

Abstract
The present study was to investigate whether sulforaphane (SFN) can prevent diabetic nephropathy in type 1 diabetic mouse model induced by multiple low-dose streptozotocin. Diabetic and age-matched control mice were given SFN at 0.5 mg/kg body weight daily for 3 months. At the end of 3-month SFN treatment, the diabetic nephropathy, shown by renal inflammation, oxidative damage, fibrosis, and dysfunction, was significantly prevented along with an elevation of renal Nrf2 expression and transcription in diabetes/SFN group compared with diabetic group. However, this renal prevention by SFN was not seen when the 3-month SFN-treated diabetic mice were aged for additional 3 months without further SFN treatment. Nrf2-mediated renal protective effects in diabetes were evaluated in human renal tubular HK11 cells transfected with control and Nrf2 siRNA and treated with 27.5 mM mannitol or high glucose plus palmitate (300 μM). Blockade of Nrf2 expression completely abolished SFN prevention of the profibrotic effect induced by high glucose plus palmitate. These results support that renal Nrf2 expression and its transcription play important roles in SFN prevention of diabetes-induced renal damage. However, the SFN preventive effect on diabetes-induced renal pathogeneses is not sustained, suggesting the requirement of continual use of SFN for its sustained effect.
1. Introduction
Diabetic nephropathy is characterized by initial oxidative stress, inflammatory response, thickening of basement membranes, expansion of mesangial matrix and interstitial fibrosis, podocytes and renal cell death, increased albuminuria, and renal dysfunction [1–3]. Reactive oxygen or nitrogen species (ROS or RNS) production in response to hyperglycemia, advanced glycosylation end products, hyperlipidemia, inflammatory cytokines including transforming growth factor (TGF)-β1, and hypertension contributes to these renal pathogenic changes [1]. However, supplying exogenous antioxidants failed to demonstrate a significant therapeutic effect on human diabetic complications [1–4]. Therefore, a strategy to upregulate endogenous multiple antioxidants, instead of exogenously supplying a single or limited combination of antioxidants, would be more efficient approach to prevent or treat diabetic complications [4].
The transcription factor NFE2-related factor 2 (Nrf2) as one member of the cap “n” collar family is a master regulator of cellular detoxification responses and redox status [5]. Under physiological conditions kelch-like ECH-associated protein 1 (KEAP1) binds to Nrf2 and sequesters it in the cytoplasm [6]. Under basal conditions, KEAP1 mediates rapid ubiquitination and subsequent degradation of Nrf2 by the proteasome [6]. Upon exposure of cells to oxidative stress or electrophilic compounds, Nrf2 dissociates from KEAP1 and translocates into the nucleus to bind to antioxidant-responsive elements in the genes encoding antioxidant enzymes, namely, NADPH quinone oxidoreductase (NQO1), heme oxygenase-1 (HO-1), glutathione S-transferase, superoxide dismutase (SOD), catalase (CAT), and γ-glutamylcysteine synthetase. Upregulation of these Nrf2-dependent antioxidants promotes detoxification, and anti-inflammatory function [6, 7]. A growing body of evidence has indicated a critical role for activator-induced Nrf2 upregulation in the prevention of diabetic complications, including nephropathy [7, 8].
Sulforaphane (SFN) is an organosulfur compound that exhibits anticancer and antidiabetic properties in experimental models and obtained from cruciferous vegetables such as broccoli, brussels sprouts, or cabbages [9]. Studies showed that SFN metabolites were detected in all tissues at 2 and 6 h after gavage, with the highest concentrations in the small intestine, prostate, lung, and kidney, suggesting that SFN is bioavailable and will be an effective dietary chemoprevention agent for these tissues [10]. SFN has garnered particular interest as an indirect antioxidant due to its extraordinary ability to induce expression of several enzymes via the KEAP1/Nrf2 pathway [9, 10]. Several studies have shown the preventive effect of SFN via induction of Nrf2 on chemical or ischemia-induced renal damage [11, 12]. A recent study has shown that after chronic treatment with SFN for 4 months diabetic mice exhibited significant renal prevention from diabetes-induced damage most likely via induction of Nrf2-mediated antioxidant pathway [13]. However, it was unclear whether a sustained renal protection would be seen in diabetic mice beyond the time-point of SFN treatment.
The present study investigated whether treatment of diabetic mice for 3 months with SFN could prevent the development of diabetic nephropathy three months after terminating SFN treatment. These would determine whether SFN has sustained protective effects in controlling diabetes-induced renal damage. We used a type 1 diabetic mouse model induced with multiple low-dose streptozotocin (MLD-STZ) as previously demonstrated by our laboratory [14]. In addition to animal experiments, we utilized in vitro cultures of human renal tubular HK 11 cells to modulate Nrf2 expression and determine a mechanistic role of Nrf2 in diabetic nephropathy.
2. Results
2.1. Effects of SFN on the Blood Glucose and Renal Function in Diabetic Mice
After the onset of hyperglycemia, diabetic and age-matched control mice were subcutaneously given SFN at 0.5 mg/kg daily for 3-months. At the end of 3 month treatment of SFN one set of mice were sacrificed to perform the experimental measurements (3-month time-point). The second set of both diabetic and control mice were aged for additional 3 months without further SFN treatment (6 month time-point). We demonstrate that after MLD-STZ induction of diabetes, blood glucose levels in diabetes (DM) groups with or without SFN treatment were significantly increased without difference between DM and DM plus SFN (DM/SFN) group (Table 1).
Table 1.
Effects of SFN on blood glucose and renal function in diabetic mice.
| 3 months | 6 months | |||||
|---|---|---|---|---|---|---|
| BG (mmol/L) | KW/TL (mg/mm) | ACR (μg/mg) | BG (mmol/L) | KW/TL (mg/mm) | ACR (μg/mg) | |
| Control | 120.3 ± 13.6 | 24.9 ± 2.0 | 101.5 ± 8.0 | 121.0 ± 9.9 | 27.9 ± 2.3 | 127.0 ± 13.4 |
| SFN | 115.7 ± 19.9 | 22.1 ± 2.5 | 111.3 ± 13.9 | 116.3 ± 10.5 | 27.4 ± 2.5 | 136.1 ± 17.6 |
| DM | 413.3 ± 23.6* | 42.9 ± 2.2* | 542.0 ± 96.9* | 282.7 ± 10.5* | 47.5 ± 3.2* | 1009.6 ± 225.8* |
| DM/SFN | 419.7 ± 29.4* | 35.5 ± 3.0∗# | 375.9 ± 62.7∗# | 347.5 ± 20.2* | 46.6 ± 3.1* | 1071.2 ± 301.8* |
Notes: Data were presented as means ± SD. *P < 0.05 versus control; # P < 0.05 versus DM group. SFN: sulforaphane; DM: diabetes mellitus; BG: blood glucose; KW/TL: kidney weight to tibia length ratio; ACR: albumin-to-creatinine ratio.
It was also shown that diabetes significantly increased albumin-to-creatinine ratio (ACR) at 3 months of diabetes and further increased it at 6-months of diabetes. Treatment with SFN for 3-month partially attenuated diabetes-increased ACR at 3-month time-point, but this partial reduction of ACR seen at 3 months time-point was diminished when it was examined at 6 month time-point. Similar to ACR, the ratio of kidney weight (KW) to tibia length (TL) was significantly increased in diabetes group compared to control animals, and SFN treatment of diabetic mice decreased this ratio at 3-month time-point, but not at 6-month time-point (Table 1).
2.2. SFN Prevented Diabetes-Induced Renal Fibrosis, Inflammation, and Oxidative Stress
Histological examination with hematoxylin and eosin (H&E) staining showed the significantly progressive changes of the renal structure, including glomerular basement membrane thickening, mesangial cell proliferation, increased mesangial matrix, and Kimmelstiel Wilson (K-W) nodules along with renal tubular epithelium damage and a large number of protein casts in DM group (Figure 1(a)). SFN treatment significantly prevented these changes in the DM/SFN group, examined at 3-month time-point, but not at 6-month time-point (Figure 1(a)).
Figure 1.
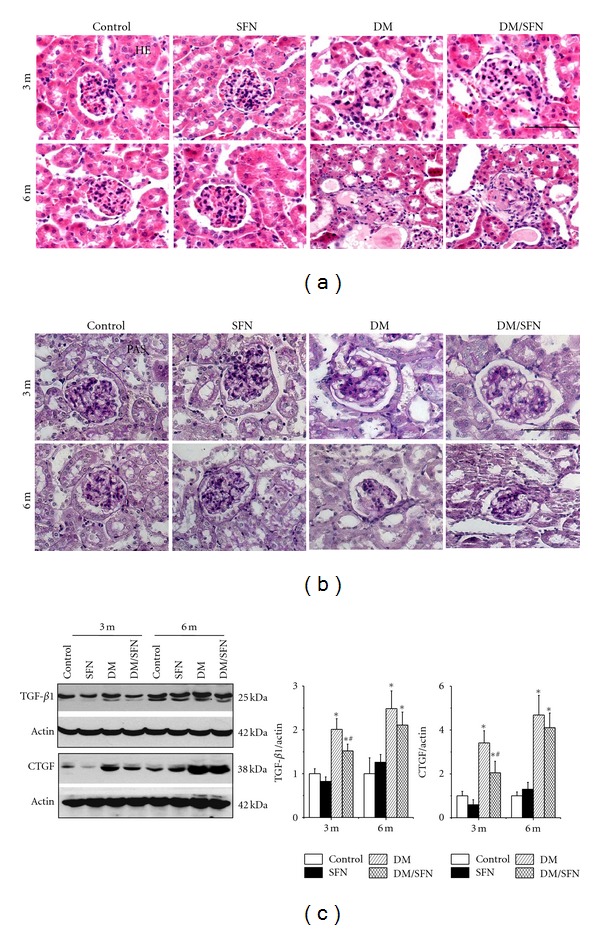
SFN prevented diabetes-induced renal structural changes and fibrosis. Diabetic and age-matched mice were treated with SFN at 0.5 mg/kg daily for five days in each week for 3 months, and then some were used to perform the experimental measurements at the end of 3-month SFN treatment (3-month time-point). Some of these diabetic and age-matched control mice with and without 3-month SFN treatment were kept for additional 3 months without further SFN treatment and then were sacrificed for study (6-month time-point). To examine renal morphology sections were stained with hematoxylin and eosin (a). Periodic acid-Schiff staining was used to examine glycogen accumulation (b). Bar = 100 μM. Western blotting assay was performed for measuring fibrosis by TGF-β1 and CTGF protein expression (c). Data were presented as means ± SD (n = 6 at least) *P < 0.05 versus control; (# P < 0.05 versus DM group).
We examined the glycogen accumulation in the kidney by periodic acid-Schiff (PAS) staining, which showed that diabetes induced a significant glycogen accumulation in a time-dependent manner from 3 to 6 months. This effect was significantly prevented by SFN treatment, examined at 3 month time-point, but not at 6 months (Figure 1(b)). Diabetes-induced renal fibrosis was further confirmed by the increased renal protein expression of transforming growth factor (TGF)-β1 and connective tissue growth factor (CTGF) as two critical profibrotic mediators (Figure 1(c)). SFN prevention of diabetes-induced TGF-β1 and CTGF expression was only observed at the 3-month time-point (Figure 1(c)).
Since inflammation and oxidative stress have been suggested to play an important role in diabetes-induced renal pathogenesis [15–18], we performed Western blot assay for the renal expression of inflammatory cytokines, plasminogen activator inhibitor (PAI)-1 (Figure 2(a)), and tumor necrosis factor (TNF)-α (Figure 2(b)), which were progressively increased in DM group and partially prevented by SFN treatment only at 3-month time-point.
Figure 2.
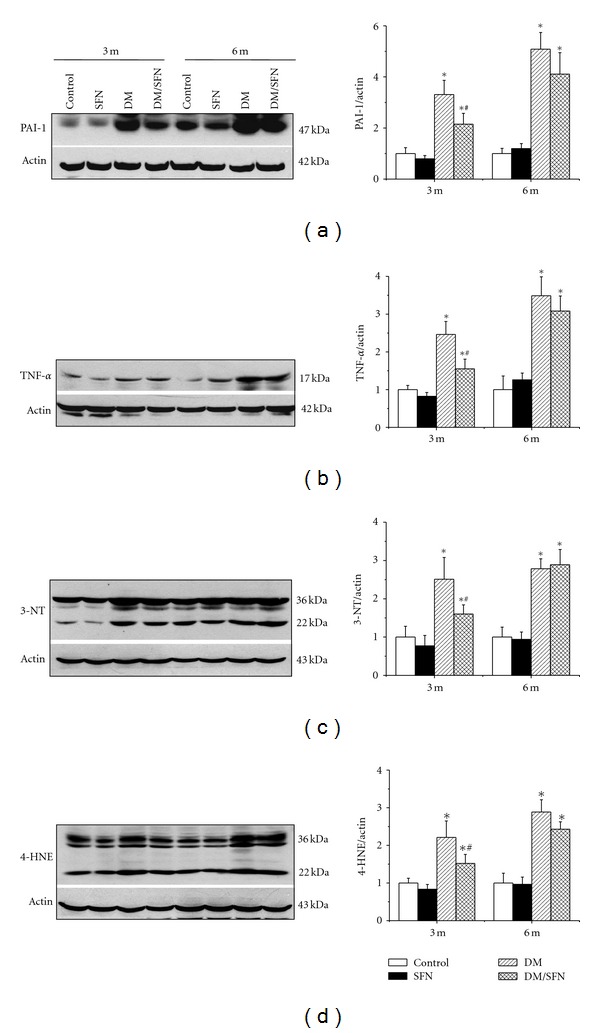
SFN prevented diabetes-induced renal inflammation and oxidative stress. Western blotting assay was performed for measuring the expression of inflammatory cytokines PAI-1 (a) and TNF-α (b), and oxidative damage accumulation of 3-NT (c) and 4-HNE (d). Data were presented as means ± SD (n = 6 at least). (*P < 0.05 versus control; # P < 0.05 versus DM group).
The next study using Western blotting assay showed the significant increase in renal oxidative damage, by detection of 3-nitrotyrosine (3-NT) accumulation as an index of nitrosative damage (Figure 2(c)) and 4-hydroxy-2-nonenal (4-HNE) accumulation as an index of lipid peroxidation (Figure 2(d)). The oxidative and nitrative damage-induced diabetes was prevented almost completely by SFN treatment only at 3-month time-point.
2.3. SFN Upregulated the Expression of Nrf2 and Its Downstream Genes In Vivo
Above results showed that SFN can protect diabetic induction of renal dysfunction, fibrosis, inflammation, and oxidative damage. One of SFN effects is induction of Nrf2; therefore, whether SFN protects the kidney from diabetes by activating Nrf2 was examined first by measuring renal Nrf2 expression and its transcription function. Nrf2 expression at mRNA (Figure 3(a)) and protein (Figure 4(a)) levels, measured by real-time PCR and Western blot, respectively, was significantly increased in the kidney of SFN-treated control mice and diabetic mice at 3-month time-point, but not at 6 months. There was a significant additive increase in the renal Nrf2 expression in DM/SFN group, compared to DM group at the end of 3-month SFN treatment.
Figure 3.
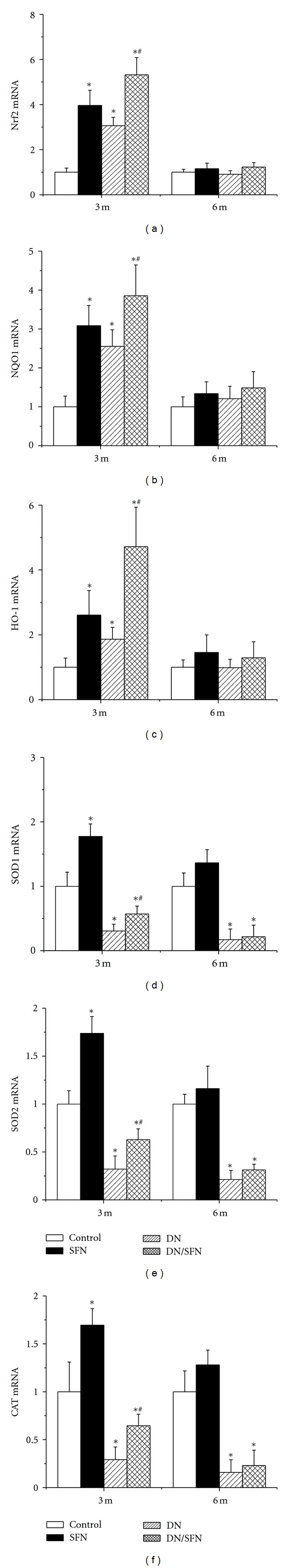
SFN upregulated renal expression of Nrf2 and its downstream genes at mRNA level. Nrf2 (a) and its downstream genes, NQO1 (b), HO-1 (c), SOD1 (d), SOD2 (e), and CAT (f) expression at mRNA level were detected by RT-PCR. Data were presented as means ± SD (n = 6 at least). (*P < 0.05 versus control; # P < 0.05 versus DM group).
Figure 4.
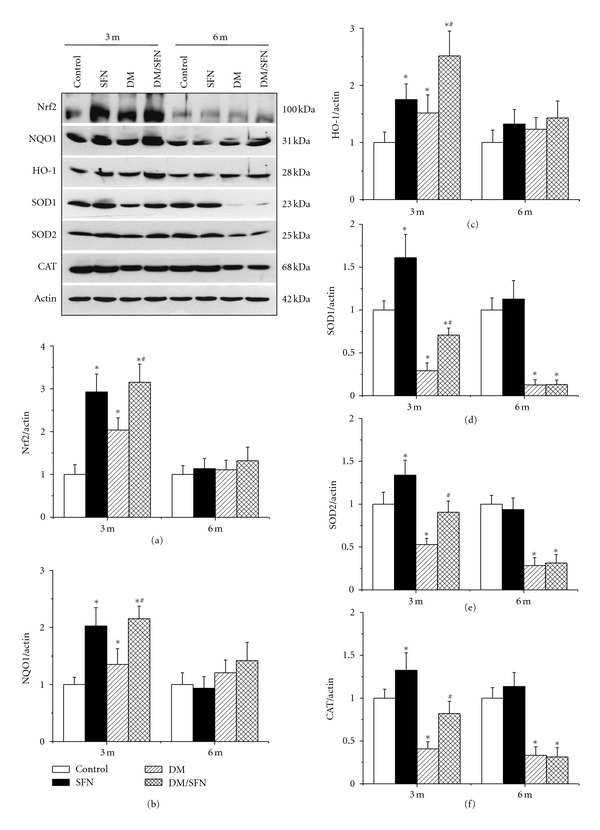
SFN upregulated renal expression of Nrf2 and its downstream genes at protein level. Nrf2 (a) and its downstream genes, NQO1 (b), HO-1 (c), SOD1 (d), SOD2 (e), and CAT (f) expression in protein level was detected by Western blotting assay. Data were presented as means ± SD (n = 6 at least). (*P < 0.05 versus control; # P < 0.05 versus DM group).
Next, we explored Nrf2 transcriptional function by examining the expression of its downstream target antioxidant genes at mRNA and protein levels. (1) At 3-month time-point, diabetes significantly increased expression of renal NQO1 and HO-1, but significantly decreased the expression of renal cytosolic SOD (SOD1), mitochondrial SOD (SOD2), and CAT at both mRNA (Figure 3) and protein levels (Figure 4). Renal expression of NQO1, HO-1, SOD1, SOD2, and CAT at mRNA and protein levels in the group of DM/SFN was significantly higher than that in DM group (Figures 3 and 4) (2) At 6-month time-point, there was no significant difference for the renal expression of NQO1 and HO-1 among groups, but there were significant decreases in renal expression of SOD1, SOD2, and CAT at both mRNA and protein levels in DM and DM/SFN groups compared to control and SFN groups (Figures 3 and 4). No significant differences were observed between DM and DM/SFN samples.
2.4. SFN Protected High-Glucose (HG)-Induced Inflammatory and Fibrotic Responses via Upregulating Nrf2 Expression and Function In Vitro
The in vivo experiments discussed above suggest a role for Nrf2 in SFN-mediated inhibition of renal damage seen in 3-month diabetic mice. In the following studies, the direct role of Nrf2 in the reduction of diabetic renal damage was further defined using human kidney proximal tubular (HK 11) cells. First we demonstrated that SFN induced Nrf2 and HO-1 protein expression in a dose-dependent manner in HK 11 cells (Figure 5(a)). HK11 cells were treated with and without HG (27.5 mM) for 48 h with palmitate (Pal, 300 μM) for the last 6 h with the presence or absence of SFN (3 μM for 12–48 h). Western blotting assay showed that HG/Pal treatment significantly increased CTGF expression, which was time-dependently prevented by pretreatment with SFN (Figure 5(b)).
Figure 5.
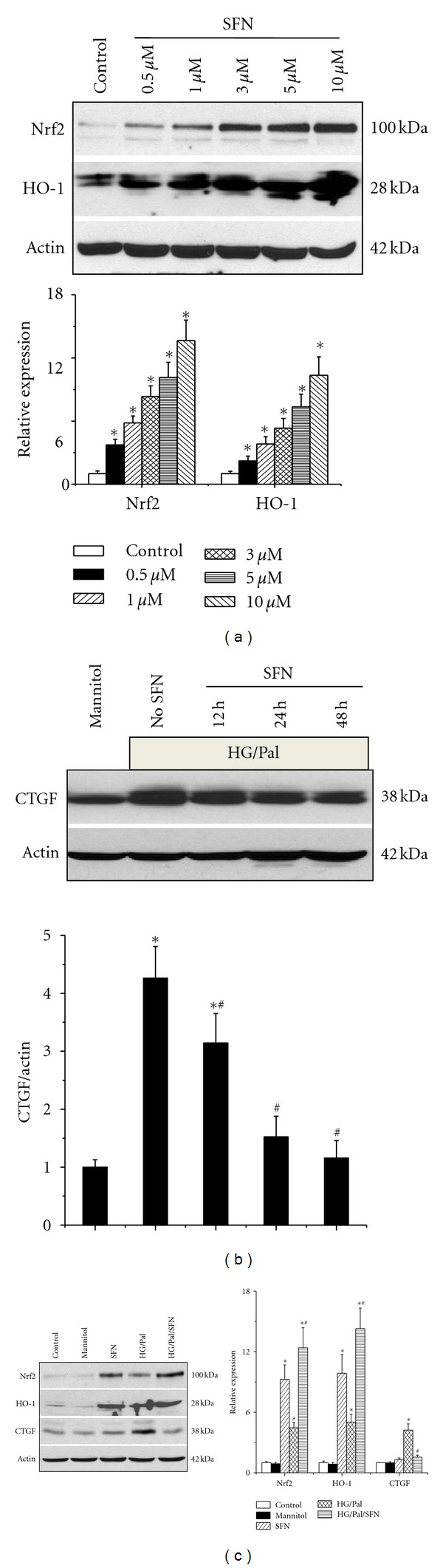
SFN protected hyperglycemia/hyperlipidemia-induced fibrotic response along with activation of Nrf2 in vitro. HK11 cells were treated with different doses of SFN (0.5 μM–10 μM) for 24 h, and then expression of Nrf2 and HO-1 at protein levels was measured by Western blotting assay (a). HK11 cells were treated with high glucose (HG, 27.5 mM) for 48 h, and, at the last 6 h, palmitate (Pal, 300 μM) was added into the culture medium. Cells were treated with SFN (3 μM) for different times (from 12 h to 48 h), and then expression of CTGF was analyzed by Western blotting assay (b). HK11 cells were treated with HG and SFN for 48 h, and, at the last 6 h, Pal was added into the culture medium as described in panel B. The expression of CTGF, Nrf2, and HO-1 at protein levels was analyzed by Western blotting assay (c). Data are presented as mean ± SD from at least three separate experiments. (*P < 0.05 versus control group; # P < 0.05 versus HG/Pal group).
Subsequently HK 11 cells were treated with HG in the presence of SFN (3 μM) for 48 h, and, at the last 6 h, Pal (300 μM) was added into the culture medium. Expression of CTGF, Nrf2, and HO-1 was analyzed by Western blotting assay (Figure 5(c)). Both SFN and HG/Pal can significantly increase Nrf2 and HO-1 expression, but only HG/Pal increased CTGF expression. Figure 5 also showed that SFN/HG/Pal can synergistically increase Nrf2 and HO-1expression, along with complete inhibition of CTGF expression induced by HG/Pal.
2.5. Silencing Nrf2 by Small Interfering RNA (siRNA) Abolished the Prevention of HG/Pal-Induced Profibrotic Effect by SFN
HO-1 and Nrf2 expression was upregulated in SFN, HG/Pal and SFN/HG/Pal groups (Figure 5(c)), suggesting upregulation of Nrf2 transcriptional function. Thus, we determined a direct role of Nrf2 in SFN-mediated prevention of HG/Pal-induced profibrotic effect by silencing Nrf2 expression. HK 11 cells were transfected with human specific Nrf2 siRNA (NFE2L2HSS181505) or control siRNA for 48 h, and then incubated with new medium containing either 5.5 mM or 27.5 mM glucose for additional 48 h. At the last 9 h and 6 h during the second 48 h incubation, SFN (3 μM) and Pal (300 μM) were added, respectively (Figure 6). Western blotting analysis showed that with the control siRNA, SFN treatment upregulated Nrf2 and HO-1 expression with a significant prevention of HG/Pal-induced CTGF; however, with the Nrf2 specific siRNA, SFN treatment could not upregulate the expression of either Nrf2 or HO-1 and also did not prevent HG/Pal-induced CTGF expression (Figure 6). This result demonstrates the requirement of Nrf2 expression and function for SFN prevention of HG/Pal-induced renal fibrotic response.
Figure 6.
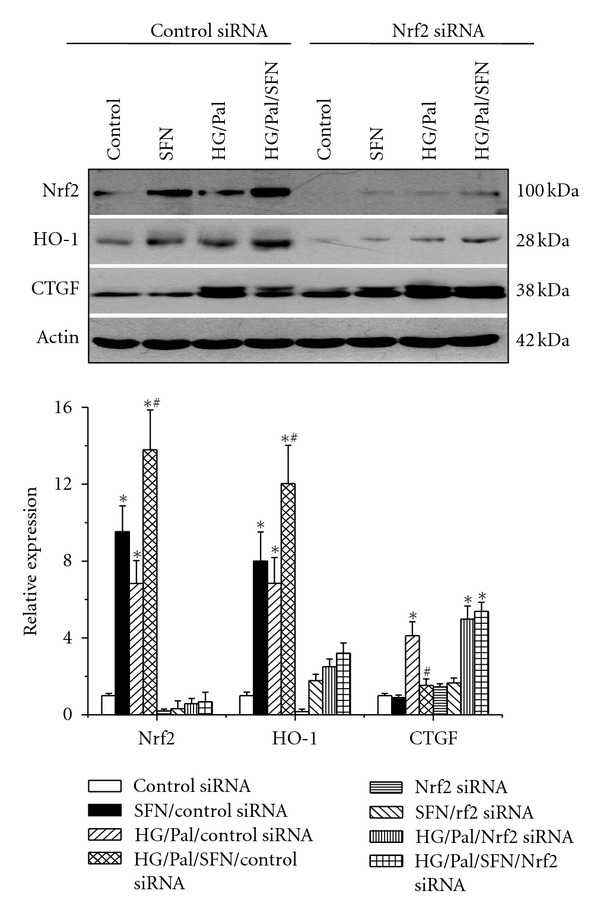
Silencing Nrf2 gene abolished SFN's prevention of HG/Pal-induced fibrosis. HK11 cells were transfected with Nrf2 siRNA and control siRNA for 48 h and then exposed to SFN (3 μM) and HG (27.5 mM) for additional 48 h. During the second 48 h, Pal (300 μM) were added at the last 6 h. Then the effect of silencing Nrf2 gene with its specific siRNA on SFN-mediated prevention of HG/Pal-induced fibrotic effect along with Nrf2 and HO-1 expression were examined by Western blotting assay. Data are presented as mean ± SD from at least three separate experiments. (*P < 0.05 versus control group; # P < 0.05 versus HG/Pal group).
3. Discussion
Using MLD-STZ-induced type 1 diabetic mouse model, we demonstrated the importance of SFN-induced Nrf2 expression in the renal protection against diabetes-induced damage. This was reflected by the significant reduction of diabetes-increased ACR and the ratio of kidney weight to tibia length as the index of renal dysfunction, and also reduction of diabetes-induced renal oxidative damage, inflammation, fibrosis, and structural abnormalities, observed at the 3-month time-point, but not at 6-month time-point.
Emerging evidence indicates that Nrf2 was upregulated in the cells in vitro and tissues in vivo when they are challenged by oxidative stress. Several studies have indicated the induction of ROS and/or RNS by HG in the cultured renal cells [19–21]. Using human mesangial cells, Jiang et al. demonstrated HG-induced elevation of nuclear protein level of Nrf2 along with an upregulation of the mRNA level of NQO1, HO-1, and glutathione S-transferase [20]. Using human renal tubular cells, we provided evidence here that exposure to HG plus palmitate also significantly increased the expression of Nrf2 and its downstream gene HO-1 (Figures 5(a) and 5(c)).
Upregulation of Nrf2 and its downstream antioxidant genes in response to hyperglycemia were found not only in cultured cells, but also in the kidney of diabetic mice. Jiang et al. have examined activation of Nrf2 in the kidneys of STZ-induced diabetic mice using MLD-STZ-induced type 1 diabetes in C57BL/6 mice. They found that, at 4 months of diabetes, Nrf2 expression in the glomeruli of diabetic mice was increased and the nuclear accumulation of Nrf2 was also increased, along with an upregulation of NQO1 [20]. Consistent with this study, we also demonstrated the increased expression of Nrf2 and its downstream genes NQO1 and HO-1 at both mRNA and protein levels, which, however, was observed only at 3-month timepoint, but not at 6-month timepoint (Figures 3 and 4). For the discrepancy of Nrf2 expression at different timepoints of diabetes, observed here, we assumed that as an adaptive mechanism, Nrf2 is quickly upregulated in cells and tissues in response to oxidative stress at early stage, but downregulated in cells or tissues exposed to overwhelming or long-lasting oxidative stress. In fact, this assumption was in an agreement with the finding from previous studies in other disease models. For instance, acute cigarette smoke exposure led to Nrf2 activation in human macrophages, but Nrf2 expression was significantly decreased in pulmonary macrophages from smokers with chronic exposure to cigarettes [22]. Rats with chronic renal failure caused by 5/6 nephrectomy exhibited significant increases in oxidative stress and inflammation in the remaining kidney. In these rats, Nrf2 expression and function was reduced mildly at six weeks but reduced markedly at 12 weeks after nephrectomy [23].
The role of Nrf2 in the prevention of diabetic nephropathy was first explored by Yoh et al. using Nrf2-KO mice [24]. They used STZ to induce diabetes in both Nrf2-KO and the wild-type (WT, C57BL/6) mice, showing that compared to WT diabetic mice, Nrf2-KO diabetic mice exhibited an increased susceptibility to the development of diabetic nephropathy, which was confirmed by Jiang et al. [20]. In addition, upregulation of Nrf2 expression with its activators was found to also provide the preventive effect on diabetes-induced renal damage. For instance, treatment of diabetic mice with resveratrol [25], 1% tBHQ [26], or MG132 [27] in STZ-induced diabetic mice, renal expression of Nrf2, and downstream antioxidants was increased along with significant decreases in renal damage and fibrosis. In addition, Zheng et al. have used SNF to treat STZ-induced diabetic model in C57 BL/6 mice for 4 months, to demonstrate the upregulation of renal Nrf2 and its downstream targets NQO1 and γ-GCS with significant alleviation of renal damage [13]. Using FVB mice, the present study also demonstrated that treatment of STZ-induced diabetic mice for 3-month provided a similar preventive effect on diabetic nephropathy to those in Zheng's study.
However, our present study here provided the new evidence to indicate that the preventive effect of SFN on diabetic nephropathy observed at the end of SFN treatment was not sustained as this protective effect was significantly diminished when examined at 3 months after terminating SFN treatment (i.e., at 6-month time-point). The lack of sustained preventive effect of SFN on renal damage is probably due to the lack of persistent upregulation of Nrf2 and its downstream target genes as shown by our results. Since in our study we used SFN at 0.5 mg/kg/day in 5 days/week for 3-months while Zheng et al. used SFN at 12.5 mg/kg/day in 3 times/week for 4 months [13], whether the dosage of SFN was too low to induce a persistent upregulation of renal Nrf2 needs to be further investigated.
In summary, we have investigated whether SFN as one of Nrf2 activators can protect diabetic nephropathy using a type 1 diabetes model. We treated diabetic and age-matched control mice with SFN at 0.5 mg/kg for five days of each week for 3 months, resulting in a significant prevention of diabetes-induced progression of renal damage and dysfunction. The renal protection was observed in the diabetic mice only at the end of 3-month SFN treatment, but not sustained for the additional 3 months without further SFN treatment. The renal prevention from diabetes was accompanied with a significant upregulation of Nrf2 expression and function in the kidney. In cultured renal tubular cell study, we define the direct role of Nrf2 in SFN protection from HG/Pal-induced fibrotic effect because silencing Nrf2 gene could completely abolish the prevention of HG/Pal effects by SFN. These results suggest that diabetic nephropathy can be prevented by SFN most likely via upregulation of Nrf2 expression and function, but, to reach this goal, SFN treatment should be continually supplemented.
4. Materials and Methods
4.1. Animals
FVB male mice, 8–10 weeks age, were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in the University of Louisville Research Resources Center at 22°C with a 12 h light/dark cycle with free access to standard rodent chow and tap water. All experimental procedures for these animals were approved by the Institutional Animal Care and Use Committee of the University of Louisville, which is compliant with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication number 85-23, revised 1996).
For induction of type 1 diabetic mouse model, mice were injected intraperitoneally with MLD-STZ (Sigma-Aldich, St. Louis, MO, USA), dissolved in 0.1 M sodium citrate buffer (pH4.5) at 50 mg/kg body weight daily for 5 consecutive days while age-matched control mice were received multiple injections of the same sodium citrate buffer. Five days after the last injection, mice with hyperglycemia (blood glucose levels ≥ 250 mg/dl) were defined as diabetic, as before [28]. SFN (Sigma-Aldich) was subcutaneously injection at 0.5 mg/kg for five days of each week for 3 months. Dose of SFN was used based on published information [29]. Mice were randomly allocated into four groups (n = 6 at least per group): normal control, SFN, DM, and DM/SFN. Since SFN was dissolved in dimethyl sulfoxide (DMSO) and diluted in PBS, mice serving as vehicle controls were given the same volume of PBS (1% DMSO).
4.2. Mouse Urinary ACR Detection
Mouse urine was collected at 3 months and 6 months of diabetes, respectively. Urinary albumin (Bethyl Laboratories Inc., Montgomery, TX, USA), and urinary creatinine (BioAssay Systems) were measured according to manufacturers' procedures provided with these kits. Mouse urinary ACR was calculated as ACR = urinary albumin/urinary creatinine (μg/mg).
4.3. Renal Histopathological Examination
After anesthesia, kidneys were isolated to take a piece of it for fixation in 10%-buffered formalin. The fixed tissue was dehydrated in graded alcohol series, cleared with xylene, embedded in paraffin, and sectioned at 5 μm thickness. To examine the basic structure change of renal tissue, sections were stained with H and E. PAS staining was also used for examination of glycogen and collagen contents, as described before [30, 31]. Sections stained for H and E and PAS then were assessed using a Nikon Eclipse E600 microscopy system.
4.4. Real-Time PCR
Collected kidney was snap frozen in liquid nitrogen and kept at −80°C. Total RNA was extracted using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). RNA concentrations and purities were quantified using a Nanodrop ND-1000 spectrophotometer. First-strand complimentary DNA (cDNA) was synthesized from total RNA according to manufacturer's protocol from the RNA PCR kit (Promega, Madison, WI, USA). Reverse transcription was performed using 1 μg of total RNA in 12.5 μL of the solution containing 4 μL 25 mM MgCl2, 4 μL AMV reverse transcriptase 5x buffer, 2 μL dNTP, 0.5 μL RNase inhibitor, 1 μL of AMV reverse transcriptase, and 1 μL of oligo dT primer, which were added with nuclease-free water to make a final volume of 20 μL. Reaction system was run at 42°C for 50 min and 95°C for 5 min. Primers of HO-1, NQO1, SOD1, SOD2, and CAT were purchased from Applied Biosystems (Carlsbad, CA, USA). Real-time PCR was carried out in a 20 μL reaction buffer that included 10 μL of TaqMan Universal PCR Master Mix, 1 μL of primer, and 9 μL of cDNA with the ABI 7300 Real-Time PCR system. Fluorescence intensity of each sample was measured at each temperature to monitor amplification of the target gene. Comparative cycle time (CT) was used to determine fold differences between samples [32].
4.5. Western Blotting Assay
Renal tissues were homogenized and HK 11 cells were sonicated in RIPA buffer (Santa Cruz). Total protein was extracted and separated on 10% SDS-PAGE gels and transferred to a nitrocellulose membranes (Bio-rad, Hercules, CA, USA). Membranes were blocked with a 5% nonfat dried milk for 1 h at room temperature and then incubated overnight at 4°C with the following antibodies: 3-NT (1 : 1000 dilution), 4-HNE (1 : 1000 dilution), TNF-α (1 : 500 dilution), PAI-1 (1 : 2000 dilution), CTGF (1 : 500 dilution), TGF-β1 (1 : 500 dilution), Nrf2 (1 : 500 dilution), HO-1 (1 : 500 dilution), NQO1 (1 : 500 dilution), SOD1 and SOD2 (1 : 2000 and 1 : 5000 dilution, resp.), CAT (1 : 5000 dilution), and β-actin (1 : 2000 dilution), all which were purchased from Santa Cruz Biotech. Inc. except for PAI-1 from BD Bioscience, TNF-α from Abcam, 3-NT from Millipore, and 4-HNE from Alpha Diagnostic International. After three washes with tris-buffered saline (pH 7.2) containing 0.05% Tween 20, membranes were incubated with the appropriate secondary antibodies for 1 h at room temperature. Antigen-antibody complexes were then visualized using an enhanced chemiluminescence kit (Thermo scientific, Rockford, IL, USA) [33].
4.6. Cell Culture, Treatments, and Measurements
HK11 cells were maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 supplemented with 5% fetal bovine serum (FBS, Invitrogen). To recapture the in vivo diabetic model in an in vitro condition, HK11 cells were exposed to D-glucose in a final concentration of 27.5 mM (HG) with 2% BSA and 1% FBS for 48 h and at the last 6 h, palmitate (300 μM) was added into the culture medium. To exclude a hyperosmotic effect, 5.5 mM D-glucose plus 22 mM D-mannitol (Sigma) was added as hyperosmotic control. To mimic the in vivo study, SFN dissolved in DMSO was added (3 μM final concentration) for 48 h. The effect of Nrf2 and its downstream gene expression on glucose-induced CTGF expression was defined with corresponding siRNA. CTGF, Nrf2, and HO-1 expression was measured by Western blotting assay.
Nrf2 expression and translocation into nuclear were monitored by fluorescent microscope. HK11 cells were cultured on chamber slides for 12 h to allow the cells to attach to the flask and then subjected to above experimental conditions. The chamber slides were incubated with rabbit anti-Nrf2 antibody (1 : 50 dilution, Abcam, Cambridge, MA) overnight at 4°C, with Cy3-conjugated goat anti-rabbit IgG antibody, and costained with Dapi for the nuclei. The fluorescent staining was analyzed under fluorescent microscope.
4.7. siRNA Transfection
HK 11 cells were transfected with either the negative control siRNA (Invitrogen) or siRNA specific to human Nrf2 (NFE2L2HSS181505, Invitrogen) by Lipofectamine TM 2000 transfection reagent (Invitrogen) according to manufacturer's instructions for 48 h followed by SFN (3 μM) and HG (27.5 mM) treatment for additional 48 h as described above. The sense and antisense sequences of the primers were 5′-CAAACUGACAGAAGUUGACAAUUAU-3′ and 5′-AUAAUUGUCAACUUCUGUCAGUUUG-3′; transfection efficiency was assessed by Western blot analysis for Nrf2 and HO-1 protein expression. Effects of siRNA knockdown of Nrf2 on the expression of CTGF were assessed by Western blot analysis at indicated time points.
4.8. Statistical Analysis
In vivo and in vitro data were collected from at least 6 animals each group and repeated cell experiments, respectively, and were presented as means ± SD. We used Image Pro Plus 6.0 software to identify the positive staining, Image Quant 5.2 to analysis western blotting. Comparisons were performed by one or two-way ANOVA for the different groups, followed by post hoc pairwise repetitive comparisons using Tukey's test with Origin 7.5 Lab data analysis and graphing software. Statistical significance was considered as P < 0.05.
Acknowledgments
This study was supported in part by a Basic Research Award from ADA (1-11-BA-0117, to L. Cai) and a Starting-Up Fund for Chinese-American Research Institute for Diabetic Complications from Wenzhou Medical College (to L. Cai and Y. Tan).
References
- 1.Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. Journal of the American Society of Nephrology. 2003;14(8, supplement 3):S241–S245. doi: 10.1097/01.asn.0000077410.66390.0f. [DOI] [PubMed] [Google Scholar]
- 2.Alpers CE, Hudkins KL. Mouse models of diabetic nephropathy. Current Opinion in Nephrology and Hypertension. 2011;20(3):278–284. doi: 10.1097/MNH.0b013e3283451901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valk EJ, Bruijn JA, Bajema IM. Diabetic nephropathy in humans: pathologic diversity. Current Opinion in Nephrology and Hypertension. 2011;20(3):285–289. doi: 10.1097/MNH.0b013e328345bc1c. [DOI] [PubMed] [Google Scholar]
- 4.Cai L. Diabetic cardiomyopathy and its prevention by metallothionein: experimental evidence, possible mechanisms and clinical implications. Current Medicinal Chemistry. 2007;14(20):2193–2203. doi: 10.2174/092986707781389646. [DOI] [PubMed] [Google Scholar]
- 5.Sykiotis GP, Habeos IG, Samuelson AV, Bohmann D. The role of the antioxidant and longevity-promoting Nrf2 pathway in metabolic regulation. Current Opinion in Clinical Nutrition and Metabolic Care. 2011;14(1):41–48. doi: 10.1097/MCO.0b013e32834136f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. Journal of Biological Chemistry. 2003;278(24):21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 7.de Haan JB. Nrf2 activators as attractive therapeutics for diabetic nephropathy. Diabetes. 2011;60(11):2683–2684. doi: 10.2337/db11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li B, Liu S, Miao L, et al. Prevention of diabetic complications by activation of nrf2: diabetic cardiomyopathy and nephropathy. Experimental Diabetes Research. 2012;2012:7 pages. doi: 10.1155/2012/216512.216512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of phase II detoxication enzymes. Food and Chemical Toxicology. 1999;37(9-10):973–979. doi: 10.1016/s0278-6915(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 10.Clarke JD, Hsu A, Williams DE, et al. Metabolism and tissue distribution of sulforaphane in NRF2 knockout and wild-type mice. Pharmaceutical Research. 2011;28(12):3171–3179. doi: 10.1007/s11095-011-0500-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin DH, Park HM, Jung KA, et al. The NRF2-heme oxygenase-1 system modulates cyclosporin A-induced epithelial-mesenchymal transition and renal fibrosis. Free Radical Biology and Medicine. 2010;48(8):1051–1063. doi: 10.1016/j.freeradbiomed.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon HY, Kang NI, Lee HK, Jang KY, Park JW, Park BH. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochemical Pharmacology. 2008;75(11):2214–2223. doi: 10.1016/j.bcp.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 13.Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60(11):3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rane MJ, Song Y, Jin S, et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. American Journal of Physiology. 2010;298(1):F49–F61. doi: 10.1152/ajprenal.00032.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovascular Toxicology. 2001;1(3):181–193. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 16.Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP. The role of oxidative stress and NF-κB activation in late diabetic complications. Biofactors. 1999;10(2-3):157–167. doi: 10.1002/biof.5520100211. [DOI] [PubMed] [Google Scholar]
- 17.Wei W, Liu Q, Tan Y, Liu L, Li X, Cai L. Oxidative stress, diabetes, and diabetic complications. Hemoglobin. 2009;33(5):370–377. doi: 10.3109/03630260903212175. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Feng W, Xue W, et al. Inactivation of GSK-3β by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes. 2009;58(6):1391–1402. doi: 10.2337/db08-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He X, Kan H, Cai L, Ma Q. Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2009;46(1):47–58. doi: 10.1016/j.yjmcc.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Jiang T, Huang Z, Lin Y, Zhang Z, Fang D, Zhang DD. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59(4):850–860. doi: 10.2337/db09-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xue M, Qian Q, Adaikalakoteswari A, Rabbani N, Babaei-Jadidi R, Thornalley PJ. Activation of NF-E2-related factor-2 reverses biochemical dysfunction of endothelial cells induced by hyperglycemia linked to vascular disease. Diabetes. 2008;57(10):2809–2817. doi: 10.2337/db06-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki M, Betsuyaku T, Ito Y, et al. Down-regulated NF-E2-related factor 2 in pulmonary macrophages of aged smokers and patients with chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology. 2008;39(6):673–682. doi: 10.1165/rcmb.2007-0424OC. [DOI] [PubMed] [Google Scholar]
- 23.Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. American Journal of Physiology. 2010;298(3):F662–F671. doi: 10.1152/ajprenal.00421.2009. [DOI] [PubMed] [Google Scholar]
- 24.Yoh K, Hirayama A, Ishizaki K, et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes to Cells. 2008;13(11):1159–1170. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- 25.Palsamy P, Subramanian S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochimica et Biophysica Acta. 2011;1812(7):719–731. doi: 10.1016/j.bbadis.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Zhang L, Wang F, et al. Attenuation of glomerular injury in diabetic mice with tert- butylhydroquinone through nuclear factor erythroid 2-related factor 2-dependent antioxidant gene activation. American Journal of Nephrology. 2011;33(4):289–297. doi: 10.1159/000324694. [DOI] [PubMed] [Google Scholar]
- 27.Luo ZF, Qi W, Feng B, et al. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sciences. 2011;88(11-12):512–520. doi: 10.1016/j.lfs.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 28.Cai L, Wang J, Li Y, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metallothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54(6):1829–1837. doi: 10.2337/diabetes.54.6.1829. [DOI] [PubMed] [Google Scholar]
- 29.Guerrero-Beltrán CE, Calderón-Oliver M, Pedraza-Chaverri J, Chirino YI. Protective effect of sulforaphane against oxidative stress: recent advances. Experimental and Toxicologic Pathology. 2012;64(5):503–508. doi: 10.1016/j.etp.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Song Y, Li C, Cai L. Fluvastatin prevents nephropathy likely through suppression of connective tissue growth factor-mediated extracellular matrix accumulation. Experimental and Molecular Pathology. 2004;76(1):66–75. doi: 10.1016/j.yexmp.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 31.Song Y, Wang J, Li V, et al. Cardiac metallothionein synthesis in streptozotocin-induced diabetic mice, and its protection against diabetes-induced cardiac injury. American Journal of Pathology. 2005;167(1):17–26. doi: 10.1016/S0002-9440(10)62949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Tan Y, Zhao F, et al. Angiotensin ii plays a critical role in diabetic pulmonary fibrosis most likely via activation of nadph oxidase-mediated nitrosative damage. American Journal of Physiology. 2011;301(1):E132–E144. doi: 10.1152/ajpendo.00629.2010. [DOI] [PubMed] [Google Scholar]
- 33.Zhou G, Li X, Hein DW, et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. Journal of the American College of Cardiology. 2008;52(8):655–666. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]