

Abstract
Aurora kinases are essential for regulation of chromosome segregation and cytokinesis during mitosis and play a role in growth and progression of human tumors, including ovarian cancer. Aurora A and Aurora B are frequently overexpressed in high-grade and low-grade ovarian cancers. Targeting Aurora kinases has great potential for improving the efficacy of chemotherapies of ovarian cancer. In this study, we investigated whether the Aurora kinase inhibitor, VE 465, can enhance the anti-tumor activity of carboplatin in human ovarian cancer cells. The antitumor activity of VE 465 was tested by MTT proliferative assay in multiple established human epithelial ovarian cancer cell lines of varying p53 status. VE 465 and carboplatin had a synergistic effect on cell viability in both platinum-sensitive and -resistant ovarian cancers. The growth-inhibitory effect was accompanied by reduction in expression of histone 3 and an increase in apoptosis. We conclude that VE 465 enhances the efficacy of carboplatin agents in ovarian carcinoma.
Keywords: ovarian cancer, Aurora kinases, Aurora kinase inhibitors, chemosensitization
Introduction
Among gynecologic malignancies, ovarian cancer is the leading cause of death. Despite extensive efforts to prolong patient survival, the overall survival rates for patients with advanced epithelial ovarian cancer have improved only slightly during the past 20 years.1-3 Combination chemotherapyconsisting of a platinum and paclitaxelis the current standard of care. Although the cancer initially responds to this treatment in most patients, the disease usually relapses because of acquired resistance to the chemotherapeutic agents. A better understanding of the molecular aspects of ovarian tumorigenesis and identification of novel molecular targets may lead to new and more effective therapeutic strategies.2-5
Human Aurora kinases A, B, and C are known to be involved in the regulation of centrosome function, bipolar spindle assembly, and chromosome segregation processes.6 Ectopic overexpression of Aurora kinases in mammalian cells induces centrosome amplification,7 chromosome instability,8 oncogenic transformation8 and chemoresistance.9 Overexpression of Aurora kinases has been detected in a wide range of human tumors,10,11 including epithelial ovarian cancers.12 Our studies suggest that 80% of human ovarian cancers examined overexpress Aurora kinase A and that its overexpression correlates with worse overall survival.13 Furthermore, some studies suggest that Aurora kinase A depletion may sensitize cancer cells to cisplatin-induced apoptosis. RNA interference (siRNA) targeting STK15 or a selective inhibitor of Aurora kinase B demonstrated an inhibitory effect on cancer cells and restore chemosensitization of cancer cells to cytotoxic therapeutic agents such as cisplatin or docetaxel.14-16 Therefore, targeting Aurora kinases has been shown to have potential for improving the effectiveness of targeted therapies in ovarian cancer.
A number of small-molecule inhibitors of Aurora kinases have been developed for the treatment of cancer14,16 including ZM447439, Hesperadin, VX-680, compound 667, a compound from the 3-amino-tetrahydropyrrolo[3,4-c]pyrazole scaffold, and AZD 1152, a highly selective inhibitor of Aurora kinase B. Treatment with one of these potent compounds resulted in arrest of proliferation of various tumor cell lines, including human ovarian cancer cell line A2780, and inhibition of phosphorylation of histone 3 (H3) on serine 10.17
Among these, VX-680 is a pan-Aurora inhibitor that target the ATP-binding site in these the Aurora kinases.18 It is a potent inhibitor of all three Aurora kinases. VX-680 causes accumulation of cells with 4N DNA content before cell death and potently inhibits the proliferation of a wide variety of tumor cell types. VX-680 markedly reduced tumor size in rodent xenograft models. Inhibition of tumor growth was paralleled by a reduction in H3 phosphorylation and a significant increase in apoptosis. VE 465, another known inhibitor of the Aurora kinases exhibits an effect similar to that of VX-680 on Aurora A, Aurora B, and Aurora C (personal communication with Merck & Co., Inc.), but it has not been tested in ovarian cancer.
In current study, we examined whether VE 465 enhances the antitumor activity of carboplatin on human ovarian cancer cells, since carboplatin is the mostly commonly used drug in ovarian cancer treatment. We found that VE 465 and carboplatin had a synergistic effect in both platinum-sensitive and -resistant ovarian cancers and that the growth-inhibitory effect was paralleled by reduction in H3 expression and increase in apoptosis. We expect that combined treatment with VE 465 and carboplatin might have the potential for improving the efficacy of treatment of ovarian cancer cell lines in the future.
Results
Growth-inhibitory effect of VE 465 on ovarian cancer cell lines
To test the antitumor activity of VE 465 in ovarian cancer cell lines varying in p53 status and sensitivity to chemotherapeutic drugs and in ER expression status, we treated seven well-established ovarian cancer cell lines with VE 465 at different concentrations for 96 h. MTT proliferative assay showed that VE 465 effectively inhibited proliferation of a number of these cell lines, with IC50 levels ranging between 0.09 and 35.9 μM (Fig. 1A and B). Among the cell lines, A2780 and 2008/C13, which express wild-type p53 (Fig. 1C), were most sensitive to VE 465. The p53 mutant NMP1 cells were most resistant to VE 465. Growth of ovarian 2008/C13 cancer cells was inhibited by 18.1% and 32.4%, respectively, after exposure to VE 465 at a concentration of 0.1 μM for 72 and 96 h and by 21.4 and 33.1%, respectively, after exposure to VE 465 at a concentration of 1 μM for 72 and 96 h. The growth-inhibitory effect of VE 465 on 2008/C13 was time dependent, but the effect on 2774 cells was not (data not shown). The sensitivity of the ovarian cancer cells to VE 465 was not related to the expression status of Aurora kinase A, Aurora kinase B, or ER-α (Fig. 1C).
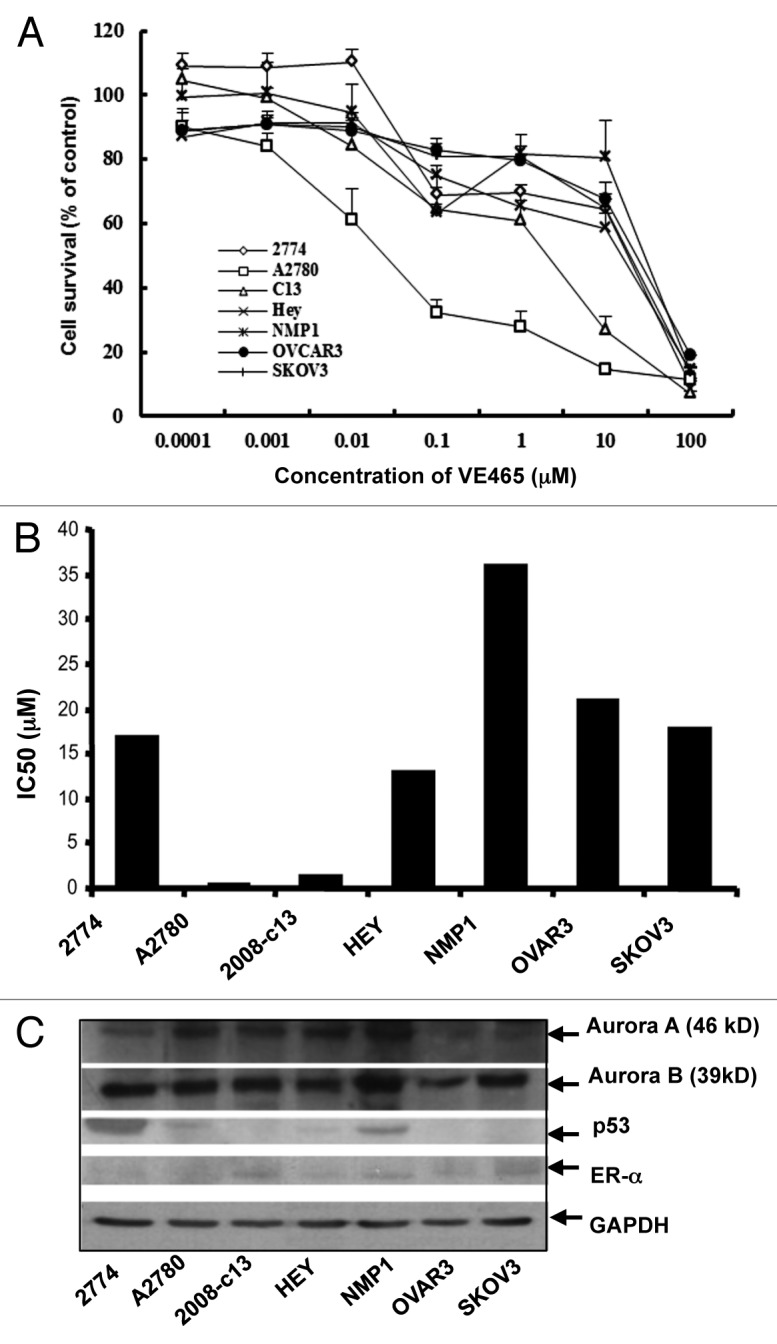
Figure 1. Growth-inhibitory effect of VE 465 on ovarian cancer cell lines as detected by MTT proliferative assay. (A) VE 465 on cell lines 2774, A2780, 2008/C13, Hey, NMP1, OVCAR3, and SKOV3. (B) IC50 of VE 465 in different ovarian cancer cell lines after treatment for 96 h. The experiments were repeated three times. (C) Western blot analysis of the expression of Aurora kinases A and B, p53, and ER-α in ovarian cancer cell lines.
Effect of VE 465 on 2008/C13 cell cycle
The cell cycle profile of 2008/C13 cells was analyzed by flow cytometry for DNA content after staining with PI. After 48 h of treatment with VE 465 at concentrations of 0.1 and 1 μM, 44.6% and 43.2% of cells were arrested at the G2/M phase, respectively, compared with 8.6% of controls (Fig. 2A and B). The expression of Aurora kinase B and the phosphorylated status of its substrate H3 were also analyzed by western blot analysis with antibodies against Aurora B and phosphorylated H3. The expression of Aurora B and phosphorylated H3 was slightly decreased in the 2008/C13 cells treated with VE 465 for 48 h (Fig. 2C, left panel), suggesting that inhibition of ovarian cancer cell growth by VE 465 is likely associated with increased G2/M arrest and inhibition of Aurora B activity.

Figure 2. The effect of VE 465 on the cell cycle of 2008/C13 cells showing arrest in G2/M phase. (A) Flow cytometry analysis of the effect of VE 465 on 2008/C13 cells. (B) Flow cytometry analysis of cells arrested in G2/M phase (left panel) and sub-G1 cells (right panel) after exposure to VE 465. (C) Western blot analysis of expression of phosphorylated H3, Aurora kinase B (left panel), and cleaved PARP (right panel) in 2008/C13 ovarian cancer cells after treatment for 48 h.
Induction of apoptosis by VE 465 in 2008/C13 cells
Interestingly, the percentage of cells with sub-G0/G1 DNA content was markedly increased in the cells treated with VE 465 at 10−7 and 10−6 M, from 1.4% in untreated cells to 26% and 38%, respectively (Fig. 2B, right panel). Cleaved PARP, a substrate of activated caspase-3, was significantly increased in ovarian cancer cells treated with VE 465 at different dosages (Fig. 2C, right panel), suggesting that VE 465 inhibited ovarian cancer cell growth through enhancement of apoptosis.
Growth-inhibitory effect of VE 465 plus carboplatin on ovarian cancer cell lines and its potential mechanisms
We further tested whether VE 465 acts as a sensitizer to carboplatin in ovarian cancer cell lines, especially in platinum-resistant ovarian cancer cell lines. We focused on the ovarian cancer cells whose growth could be inhibited by VE 465. The 2008/C13 cells were treated with carboplatin at different concentrations in combination with VE 465 at concentrations of 0.1, 1, and 10 μM for 72 h. The sensitivity of these platinum-resistant cells to carboplatin at 62.5 μg/ml in the presence of VE 465 at 10−6 M was 4.3 and 4.7 times greater than their sensitivity to carboplatin or VE 465 alone (p < 0.001; Fig. 3A).

Figure 3. Growth-inhibitory effects of VE 465 or carboplatin or a combination of the two for 72 h on ovarian cancer cell lines 2008/C13 (A) and A2780 (B) as detected by MTT proliferative assay.
The growth inhibition resulting from combined treatment with VE 465 and carboplatin was analyzed for synergistic and additive effects. Synergistic effects were determined by calculating the ratio between the percentage of growth inhibition expected assuming an additive interaction and the actual growth inhibition observed when VE 465 and carboplatin were combined (values > 1 indicate synergistic action; values = 1 indicate additive action). Using this method, the effect of the two drugs in cisplatin-resistant 2008/C13 cells was determined to be synergistic (Table 1). A similar effect was observed in platinum-sensitive A2780 cell line (Fig. 3B). Our preliminary data suggest that combined carboplatin and VE 465 significantly inhibit growth of platinum-sensitive and -resistant ovarian cancer cells.
Table 1. Percentage of surviving C13 cells (combination index*) after treatment with different combinations of carboplatin and VE465.
| Carboplatin concentration (µg/ml) | |||||||
|---|---|---|---|---|---|---|---|
| |
3.9 |
7.8 |
15.6 |
31.25 |
62.5 |
125 |
250 |
| Carboplatin + VE−7 |
72% (1.15) |
65% (1.24) |
67% (1.21) |
41% (1.74) |
17% (3.12) |
9% (2.68) |
7% (1.49) |
| Carboplatin + VE−6 |
72% (1.11) |
69% (1.13) |
62% (1.27) |
38% (1.81) |
15% (3.44) |
10% (2.48) |
7% (1.42) |
| Carboplatin + VE−5 | 33% (1.41) | 33% (1.36) | 26% (1.76) | 19% (2.13) | 11% (2.62) | 9% (1.61) | 7% (0.73) |
CI (combination index) values greater than 1 indicate synergistic action of the two combined drugs.
We next attempted to identify cellular changes in the 2008/C13 cells after exposure to both drugs. Flow cytometry analysis showed that the combination of 0.1 μM VE 465 and 16 μg/mL carboplatin induced 34.9% of cells to arrest at G2/M phase, while VE 465 or carboplatin alone (at the same concentrations) caused arrest in 24 and 11.9% of cells, respectively (Fig. 4). Western blot analysis showed that expression of Aurora kinase B in 2008/C13 cells was induced by carboplatin at concentrations of 16 and 32 μg/mL, while it was slightly reduced by the combination of carboplatin with 1 μM VE 465. This result was consistent with the finding that phosphorylated H3 expression was significantly reduced by VE 465 alone and by co-treatment with VE 465 and carboplatin, suggesting that the growth-inhibitory effect of combined treatment on ovarian cancer cell lines was associated with inhibition of Aurora B activity (Fig. 5A).

Figure 4. Flow cytometry analysis of the effects of VE 465 plus carboplatin on the cell cycle of 2008/C13 ovarian cancer cells. (A) The effects of VE 465 plus carboplatin on 2008/C13 cells. (B) Percentage of cells arrested in G2/M phase after exposure to VE 465 and carboplatin.

Figure 5. Western blot analysis of expression of phosphorylated H3 and Aurora kinase B (A) and ER-α (B) in 2008/C13 ovarian cancer cells after treatment with VE 465 and carboplatin for 48 h. V6 and V7: VE 465 at concentration of 10−6 and 10−7 M; C16 and C32: Carboplatin at concentration of 16 and 32 μg/mL.
Since it has been suggested that there is an interaction between estrogen and H3 in the presence of Aurora B in ovarian follicle and mammary epithelial tumor cells,28 we investigated whether ER could be modulated by VE 465 in platinum-resistant ovarian cancer cells. We found that expression of ER-α was reduced in cells following exposure to the combination of 0.1 μM VE 465 and 16 μg/mL carboplatin, compared with either agent alone (Fig. 5B), suggesting that the synergistic effect of VE 465 and carboplatin may be associated with inhibition of H3 in the presence of ER.
Notably, flow cytometry analysis showed that the percentage of cells with sub-G0/G1 DNA content was markedly increased in cells treated with VE 465 at 10−7 M alone or in combination with carboplatin at 16 μg/mL (Fig. 6A and B). Cleaved PARP, a substrate of activated caspase-3, was significantly increased in ovarian cancer cells treated with this combination of VE 465 and carboplatin, compared with either drug alone (Fig. 6C), suggesting that VE 465 sensitized platinum-resistant ovarian cancer cells (2008/C13) to carboplatin through induction of apoptosis, possibly in a caspase-3–dependent pathway.
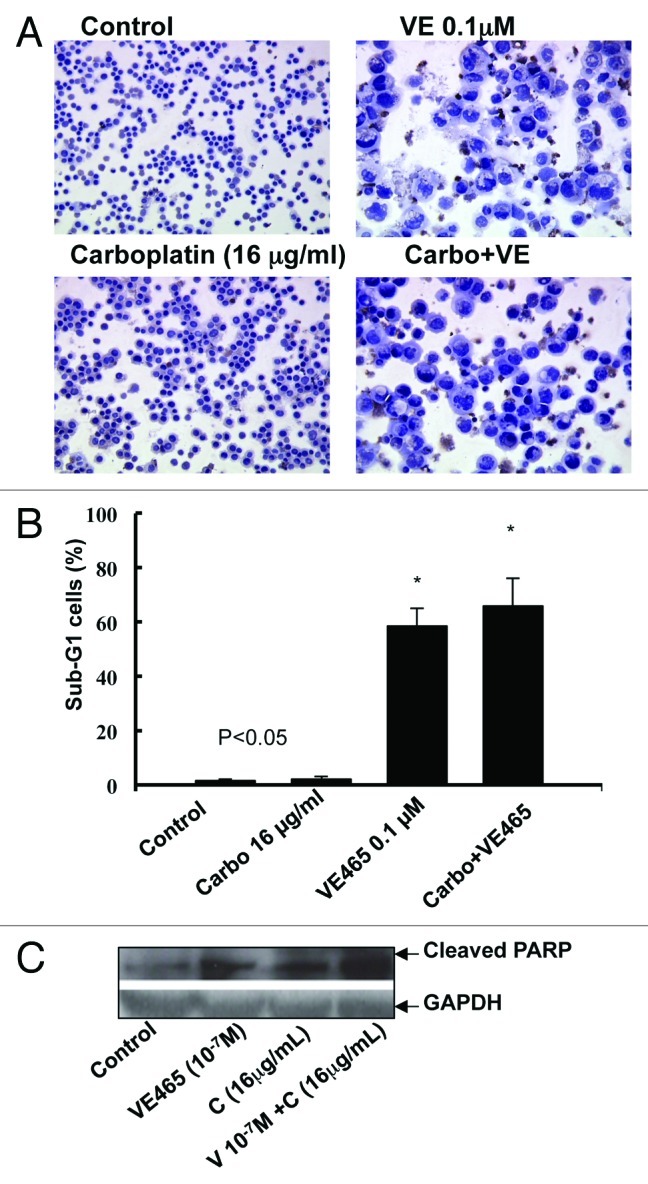
Figure 6. Effects of treatment with VE 465 plus carboplatin for 48 h on apoptosis in ovarian cancer cell line 2008/C13 as detected by TUNEL (A), flow cytometry (B), and western blot analysis of cleaved PARP expression.
Discussion
In the present study, we demonstrated that VE 465 has selective antitumor activity in ovarian cancer cells and a synergistic effect with carboplatin on both platinum-sensitive and -resistant ovarian cancer cells. The growth-inhibitory effect of VE 465 was paralleled by a reduction in H3 expression and an increase in apoptosis, suggesting that VE 465 could be a chemosensitizer, enhancing the antitumor activity of carboplatin on human ovarian cancer cells.
VX-680 is a potent inhibitor of all three Aurora kinases, with apparent inhibition constant (Ki, app) values of 0.6, 18, and 4.5 nM for Aurora A, Aurora B, and Aurora C, respectively29; it has been confirmed to have antitumor activity in a diverse range of tumor types. VE 465 has exhibited effects similar to those of VX-680 on Aurora A, Aurora B, and Aurora C (personal communication with Merck & Co, Inc.). Our findings indicate that sensitivity to VE 465 varied in a panel of ovarian cancer cell lines. Among the seven ovarian cancer cell lines tested in this study, A2780 and 2008/C13 cells, which both express wild-type p53, were most sensitive to VE 465. NMP1, which expresses mutant p53, was most resistant to VE 465, which is likely consistent with the published observation that Aurora A induced chemoresistance in ovarian cancer cells expressing wild-type p53.27 The sensitivity of the ovarian cancer cells tested to VE 465 was not related to the expression status of Aurora A, Aurora B, or ER-α.
Which Aurora kinase is responsible for the effects observed after treatment with Aurora kinase inhibitors remains unknown to researchers in the field. In this study, we found that Aurora A and Aurora B were expressed in all ovarian cancer cell lines tested. The finding that the anti-proliferative activity of VE 465 was not correlated with the expression status of Aurora A and Aurora B contradicts the finding that the antitumor activity of VE 465 was closely related to expression status of Aurora kinases in prostate cancer.30 Further study showed that the expression of Aurora A in 2008/C13 cells was not significantly changed after exposure to VE 465 in various doses, whereas the expression of Aurora B decreased in cells exposed to higher doses of VE 465. Moreover, phosphorylation of H3 on serine 10, a functional substrate of the Aurora kinases, was slightly inhibited by VE 465 in 2008/C13 cells, suggesting that the anti-tumor activity of VE 465 might be correlated with its enzyme activity on Aurora B. It has been suggested that the intrinsic antitumor activity of many of these inhibitors correlates principally with inhibition of Aurora B, since H3 is the functional substrate for Aurora B.31
A number of studies have suggested that knockdown of Aurora kinase A by siRNA and small molecule inhibitors enhances sensitivity to chemotherapeutic agents of a wide range of human cancers.9,32,33 It has also been suggested that Aurora A activates Akt and induces cell survival and chemoresistance in a p53-dependent manner in ovarian cancer.27 This prompted us to investigate whether VE 465 could sensitize ovarian cancer cells, especially in platinum-resistant ovarian cancer cells to chemotherapy. In this study, we found that VE 465 and carboplatin had a synergistic effect in platinum-resistant human ovarian cancer cells. In addition, flow cytometry analysis and TUNEL assay showed a marked increase in apoptosis in cells treated with VE 465 at 10−7 M or the combination of VE 465 with carboplatin at 16 μg/mL. Cleaved PARP, a substrate of activated caspase-3, was detected in significantly greater levels in ovarian cancer cells treated with a combination of VE 465 and carboplatin than in those treated with either drug alone, suggesting that VE 465 sensitized platinum-resistant ovarian cancer cells (2008/C13) to carboplatin through induction of apoptosis, possibly in a caspase-3-dependent pathway. This finding is consistent with those of other studies of Aurora kinase inhibitors in human cancers.15,29,34,35
Further study on the potential mechanisms of action of VE 465, alone and in combination with carboplatin, showed that the expression of phosphorylated H3 was significantly reduced by VE 465 alone and by co-treatment with VE 465 and carboplatin, suggesting that the growth- inhibitory effect of combined treatment on ovarian cancer cell lines was mediated by inhibition of Aurora kinase activity.
It has been suggested that cell cycle-dependent phosphorylation of H3 in both ovarian granulosa and breast cancer cells is driven by estrogen through the action of oncogenic kinase Aurora B on ER-α. We therefore attempted to address whether the observed anti-tumor activity of VE 465 was related to ER expression status. Interestingly, the expression of ER-α in these cells was significantly lower after exposure to the combination of 0.1 μM VE 465 and 16 μg/mL carboplatin than after exposure to either drug alone. However, the potential mechanism by which the Aurora kinase inhibitor directly or indirectly affected ER expression is not clearly understood. One recent study showed that Aurora kinase A phosphorylated BRCA1 in Hela S3 cells36, and it has also been suggested that BRCA1 knockdown caused an increase in ER-α phosphorylation on serine-167 depending on PI3k/Akt signaling. It is reasonable to postulate that the Aurora kinase inhibitor might have the effect on reactivating BRCA1, thereby subsequently inhibiting estrogen receptor activity. The exact mechanisms need to be further investigated.
Materials and Methods
Chemicals and reagents
VE 465, kindly provided by Merck & Co., Inc., was freshly dissolved in dimethyl sulfoxide (DMSO) at a stock concentration of 10−2 M. Carboplatin was obtained from the pharmacy of The University of Texas MD Anderson Cancer Center at a stock concentration of 10 mg/mL and divided into aliquots. Monoclonal antibodies against Aurora kinases A and B and polyclonal antibody against estrogen receptor α antibody (ab3575,68 kDa) were obtained from Abcam. Monoclonal antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was obtained from Santa Cruz Biotechnology, Inc. Polyclonal antibodies against cleaved caspase-3 and phosphorylated H3 were obtained from Cell Signaling Technology. Polyclonal antibody against the 85-kDa caspase-cleaved fragment of human poly (ADP-ribose) polymerase (PARP) was obtained from Promega Corporation. Monoclonal antibody against p53 (DO-7) was obtained from Calbiochem EMD Chemicals, Inc.
Cell cultures
Seven established human epithelial ovarian cancer cell lines were used in this study. SKOV3 (cisplatin resistant; p53 null) and OVCAR3 (cisplatin resistant; mutant p53)19 cell lines were obtained from American Type Culture Collection. The 2774 [cisplatin sensitive; mutant p53 and estrogen receptor (ER) negative] cell line20-22 was established in the laboratory of J. Sinkovics (St. Joseph's Hospital) and A2780 (cisplatin sensitive; wild-type p53)23 were obtained from Dr Ralph S. Freedman of MD Anderson Cancer Center. The NMP-1cell line (cisplatin resistant; mutant p53)24,25 was developed from its parental OVCAR3 line following in vitro culture of the cells with increasing concentrations of cisplatin and subsequent passage in the nude mouse in the laboratory of Dr Jim Klostergaard at MD Anderson. The Cell lines HEY (wild-type p53) and NMP-1 were obtained from Dr Klostergaard.26 The platinum-resistant 2008/C13 cell line (wild-type p53)27 was obtained from Dr Zahid Siddik (MD Anderson Cancer Center). All cell lines were grown in RPMI 1640 medium (GIBCO Laboratories) supplemented with 10% fetal calf serum (Sigma) at 37°C and in an atmosphere containing 5% CO2 and were maintained in monolayer cultures. No human samples were used in this study.
MTT proliferative assay
Inhibition of cell growth by VE 465, carboplatin, and combination of the two was detected by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell proliferation assay in quadruplicate. Briefly, cells were seeded into 96-well plates (4,000 cells/well) and maintained in RPMI 1640 medium for 24 h. 2008/C13 and 2774 cells then were exposed to VE 465 at concentrations of 0.001, 0.01, 0.1, 1, 10, or 100 μM for 24, 48, 72, or 96 h. The other ovarian cancer cell lines, including A2780, HEY, NMP1, SKOV 3, and OVCAR3, were treated with VE 465 at the same concentrations for 96 h. Cells were also treated with a combination of VE 465 at concentrations of 0.1, 1, or 10 μM and carboplatin at concentrations of 4, 8, 16, 31, 63, 125 and 250 μg/ml for 72 h. Controls were treated with an equal volume of vehicle. After treatment, cell growth and viability were determined by incubating the cells for 1 to 2 h at 37°C with 20 μL of MTT substrate. Absorbency was measured at 450 nm by a 96-well Synergy HT-microplate reader (Biotek). The MTT assay was performed at different time points after treatment. Data are expressed as percentage of control (number of cells after treatment/number of cells before treatment). The experiments were repeated separately three times. The median inhibitory concentrations (IC50) were calculated by the Sigma plot 8.0 program.
Cell cycle analysis
The cell cycle distribution of cells was analyzed by flow cytometry. 2008/C13 cells (5 × 105 were treated with VE 465 at concentrations of 0.1, 1, or 10 μM or carboplatin at concentrations of 4, 8, 16, 31, 63, 125, or 250 μg/ml for 48 h. Controls were treated with DMSO. Following treatment, the cells were subjected to trypsinization and to centrifugation at 2,500 rpm for 5 min. Cells were then washed twice with 1 × phosphate-buffered saline solution (PBS), fixed with 70% ethanol overnight, washed twice again with 1 × PBS, and resuspended in 0.5 mL of propidium iodide (PI) solution consisting of 50 μg/mL PI and 20 μg/mL ribonuclease in PBS (1 × 106 cells per mL of PI buffer). The samples were incubated at 37°C for 15 min, and PI fluorescence was analyzed by using an EPIS XL analyzer (Beckman-Coulter). The software used for cell cycle analysis was Multicycle from Phoenix Flow Systems.
TUNEL assay
Platinum-resistant 2008/C13 cells were treated with VE 465 at 0.1 μM and carboplatin at 16 µg/mL for 48 h, after which the floating and adherent cells were collected. After cytospinning, the 2008/C13 cells were fixed with 4% paraformaldehyde at room temperature for 20 min and air-dried. The slides were stored at -20°C until use. Apoptotic cells were evaluated by the terminal deoxynucleotidyl transferase-mediated dUTP-biotin end labeling of fragmented DNA (TUNEL) assay, following the manufacturer’s protocol (APO-BRDU-IHC kit; Phoenix Flow Systems).
Immunoblot analysis
Extracts from the 2008/C13 cells treated with VE 465 or carboplatin at different concentrations were prepared from PBS-washed cell pellets for protein analysis. Pellets were subjected to lysis in lysis buffer [1 × PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), and freshly added proteinase inhibitors]. Cell lysates were subjected to centrifugation at 14,000 rpm for 10 min, and the supernatants were analyzed. The protein concentrations were estimated by using the protein assay (Pierce Chemical). Next, 35 μg of protein was loaded into each lane of and fractionated on 10–12% SDS-polyacrylamide gels, which were then transferred to a nitrocellulose membrane. Blots were probed with antibodies against the 85-kDa caspase-cleaved fragment of PARP, cleaved caspase-3, phosphorylated hH3, and GAPDH (dilution, 1:200–1:500). Anti-rabbit IgG and anti-mouse IgG conjugated with horseradish peroxidase were used as the secondary antibodies. GAPDH was used as a protein loading control. The signal was developed with an enhanced chemiluminescence detection system (Amersham Corporation). We used the NIH Image program (Scion Image for Windows) for image analysis.
Statistical analysis
The differences in cell growth inhibition and induction of apoptosis in 2008/C13 cells treated with both or VE 465 and carboplatin alone were assessed using the Student’s t-test. Statistical significance was set at an α of < 0.05.
Conclusions
In summary, we demonstrated that VE 465 has selective antitumor activity in ovarian cancer cells and that VE 465 and carboplatin had a synergistic effect on both platinum-sensitive and -resistant ovarian cancer cells. The growth-inhibitory effect was paralleled by a reduction in H3 and increase in apoptosis, suggesting that VE 465 could act as a chemosensitizer to enhance the antitumor activity of carboplatin on human ovarian cancer cells. Confirmation of the in vitro antitumor activity of VE 465 in nude mice bearing cisplatin-resistant ovarian cancer cell lines is warranted. We suggest that VE 465 enhances the antitumor activity of carboplatin on human ovarian cancer cells and that combined treatment with VE 465 and carboplatin might have the potential to be an effective treatment of ovarian cancer in the future.
Acknowledgments
The authors would like to thank Kathryn Halein the Department of Scientific Publications at MD Anderson Cancer Center for editing our manuscript and Merck & Co., Inc. and Vertex Pharmaceuticals for kindly providing VE 465. This work was supported by Ovarian SPORE grant P50 CA083639 (PP-DRP7) and the institution’s core grant (NIH grant no. 5P30CA016672-32).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/21045
References
- 1.Jemal A, Clegg LX, Ward E, Ries LA, Wu X, Jamison PM, et al. Annual report to the nation on the status of cancer, 1975-2001, with a special feature regarding survival. Cancer. 2004;101:3–27. doi: 10.1002/cncr.20288. [DOI] [PubMed] [Google Scholar]
- 2.Kavanagh JJSS, Pecoreli S, Dipertrillo A, Einhorn N. Chemotherapy in advanced ovarian cancer. Cambrige, MA: Blackwell Scientific Publications, 1998. [Google Scholar]
- 3.McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334:1–6. doi: 10.1056/NEJM199601043340101. [see comments] [DOI] [PubMed] [Google Scholar]
- 4.Verschraegen CFDK, Johnes LA. Biology of gynecologic cancers. Cambrige, MA: Blackwell Scientific Publications, 1998. [Google Scholar]
- 5.Kudelka AP, Verschraegen CF, Shen Y, Gonzalez De Leon C, Edwards CL, Freedman RS, et al. Long-term results and pharmacokinetics of high-dose paclitaxel in patients with refractory epithelial ovarian carcinoma. Int J Gynecol Cancer. 1999;9:44–53. doi: 10.1046/j.1525-1438.1999.09856.x. [DOI] [PubMed] [Google Scholar]
- 6.Fu S, Hu W, Kavanagh JJ, Bast RC., Jr. Targeting Aurora kinases in ovarian cancer. Expert Opin Ther Targets. 2006;10:77–85. doi: 10.1517/14728222.10.1.77. [DOI] [PubMed] [Google Scholar]
- 7.Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- 8.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 9.Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-kappaB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352:220–5. doi: 10.1016/j.bbrc.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Sakakura C, Hagiwara A, Yasuoka R, Fujita Y, Nakanishi M, Masuda K, et al. Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation.PG - 824-31. Br J Cancer. 2001;12:84. doi: 10.1054/bjoc.2000.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9:991–7. [PubMed] [Google Scholar]
- 12.Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, et al. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003;9:1420–6. [PubMed] [Google Scholar]
- 13.Landen CN, Jr., Lin YG, Immaneni A, Deavers MT, Merritt WM, Spannuth WA, et al. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007;13:4098–104. doi: 10.1158/1078-0432.CCR-07-0431. [DOI] [PubMed] [Google Scholar]
- 14.Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–8. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 15.Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007;110:2034–40. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- 16.Lin YG, Immaneni A, Merritt WM, Mangala LS, Kim SW, Shahzad MM, et al. Targeting aurora kinase with MK-0457 inhibits ovarian cancer growth. Clin Cancer Res. 2008;14:5437–46. doi: 10.1158/1078-0432.CCR-07-4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fancelli D, Berta D, Bindi S, Cameron A, Cappella P, Carpinelli P, et al. Potent and selective Aurora inhibitors identified by the expansion of a novel scaffold for protein kinase inhibition. J Med Chem. 2005;48:3080–4. doi: 10.1021/jm049076m. [DOI] [PubMed] [Google Scholar]
- 18.Cheetham GM, Knegtel RM, Coll JT, Renwick SB, Swenson L, Weber P, et al. Crystal structure of aurora-2, an oncogenic serine/threonine kinase. J Biol Chem. 2002;277:42419–22. doi: 10.1074/jbc.C200426200. [DOI] [PubMed] [Google Scholar]
- 19.Husain A, Chi DS, Prasad M, Abu-Rustum N, Barakat RR, Brown CL, et al. The role of laparoscopy in second-look evaluations for ovarian cancer. Gynecol Oncol. 2001;80:44–7. doi: 10.1006/gyno.2000.6036. [DOI] [PubMed] [Google Scholar]
- 20.Modesitt SC, Ramirez P, Zu Z, Bodurka-Bevers D, Gershenson D, Wolf JK. In vitro and in vivo adenovirus-mediated p53 and p16 tumor suppressor therapy in ovarian cancer. Clin Cancer Res. 2001;7:1765–72. [PubMed] [Google Scholar]
- 21.Freedman RS, Pihl E, Kusyk C, Gallager HS, Rutledge F. Characterization of an ovarian carcinoma cell line. Cancer. 1978;42:2352–9. doi: 10.1002/1097-0142(197811)42:5<2352::AID-CNCR2820420536>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 22.Thompson MA, Adelson MD, Kaufman LM. Lupron retards proliferation of ovarian epithelial tumor cells cultured in serum-free medium. J Clin Endocrinol Metab. 1991;72:1036–41. doi: 10.1210/jcem-72-5-1036. [DOI] [PubMed] [Google Scholar]
- 23.Heise C, Lemmon M, Kirn D. Efficacy with a replication-selective adenovirus plus cisplatin-based chemotherapy: dependence on sequencing but not p53 functional status or route of administration. Clin Cancer Res. 2000;6:4908–14. [PubMed] [Google Scholar]
- 24.Mujoo K, Zhang L, Klostergaard J, Donato NJ. Emergence of cisplatin-resistant cells from the OVCAR-3 ovarian carcinoma cell line with p53 mutations, altered tumorigenicity, and increased apoptotic sensitivity to p53 gene replacement. Int J Gynecol Cancer. 2000;10:105–14. doi: 10.1046/j.1525-1438.2000.00018.x. [DOI] [PubMed] [Google Scholar]
- 25.Auzenne E, Donato NJ, Li C, Leroux E, Price RE, Farquhar D, et al. Superior therapeutic profile of poly-L-glutamic acid-paclitaxel copolymer compared with taxol in xenogeneic compartmental models of human ovarian carcinoma. Clin Cancer Res. 2002;8:573–81. [PubMed] [Google Scholar]
- 26.Buick RN, Pullano R, Trent JM. Comparative properties of five human ovarian adenocarcinoma cell lines. Cancer Res. 1985;45:3668–76. [PubMed] [Google Scholar]
- 27.Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304–12. doi: 10.1002/ijc.22154. [DOI] [PubMed] [Google Scholar]
- 28.Ruiz-Cortés ZT, Kimmins S, Monaco L, Burns KH, Sassone-Corsi P, Murphy BD. Estrogen mediates phosphorylation of histone H3 in ovarian follicle and mammary epithelial tumor cells via the mitotic kinase, Aurora B. Mol Endocrinol. 2005;19:2991–3000. doi: 10.1210/me.2004-0441. [DOI] [PubMed] [Google Scholar]
- 29.Harrington EA, Bebbington D, Moore J, Rasmussen RK, Ajose-Adeogun AO, Nakayama T, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 30.Lee EC, Frolov A, Li R, Ayala G, Greenberg NM. Targeting Aurora kinases for the treatment of prostate cancer. Cancer Res. 2006;66:4996–5002. doi: 10.1158/0008-5472.CAN-05-2796. [DOI] [PubMed] [Google Scholar]
- 31.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res. 2006;66:7668–77. doi: 10.1158/0008-5472.CAN-05-3353. [DOI] [PubMed] [Google Scholar]
- 32.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/S1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 33.Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005;65:2899–905. doi: 10.1158/0008-5472.CAN-04-3981. [DOI] [PubMed] [Google Scholar]
- 34.Huang XF, Luo SK, Xu J, Li J, Xu DR, Wang LH, et al. Aurora kinase inhibitory VX-680 increases Bax/Bcl-2 ratio and induces apoptosis in Aurora-A-high acute myeloid leukemia. Blood. 2007 doi: 10.1182/blood-2007-07-099325. [DOI] [PubMed] [Google Scholar]
- 35.Tyler RK, Shpiro N, Marquez R, Eyers PA. VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell Cycle. 2007;6:2846–54. doi: 10.4161/cc.6.22.4940. [DOI] [PubMed] [Google Scholar]
- 36.Ouchi M, Fujiuchi N, Sasai K, Katayama H, Minamishima YA, Ongusaha PP, et al. BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J Biol Chem. 2004;279:19643–8. doi: 10.1074/jbc.M311780200. [DOI] [PubMed] [Google Scholar]