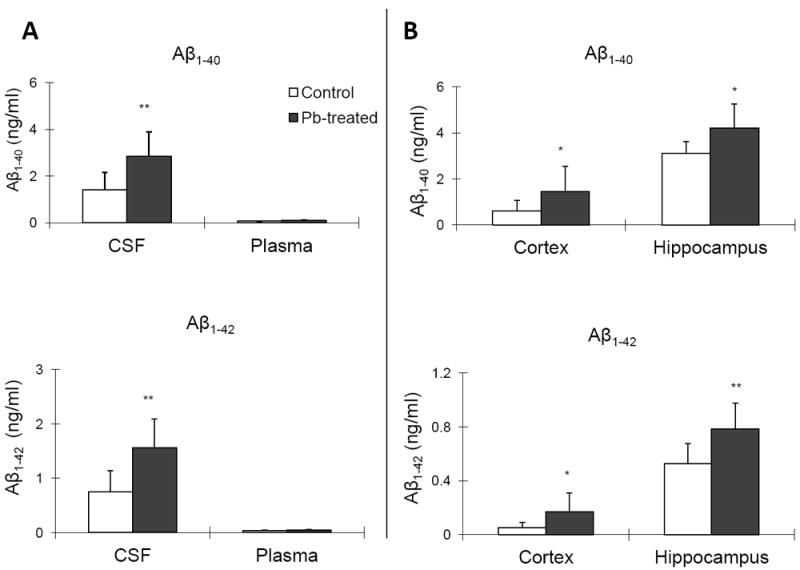
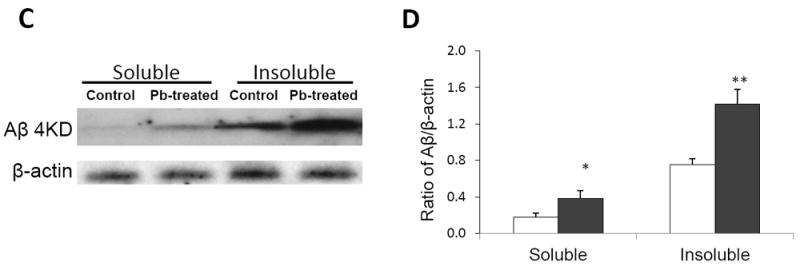
Pb exposure increased levels of Aβ1-40 and Aβ1-42 in CSF and brain tissues. Tg-SWDI transgenic mice received oral gavage of 50 mg/kg as Pb acetate once daily for 6 weeks. Concentrations of Aβ1-40 and Aβ1-42 in plasma, CSF, and different brain regions were determined by ELISA. (A). Concentrations of Aβ1-40 and Aβ1-42 in the CSF and plasma in Pb-treated APP transgenic mice as compared to controls. (B). Concentrations of Aβ1-40 and Aβ1-42 in brain cortex and hippocampus in Pb-treated APP transgenic mice as compared to controls. (C). Western blot analysis of soluble and insoluble Aβ expression in mouse brain. (D). Band intensities by Western blot were quantified by normalizing to β-actin densities. Data represent mean ± SD, n=7. *: p<0.05, **: p<0.01.
To examine the formation of amyloid plagues in mouse brains, we performed the immunohistochemistry (IHC) staining on control and Pb-treated brain sections by using anti-Aβ antibody 3D6. The data in Fig. 2A displayed the expression of amyloid plagues in control Tg-SWDI transgenic mouse brain; the plaque staining, however, became more frequent and wider spread in Pb-treated animals. By counting the number of amyloid plaques in selected regions in all animals (n=7), Pb treatment increased the amyloid plaques by 64.3% as compared to controls (p<0.05) (Fig. 2B). The amyloid plaque load in the hippocampus was also quantified. After Pb-treated, the amyloid plaque load increased from 0.84% to 1.68% (p<0.05) (Fig. 2C). Moreover, experiments with thioflavin S staining confirmed that Pb exposure increased not only the diffusive immune-reactive signals, but also the thioflavin-positive compact amyloid plaques in Tg-SWDI mice (Fig. 2D).
Fig. 2.
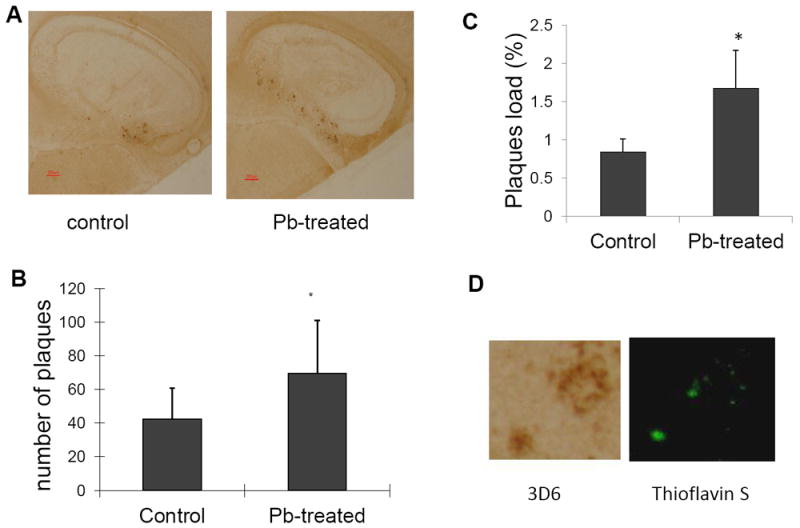
Formation of amyloid plaques in APP mouse brains following in vivo Pb exposure. The Pb exposure dose regimen was the same as that described in the legend to Fig. 1. At the end of Pb exposure, mouse brain sections were stained with 3D6 antibody to detect amyloid plaques. (A). Representative brain sections from Pb-treated and control mouse brain. Scale bar denotes 200 μm. (B). Numbers of amyloid plaques counted in the designated area. (C). Quantification of amyloid load in hippocampus. Surface 3D6 staining was expressed as a percentage of the total hippocampus surface. Data represent mean ± SD, n=7. *: p<0.05. (D). 3D6 and thioflavine S positive plaques.
To further examine whether Pb exposure shortened the onset of plague formation, we treated 4-wk-old APP transgenic mice for 4 wk with the same Pb dose regimen or equivalent molar concentration of Na acetate as control (n=5 per group). Two mice in the Pb group and 3 mice in the control group developed 2-3 amyloid plaques. The onset of the plague formation, however, was not evidently shortened in Pb-treated mice (data not shown).
3.2. Impaired spatial learning in chronic Pb-exposed mice
Pb exposure-induced Aβ and amyloid plaque deposition may cause the neurobehavioral chances in Tg-SWDI mice. To test this hypothesis, we used the Morris water maze test to evaluate the spatial learning ability in these mice at 12 wk of age. After the pre-training for 3 days, the control mice required 3.7±1.0 (SD) sec to find the visible platform during the initial trial (day 4) and were improved to 3.2±0.5 sec by the third trial (day 6). In contrast, the Pb-treated mice initially required 5.0±1.8 sec to find the platform and then 4.3±1.6 sec by the third attempt (Fig. 3A). In the submerged (invisible) platform test, the control mice found the platform in 5.1±1.8 sec upon the first trial and needed 4.7±2.5 sec for the same task by the third test, whereas the Pb-treated mice required 8.9±4.3sec for initial trial and 10.0±5.5 sec by the third attempt (Fig. 3B). Clearly, in both testings, the control mice performed significantly better than did the Pb-treated mice (p < 0.05), suggesting an impaired spatial learning in Tg-SWDI mice treated with Pb.
Fig. 3.
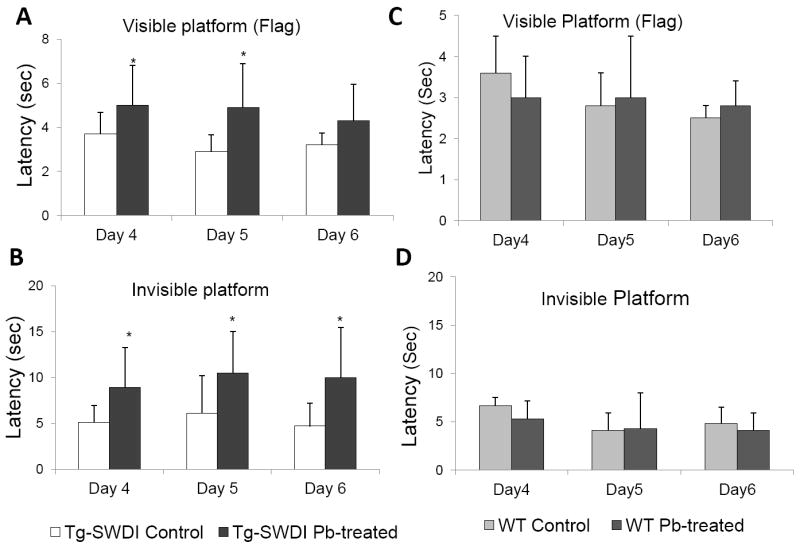
Pb exposure impaired mouse spatial learning ability in vivo. After 4-wk Pb exposure (at 12 wk of age), control and Pb-treated Tg-SWDI and WT mice were evaluated for cognitive and motor skill performance using the Morris water maze test. Time interval between the start of the test and the learning to reach the platform (latency) for a visible platform test in Tg-SWDT mice (A) and WT mice (C). The latency for a hidden platform test in Tg-SWDI mice (B) and WT mice (D). Data represent mean ± SD, n=7. *: p<0.05.
Groups of WT mice (n=7 per group) also were treated with Pb or Na acetate in the same dose regimen by oral gavage, followed by the Morris water maze test. There were no significant differences between the WT Pb-treated mice and control mice (Fig. 3C&D). The data support the view that Pb-induced neurobehavioral changes in Tg-SWDI mice may be due largely to amyloid plaque deposition in brain.
3.3. APP level and activities of β– and γ– secretases in Pb-treated mice were not significantly changed following chronic Pb exposure
Increased accumulation of Aβ in Tg-SWDI transgenic mice brain could result from the overproduction of APP and/or Aβ in brain. Thus, we set out to determine brain levels of APP, a precursor of Aβ, and the activity of β– and γ– secretases, which are responsible for generation of Aβ from APP. After 6 wks of Pb treatment, there was no significant difference in APP levels between control and Pb-treated mice (Fig.4A), nor was the difference in activities of β– and γ– secretases between control and Pb-treated mice observed (Fig.4B.C). These results suggested that Pb exposure seemed unlikely to affect the Aβ production in these transgenic mouse brains.
Fig. 4.
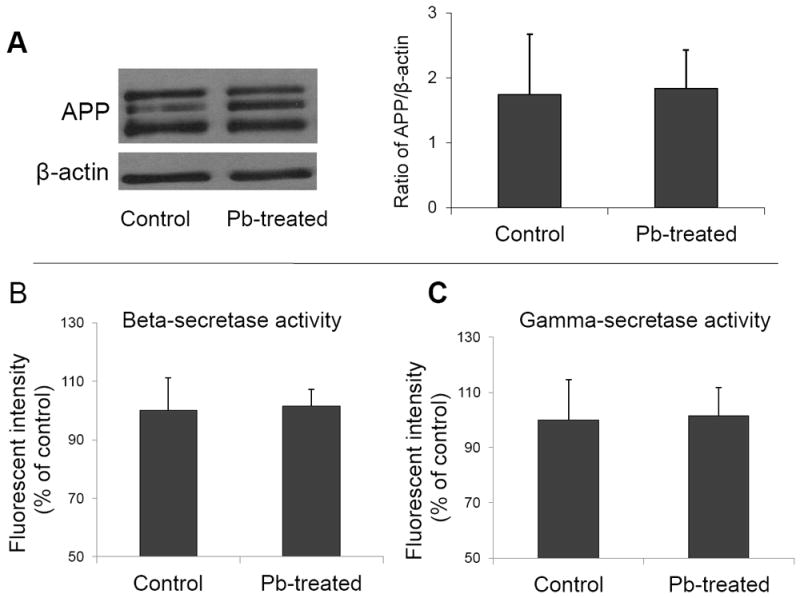
Effect of in vivo Pb exposure on brain APP levels and β– and γ–secretases activities. The Pb exposure dose regimen was the same as that described in the legend to Fig. 1. Assays were performed at the end of exposure. (A). APP protein expression in mouse brain by Western blot. The band intensities were quantified and normalized to β-actin densities. (B). Activities of β–secretase in mouse brain. (C). Activities of γ–secretase in mouse brain. Activities of both β– and γ– secretases in mouse brain were determined by a fluorogenic substrate assay in brains freshly isolated from animals. Data represent mean ± SD, n=3-4 per group.
3.4. Aβ fibril formation were accelerated following Pb exposure in vitro
From the mechanistic point of view, the fibril form of Aβ constitutes the amyloid plaques. Thus, it was reasonable to postulate that the increased amyloid plaque formation in Pb-treated brain may result from an increased rate of fibril formation. To test this hypothesis, we incubated the soluble fresh synthetic Aβ1-40 (50 μM) with Pb in vitro and used the ThT reagent, which binds specifically to the fibrillary structure and then emits the fluorescent signal, to detect Aβ fibril formation. The data presented in Fig. 5 showed a Pb concentration-dependent increase in fibril formation by more than 30% in comparison to controls (p<0.05).
Fig. 5.
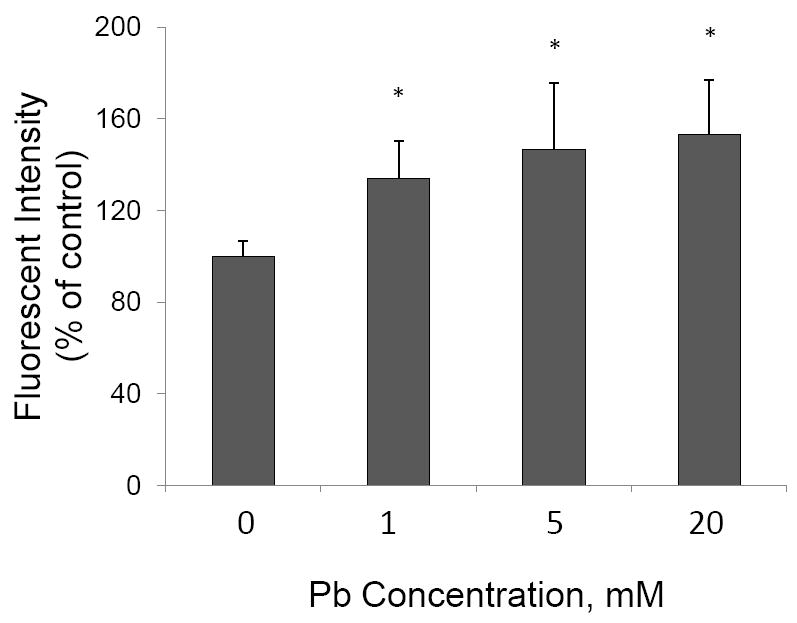
Increased Aβ fibril formation following Pb treatment in vitro. Soluble synthetic Aβ1-40 molecules were incubated with Pb at the concentrations specified. The formation of Aβ fibril was determined by using ThT reagent. Data represent mean ± SD, n=3 per group repeated three times with similar results. *: p<0.05.
3.5. Co-localization of Pb with amyloid plaques in Pb-treated mouse brain
To obtain an image of Pb distribution, a thin tissue section was raster scanned across the focused X-Ray beam. An energy resolving detector recorded the X-Ray fluorescence spectrum on a pixel-by-pixel basis. The collected spectrum was then subsequently fitted to quantify the metal concentration. We chose an X-Ray energy of 13.3keV to excite the Pb Lα transition for control and Pb-treated samples (Fig. 6A). The ratio of Pb concentrations in non-plaque region in Pb-treated mice to those in control mice was 1.19±0.01. In the plaque region, however, this ratio was increased to 1.78± 0.28 (p<0.05, n=3) (Fig.6B). More interestingly, our data showed that high Pb signals were co-localized with the amyloid plaques (Fig. 6C), further supporting the view that Pb accumulation in mouse brain induced the amyloid plaque formation.
Fig. 6.
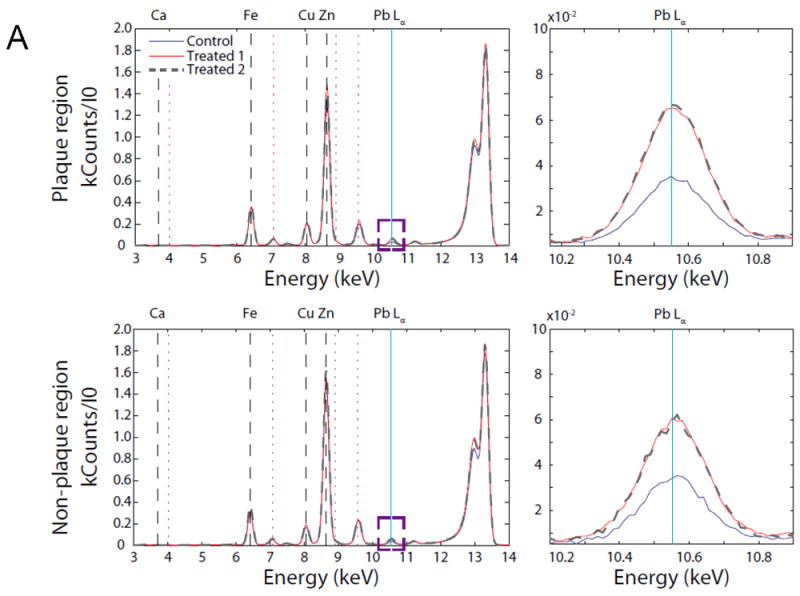
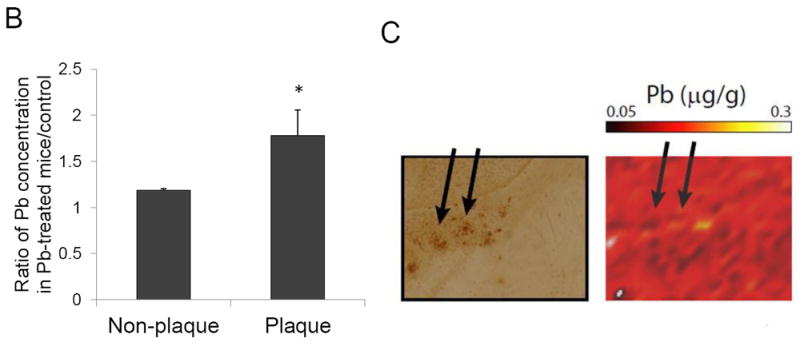
Co-localization of Pb with amyloid plaques. The Pb exposure dose regimen was the same as that described in the legend to Fig. 1. After 6-wk Pb exposure, brain slices were made for XRF measurements. (A). Average pixel spectrum for plaque and non-plaque regions. (B). the ratio of Pb concentrations in plaque and non-plaque regions in Pb-treated mice to those of control mice. Data represent mean ± SD, n=3. *: p<0.05. (C). Arrows indicate that high Pb signals were co-localized with amyloid plaques.
4. Discussion
Recent animal studies have established a relationship between Pb exposure and the increased risk of AD (Basha et al., 2005a, b; Huang et al., 2011). Our previous work on transgenic APP mice, which over-express APP, also indicates a significant increase of Aβ in the CSF and brain tissues following acute Pb exposure (Gu et al., 2011). The studies presented in this report took the advantage of the transgenic Tg-SWDI mouse line, which possesses the unique ability to form the amyloid plaques in brain tissues, allowing to study the onset as well as the severity of the plaque formation upon exogenous environmental insults. Our data clearly show that 6-wk Pb exposure in Tg-SWDI mice has led to a detectable formation of amyloid plaques in brain regions, particularly in hippocampus. The Pb-induced plaque formation is characterized by the following features: (1) There is an evident elevation of Aβ in the CSF and selected brain regions after in vivo Pb exposure; (2) the increased Aβ is unlikely due to the overproduction of Aβ from its precursor APP; (3) the amyloid plaques contain a significantly high amount of Pb; and (4) the Tg-SWDI mice exposed to Pb and with the amyloid plaques display the learning deficits.
The Pb concentration in human blood considering neurotoxic is between 10 μg/dL and more than 70 μg/dL. The CDC National Surveillance Data (1997–2007) show that some children have elevated blood lead level of ≥ 70μg/dL. In the current study, Pb exposure resulted in a blood Pb concentration of 29 μg/dL with elevated Pb concentrations in brain tissues about 0.3 μg/g. The Pb treatment dose regimen resulted in a brain Pb concentration about 6-16 times higher than the controls. Under such an exposure condition, we found a significant increase of Aβ in Tg-SWDI mouse brain.
Our findings on the increased Aβ levels and plaques formation are consistent with the previous report on monkey (Wu et al., 2008). While the clinical data are still limited, one early case report has indeed shown that a patient who was exposed to Pb at age of 2.24 and died at 42 had evident Alzheimer’s neurofibrillary tangles with the gramulo-vacuolar degeneration in hippocampus (Niklowitz and Mandybur, 1975). Amyloid plaques are composed of a tangle of regularly ordered fibrillar aggregates, namely amyloid fibers which are rather insoluble, difficult to remove from the brain, and believed to be a highly neurotoxic form of Aβ. Basha et al (2005b) report that Pb can promote the formation of Aβ aggregates. Recent studies by Zhu et al. (2011) show that Pb can also enhance Tau filament formation. Our in vitro data clearly show an accelerated Aβ fibril formation when the Aβ molecules were incubated with Pb, suggesting a direct interaction between Pb and Aβ molecules. More convincingly, our XFR data provide the first-hand evidence that Pb was co-localized within the amyloid plaques after in vivo Pb exposure. Thus, it seems likely that Pb facilitates the process of plaque formation, which is brought by the transformation of Aβ from a soluble form to neurotoxic aggregated, fibrillary structure. The formation of amyloid fibril is known to require a chemically discriminating nucleation event (Hughes et al., 1998). Whether Pb directly affects nucleation event in amyloid fibril formation is unknown and will be a highly interesting research subject for future investigation.
Pb exposure is associated with both peripheral and central neurological effects including memory deficits (Shih et al., 2007). Reports indicate that exposure to low levels of Pb during early development is sufficient to cause cognitive deficits in human and experimental animals (Moreira et al., 2001; Murphy and Regan, 1999; White et al., 1993). In a rat model, Pb-induced learning deficits may be restricted to young rats, since chronic Pb exposure in adult rat does not impair spatial learning (Gilbert et al., 2005). The data from this study showed that Pb impaired the spatial learning in adult Tg-SWDI mice but not in adult WT mice when Morris water maze test was performed. It is therefore possible that Pb-induced neurobehavioral alteration in Tg-SWDI mice was due mainly to Pb-induced amyloid plaques formation in brain.
Reports in literature show that Pb exposure can increase APP production (Wu et al., 2008; Huang et al., 2011). Our results did not reveal any significant effect of Pb on APP production, nor did we find any significant changes in the activities of two enzymes critical to APP proteolysis in Pb-treated mice. These observations are consistent with the reports by others (Basha et al., 2005a). Hence, these data indicate that Pb-induced Aβ accumulation in brain seems unlikely to be due to its effect on Aβ production. In our previous studies, Pb was found to disrupt the LRP1-mediated removal of Aβ from the CSF; a slowed clearance thus may be responsible for the Aβ accumulation in brain (Behl et al., 2009a,b; Behl et al., 2010; Gu et al., 2011). A similar mechanism may apply for the current Pb exposure model. In addition, our early data have established that Aβ in the CSF is actively removed by the choroid plexus in brain ventricles, a brain tissue where the blood-CSF barrier reside (Crossgrove et al., 2005). The process involves the efflux transport of Aβ and enzymatic degradation by insulin degrading enzyme (IDE), endothelin-converting enzyme-1 (ECE1), and neprilysin (Crossgrove et al., 2005, 2007). The interactions between Pb and these Aβ degradation enzymes are currently unknown. In addition, since human APP is controlled by thymus cell antigen 1 and theta (Thy1) promoter in the Tg-SWDI mouse line (Davis et al., 2004), it is possible that Pb may increase mouse APP expression by affecting its own promoter but not the Thy1 promoter fragment used in this transgenic mouse line. These hypotheses need further experimentation to approve.
In summary, the current data report that in vivo Pb exposure results in Aβ accumulation and amyloid plaque formation in a transgenic mouse model whose expression of amyloid plaques are genetically accelerated. The formation of amyloid plaques may underlie an impaired spatial learning ability in these mice. Chronic Pb exposure does not appear to affect the production of Aβ, but rather it may directly facilitate Aβ fibril formation. The co-localization of Pb within amyloid plaques in Pb-treated animal provides a great opportunity to uncover the mechanism of Pb-induced amyloid plaques formation.
Highlights.
Chronic in vivo Pb exposure in Tg-SWDI APP mice increases amyloid plaques in brain
Morris water maze test reveals impaired spatial learning ability in Pb-treated mice
In vitro studies discover that incubation with Pb facilitates Aβ fibril formation
Synchrotron XRF studies display a high level of Pb in brain amyloid plaques in vivo
We provide the firsthand evidence of involvement of Pb in amyloid plaque formation
Acknowledgments
The authors greatly appreciate the assistance of Taisiya Zakharova in sample preparation for XRF studies. This work was supported in part by National Institute of Health/National Institute of Environmental Health Sciences [ES017055 to Y.D. and W.Z. and ES008146 to W.Z.]. The use of the APS at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences (under Contract No. DE-AC02-06CH11357). BioCAT is an NIH-supported Research Center RR-08630. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Huiying Gu, Email: huiygu@iupui.edu.
Gregory Robison, Email: grobison@purdue.edu.
Lan Hong, Email: hongl@purdue.edu.
Raul Barrea, Email: rbarrea@iit.edu.
Xing Wei, Email: xinwei@iupui.edu.
Martin R. Farlow, Email: mfarlow@iupui.edu.
Yulia N Pushkar, Email: ypushkar@purdue.edu.
References
- Barrea RA, Gore D, Kujala N, Karanfil C, Kozyrenko S, Heurich R, Vukonich M, Huang R, Paunesku T, Woloschak G, Irving TC. Fast-scanning high-flux microprobe for biological X-ray fluorescence microscopy and microXAS. J Synchrotron Radiat. 2010;17:522–529. doi: 10.1107/S0909049510016869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha MR, Murali M, Siddiqi HK, Ghosal K, Siddiqi OK, Lashuel HA, Ge YW, Lahiri DK, Zawia NH. Lead (Pb) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005a;19:2083–2084. doi: 10.1096/fj.05-4375fje. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005b;25:823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Zhang YZ, Zheng W. Involvement of insulin degrading enzyme in the clearance of β-amyloid at the blood-CSF barrier: Consequences of lead exposure. Cerebrospinal Fluid Res. 2009a;6:11. doi: 10.1186/1743-8454-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Zhang YS, Monnot AD, Jiang W, Zheng W. Increased β-amyloid levels in the choroid plexus following lead exposure and the involvement of low density lipoprotein receptor protein-1. Toxicol Appl Pharmacol. 2009b;240:245–254. doi: 10.1016/j.taap.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl M, Zhang YS, Shi YZ, Cheng JX, Du YS, Zheng W. Lead-induced increase in β-amyloid accumulation in the choroid plexus: Role of low density lipoprotein receptor protein-1 and protein kinase C activity. Neurotoxicology. 2010;31:524–532. doi: 10.1016/j.neuro.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Li GJ, Zheng W. The choroid plexus removes beta-amyloid from the cerebrospinal fluid. Exp Biol Med. 2005;230:771–776. doi: 10.1177/153537020523001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossgrove JS, Smith EL, Zheng W. Macromolecules involved in production and metabolism of beta-amyloid at the brain barriers. Brain Res. 2007;1138:187–195. doi: 10.1016/j.brainres.2006.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Du YS, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M. Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicity. Brain. 2003;126:1935–1939. doi: 10.1093/brain/awg191. [DOI] [PubMed] [Google Scholar]
- Fischetti R, Stepanov S, Rosenbaum G, Barrea R, Black E, Gore D, Heurich R, Kondrashkina E, Kropf AJ, Wang S, Zhang K, Irving TC, Bunker GB. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. J Synchrotron Radiat. 2004;11:399–405. doi: 10.1107/S0909049504016760. [DOI] [PubMed] [Google Scholar]
- Gilbert ME, Kelly ME, Samsam TE, Goodman JH. Chronic developmental lead exposure reduces neurogenesis in adult rat hippocampus but does not impair spatial learning. Toxicol Sci. 2005;86:365–374. doi: 10.1093/toxsci/kfi156. [DOI] [PubMed] [Google Scholar]
- Gu HY, Wei X, Monnot AD, Fontanilla CV, Behl M, Farlow MR, Zheng W, Du Y. Lead exposure increases levels of beta-amyloid in the brain and CSF and inhibits LRP1 expression in APP transgenic mice. Neurosci Lett. 2011;490:16–20. doi: 10.1016/j.neulet.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Hu H, Tellez-Rojo MM, Bellinger D, Smith D, Ettinger AS, Lamadrid-Figueroa H, Schwartz J, Schnaas L, Mercado-Garcia A, Hernandez-Avila M. Fetal lead exposure at each stage of pregnancy as a predictor of infant mental development. Environ Health Perspect. 2006;114:1730–1735. doi: 10.1289/ehp.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Buttini M, Wyss-Coray T, McConlogue L, Kodama T, Pitas RE, Mucke L. Elimination of the class a scavenger receptor does not affect amyloid plaque formation or neurodegeneration in transgenic mice expressing human amyloid protein precursors. Am J Pathol. 1999;155:1741–1747. doi: 10.1016/S0002-9440(10)65489-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Bihaqi SW, Cui L, Zawia NH. In vitro Pb exposure disturbs the balance between Abeta production and elimination: the role of AbetaPP and neprilysin. Neurotoxicology. 2011;32:300–306. doi: 10.1016/j.neuro.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S. Alpha2-macroglobulin associates with beta-amyloid peptide and prevents fibril formation. Proc Natl Acad Sci U S A. 1998;95:3275–3280. doi: 10.1073/pnas.95.6.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyslop PA, Bender MH. Methods for sample preparation for direct immunoassay measurement of analytes in tissue homogenates: ELISA assay of amyloid beta-peptides. Curr Protoc Neurosci. 2002;Chapter 7(Unit 7):20. doi: 10.1002/0471142301.ns0720s18. [DOI] [PubMed] [Google Scholar]
- Jiang YM, Long LL, Zhu XY, Zheng H, Fu X, Ou SY, Wei DL, Zhou HL, Zheng W. Evidence for altered hippocampal volume and brain metabolites in workers occupationally exposed to lead: A study by magnetic resonance imaging and H-1 magnetic resonance spectroscopy. Toxicol Lett. 2008;181:118–125. doi: 10.1016/j.toxlet.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Mook-Jung I, Koh JY. Histochemically reactive zinc in plaques of the Swedish mutant beta-amyloid precursor protein transgenic mice. J Neurosci. 1999;19:RC10. doi: 10.1523/JNEUROSCI.19-11-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovjan AC, Lanzirotti A, Miller LM. Amyloid plaques in PSAPP mice bind less metal than plaques in human Alzheimer’s disease. Neuroimage. 2009;47:1215–1220. doi: 10.1016/j.neuroimage.2009.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Coppede F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667:82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J. Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with beta-amyloid deposits in Alzheimer’s disease. J Struct Biol. 2006;155:30–37. doi: 10.1016/j.jsb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Moreira EG, Vassilieff I, Vassilieff VS. Developmental lead exposure: behavioral alterations in the short and long term. Neurotoxicol Teratol. 2001;23:489–495. doi: 10.1016/s0892-0362(01)00159-3. [DOI] [PubMed] [Google Scholar]
- Murphy KJ, Regan CM. Low-level lead exposure in the early postnatal period results in persisting neuroplastic deficits associated with memory consolidation. J Neurochem. 1999;72:2099–2104. doi: 10.1046/j.1471-4159.1999.0722099.x. [DOI] [PubMed] [Google Scholar]
- Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- Niklowitz WJ, Mandybur TI. Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol. 1975;34:445–455. doi: 10.1097/00005072-197509000-00006. [DOI] [PubMed] [Google Scholar]
- Ogomori K, Kitamoto T, Tateishi J, Sato Y, Suetsugu M, Abe M. Beta-protein amyloid is widely distributed in the central nervous system of patients with Alzheimer’s disease. Am J Pathol. 1989;134:243–251. [PMC free article] [PubMed] [Google Scholar]
- Schnaas L, Rothenberg SJ, Flores MF, Martinez S, Hernandez C, Osorio E, Velasco SR, Perroni E. Reduced intellectual development in children with prenatal lead exposure. Environ Health Perspect. 2006;114:791–797. doi: 10.1289/ehp.8552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih RA, Hu H, Weisskopf MG, Schwartz BS. Cumulative lead dose and cognitive function in adults: A review of studies that measured both blood lead and bone lead. Environ Health Perspect. 2007;115:483–492. doi: 10.1289/ehp.9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology. 2006;66:1476–1484. doi: 10.1212/01.wnl.0000216138.69777.15. [DOI] [PubMed] [Google Scholar]
- Tan J, Ma Z, Han L, Du R, Zhao L, Wei X, Hou D, Johnstone BH, Farlow MR, Du YS. Caffeic acid phenethyl ester possesses potent cardioprotective effects in a rabbit model of acute myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2005;289:H2265–2271. doi: 10.1152/ajpheart.01106.2004. [DOI] [PubMed] [Google Scholar]
- Tong S, von Schirnding YE, Prapamontol T. Environmental lead exposure: a public health problem of global dimensions. Bull World Health Organ. 2000;78:1068–1077. [PMC free article] [PubMed] [Google Scholar]
- Vogt S. MAPS: A set of Python software tools for analysis and visualization of 3D X-ray fluorescence data sets. J Physique IV. 2003;104:635–638. [Google Scholar]
- Wei X, Du Z, Zhao LM, Feng DN, Wei G, He Y, Tan J, Lee WH, Hampel H, Dodel R, Johnstone BH, March KL, Farlow MR, Du YS. IFATS collection: the conditioned media of adipose stromal cells protect against hypoxiaischemia-induced brain damage in neonatal rats. Stem Cells. 2009;27:478–488. doi: 10.1634/stemcells.2008-0333. [DOI] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, Rossi-George A, Lasley SM, Qian YC, Basha MR. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- White RF, Diamond R, Proctor S, Morey C, Hu H. Residual cognitive deficits 50 years after lead poisoning during childhood. Br J Ind Med. 1993;50:613–622. doi: 10.1136/oem.50.7.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan XX, Xiong K, Luo XG, Struble RG, Clough RW. beta-Secretase expression in normal and functionally deprived rat olfactory bulbs: Inverse correlation with oxidative metabolic activity. J Comp Neurol. 2007;501:52–69. doi: 10.1002/cne.21239. [DOI] [PubMed] [Google Scholar]
- Zhu HL, Meng SR, Fan JB, Chen J, Liang Y. Fibrillization of Human Tau Is Accelerated by Exposure to Lead via Interaction with His-330 and His-362. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025020. [DOI] [PMC free article] [PubMed] [Google Scholar]