

Abstract
Accumulation of misfolded α-synuclein (αS) is mechanistically linked to neurodegeneration in Parkinson's disease (PD) and other α-synucleinopathies. However, how αS causes neurodegeneration is unresolved. Because cellular accumulation of misfolded proteins can lead to endoplasmic reticulum stress/unfolded protein response (ERS/UPR), chronic ERS could contribute to neurodegeneration in α-synucleinopathy. Using the A53T mutant human αS transgenic (A53TαS Tg) mouse model of α-synucleinopathy, we show that disease onset in the αS Tg model is coincident with induction of ER chaperones in neurons exhibiting αS pathology. However, the neuronal ER chaperone induction was not accompanied by the activation of phospho-eIF2α, indicating that α-synucleinopathy is associated with abnormal UPR that could promote cell death. Induction of ERS/UPR was associated with increased levels of ER/microsomal (ER/M) associated αS monomers and aggregates. Significantly, human PD cases also exhibit higher relative levels of ER/M αS than the control cases. Moreover, αS interacts with ER chaperones and overexpression of αS sensitizes neuronal cells to ERS-induced toxicity, suggesting that αS may have direct impact on ER function. This view is supported by the presence of ERS-activated caspase-12 and the accumulation of ER-associated polyubiquitin. More important, treatment with Salubrinal, an anti-ERS compound, significantly attenuates disease manifestations in both the A53TαS Tg mouse model and the adeno-associated virus-transduced rat model of A53TαS-dependent dopaminergic neurodegeneration. Our data indicate that the accumulation αS within ER leads to chronic ER stress conditions that contribute to neurodegeneration in α-synucleinopathies. Attenuating chronic ERS could be an effective therapy for PD and other α-synucleinopathies.
Introduction
Parkinson's disease (PD) is the second most common neurodegenerative disease after Alzheimer's disease. While the etiology of PD is unknown in most cases, degenerating neuronal populations in PD exhibit α-synuclein (αS) abnormalities and mutations in the αS gene cause familial PD, indicating that the αS abnormalities are mechanistically linked to pathogenesis of PD and other α-synucleinopathies (Vila and Przedborski, 2004; Obeso et al., 2010). While abnormal oligomerization/aggregation of αS is most often implicated as a pathogenic event in α-synucleinopathy, how αS causes neurodegeneration in vivo is poorly understood. Consequently, approaches to halt or prevent α-synucleinopathy and related neurodegeneration are currently lacking.
Cellular accumulation of misfolded proteins can lead to chronic endoplasmic reticulum stress (ERS) and trigger an integrated cellular response called unfold protein response (UPR), which attempts to protect cells from accumulation of toxic misfolded proteins. However, chronic unabated ERS leads to the activation of cell death cascade (Ron and Walter, 2007). Potential involvement of chronic ERS in αS-dependent neurodegeneration was first demonstrated in a PC12 cell model of αS toxicity (Smith et al., 2005). A recent series of reports suggests that increased αS expression can cause ER stress in yeast and other cells by interrupting Rab-dependent ER to Golgi membrane trafficking (Cooper et al., 2006). However, except for a limited number of neuropathological studies suggesting the activation of UPR in human PD cases (Hoozemans et al., 2007), it is unknown if αS abnormalities can directly cause ER stress in vivo. More important, it is not known whether ERS is important for onset/progression of disease manifestation in vivo.
To determine whether ERS is involved in αS-dependent neurodegeneration in vivo, we analyzed the activation of ERS pathways as a function of αS expression and α-synucleinopathy in the transgenic (Tg) mouse model expressing various human αS variants. We show that α-synucleinopathy is coincident with induction of ERS, abnormal UPR signal, and activation of ERS-induced cell death pathway in vivo. Significantly, α-synucleinopathy was also associated with increase in ER/microsomal (ER/M) αS aggregates and polyubiquitin. More important, Salubrinal, an anti-ERS agent, attenuates disease manifestations in the A53TαS Tg mouse model and in a rat adeno-associated virus (AAV) model of αS toxicity. We propose that increased ER accumulation of αS and αS aggregates triggers the chronic ERS that contributes to neurodegeneration. Further, agents that protect cells from chronic ERS could be developed as disease-modifying therapeutics for PD and other α-synucleinopathies.
Materials and Methods
Tg mouse models
Tg mice expressing high levels of WT or mutant (A53T and A30P) αS under the control of the mouse prion protein promoter have been described previously (Lee et al., 2002; Martin et al., 2006; Wang et al., 2008). Mice expressing A53TαS [line G2–3(A53T)] develop fatal neurological disease at ∼12 months of age which rapidly progresses to end state within 14–21 d of onset. At disease onset, the mice exhibit neuronal α-Syn and ubiquitinated aggregates/inclusions(Lee et al., 2002), degeneration of axons (Martin et al., 2006; Wang et al., 2008), and neuronal loss (Martin et al., 2006). For this study, early-stage-affected A53TαS Tg mice exhibit slight instability, bradykinesia, and ataxia. The end-stage mice were defined by the onset of the paralysis. Presymptomatic mice were 10–14 months old and free from any motor dysfunction. Age-matched nTg littermates, A30PαS and WTαS Tg mice, were also used. SOD1(G37R) Tg mice were provided by Dr. D.R. Borchelt (Department of Neuroscience, University of Florida).
For the Salubrinal treatment, a cohort of G2–3(A53T) Tg mice (n = 27) was randomly assigned to either Vehicle (n = 12) or Salubrinal (n = 14) group using GraphPad StatMate. At 12 months of age, six Tg mice developed neurological symptoms. Remaining asymptomatic G2–3(A53T) Tg mice were administered 1.5 mg/kg of Salubrinal (Axxora) (n = 11) or vehicle (n = 10), three times per week via oral gavages for ∼6 months by a lab staff blinded to the experimental conditions. Salubrinal was first dissolved in DMSO and then diluted 20 times with milk. Mice that became sick during the treatment were taken at end stage as described above. All animal study methods were approved in full by the Institutional Animal Care and Use Committee of the Johns Hopkins University and consistent with the requirements of the National Institutes of Health Office of Laboratory Animal Welfare Policy.
Tissues from human PD and control cases
Brain tissues were obtained from the Brain Resource Center (Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD). The pathological characterizations of the tissues were done as described previously (Pletnikova et al., 2005). The diagnosis and postmortem delay times for the human tissues are listed in the Table 1.
Table 1.
Diagnosis, age, and postmortem delay of human brain tissues used
| Diagnosis | Age (years) | PMD (h) | |
|---|---|---|---|
| Controls without BrSt LBDs | Control | 66 | 10 |
| Control | 74 | 4 | |
| AD | 78 | 4 | |
| AD | 88 | 14 | |
| AD | 80 | 9 | |
| AD | 92 | 7 | |
| AD | 78 | 8 | |
| AD | 69 | 10.5 | |
| PD with BrSt LBDs | PD w/dementia | 75 | 9 |
| PD w/dementia | 83 | 5 | |
| PD w/dementia/AD | 80 | 16 | |
| PD/AD probable | 82 | 5 | |
| PD/AD possible | 75 | 9 | |
| PD/AD possible | 85 | 14 | |
| PD/diffuse Aβ deposits | 77 | 16 | |
| PD/LBD | 90 | 16 | |
| PD/AD/LBD | 84 | 12 | |
| PD/AD | 64 | 21 | |
| PD | 85 | 14 | |
| PD | 76 | 18 |
All PD cases exhibit brainstem (BrSt) α-synuclein pathology level of at least 10 where as the controls are 2 or less (Pletnikova et al., 2005). AD, Alzheimer's disease; LBD, Lewy body dementia; MID, mutiple infarct dementia; PD, Parkinson's disease; PMD, postmortem delay.
Cell lines
Inducible BE(2)-M17 neuroblastoma cell line was created using the Tet-responsive system. Briefly, full-length cDNA for WT or A53T mutant αS was cloned into pcDNA4/TO tetracycline-regulated expression vector (Invitrogen). Constructs including control plasmid pcDNA4/TO/lacZ were cotransfected into BE(2)-M17 Tet-on cells with pcDNA6/TR (Invitrogen) and selected using 10 μg/ml blasticidin and 200 μg/ml Zeocin. Clones of cells were induced to express αS or LacZ by addition of 1 μm doxycycline. For the toxicity studies, M17 cells were induced to express the transgene (αS or LacZ) by treating with doxycycline for 3 d, followed by increasing concentrations of tunicamycin and thapsigargin (Sigma-Aldrich). Cell toxicity was assayed using the Cell Proliferation Kit II (XTT) (Roche). SH-SY5Y cell lines expressing mouse αS or βS were also used (Li et al., 2004b; Li and Lee, 2005).
Microsomes preparation and membrane floatation
For subcellular fractionation of ER membrane-enriched microsomes (Cox and Emili, 2006), fresh tissues were homogenized in a 1:10 (w/v) volume of lysis buffer with the following (in mm): 250 sucrose, 20 HEPES, 10 KCl, 1.5 MgCl2, 2 EDTA, protease-inhibitor mixture) using a Teflon pestle homogenizer. Initial homogenates were centrifuged at 1000 × g to remove nuclei and unbroken cells. The resulting supernatant was centrifuged at 10,000 × g to remove mitochondria and the postmitochondrial supernatant was centrifuged at 100,000 × g. The pellet was used as microsome fraction while the supernatant was used as pure cytosol. The microsome pellets were washed once with lysis buffer and resuspended in 100 μl of lysis buffer. To further enrich for the ER content, the microsome preparation were applied to a 0.2 m/0.8 m/2 m (v/v ratio 3:4:4) discontinuous sucrose gradient (Croze and Morré, 1984) and centrifuged at 90,000 × g for 2 h in a swinging bucket rotor (Sorvall TH-641). The interface between 0.8 and 2 m was collected, diluted with sucrose-free lysis buffer, and centrifuged at 110,000 × g for 45 min, and the final pellet was collected (ER-enriched microsome).
Pure mitochondria were obtained by discontinuous gradient fractionation of the crude mitochondria pellet (P10) (Vijayvergiya et al., 2005). The pellet was resuspended in lysis buffer and then layered on top of a chilled 7.5/10% (w/v) discontinuous Ficoll gradient. The samples were centrifuged at 24,000 rpm for 24 min at 4°C in a swinging bucket rotor (Sorvall TH-641). The purified mitochondrial pellet was resuspended in 200 μl of lysis buffer.
Pure nuclei were isolated starting from the crude nuclei pellet (P1) (Cox and Emili, 2006) using a sucrose gradient. Briefly, crude nuclei pellet were washed once and then resuspended in a 2 m sucrose solution made in sucrose-free lysis buffer. This pellet was then layered at the top of a 2 m sucrose gradient and spun in a swinging bucket ultracentrifuge at 80,000 × g for 35 min. After aspirating the supernatant, the pellet that includes pure nuclei was resuspended in the original lysis buffer.
For fractionation by membrane floatation (Ding et al., 2002), microsomes (P100) were resuspended in 0.42 ml of 60% iodixanol solution (Sigma-Aldrich) and overlayered with a discontinuous gradient containing 2.5 ml of 25% and 0.1 ml of 5% iodixanol. Samples were centrifuged at 200,000 × g for 2 h in a swinging bucket rotor (Sorvall TH-641) and the fractions were collected from the 5/25% (membrane-bound) and 25/60% (membrane-free) interfaces and analyzed.
Proteinase K protection assay
The microsome fractions were treated with or without 50 μg/ml proteinase K (PK; Roche) and 1% Triton X-100 for 20 min on ice. The reaction was stopped by the addition of 2 mm (final concentration) phenylmethylsulfonyl fluoride.
Immunoblotting and dot-blot analysis
Immunoblot and dot-blot analysis of mice and human brain tissues were performed as previously described (Lee et al., 2002; Li et al., 2004b; Li et al., 2005; Martin et al., 2006; Wang et al., 2008). For semiquantitative analysis of protein expression, the chemiluminescence signal associated with antibody binding (Pierce) was captured using the Bio-Rad Molecular Imager ChemiDoc XRS+ System or on x-ray films. The intensities of the immunoreactive bands were determined using the Quantity One software (Bio-Rad). For dot-blot analysis, lysates were spotted directly on the nitrocellulose membrane and allowed to dry completely. Immunoreactivity was visualized using chemiluminescence detection (Pierce) after incubations with the appropriate horseradish peroxidase-conjugated secondary antibody, using a CCD-based Bio-Rad Molecular Imager ChemiDoc XRS+ System or x-ray films. The intensities of the immunoreactive bands were determined by densitometry and the Quantity One software (Bio-Rad).
The following antibodies were used: grp78, grp94, proline disulfide isomerase (PDI), calnexin, syntaxin 6 (Stressgen); p-eIF2α, eIF2α, caspase-9, caspase-3, synaptotagmin (Cell Signaling Technology); syn-1, cytochrome c (BD Transduction Laboratories); ATF4, CHOP, p38, caspase-12, ubiquitin, cathepsin D (Santa Cruz Biotechnology); VDAC, NeuN, (Abcam); pser129-αS (Fujiwara et al., 2002); syn303 (Duda et al., 2000); and βS (Oncogene).
Immunoprecipitation
Immunoprecipitation was conducted using Seize X Protein G Immunoprecipitation Kit (Pierce) as previously described (Li et al., 2004a,b). Briefly, 1 μg of syn-1 or grp78 antibody was cross-linked using DSP 2 mm to protein G agarose and used for immunoprecipitation. Bound proteins were freed from the beads by SDS-sample buffer before fractionation by SDS-PAGE.
Immunohistochemistry and confocal microscopy
For immunohistochemical analysis, mice were fixed with 4% paraformaldehyde (PFA), serially frozen-sectioned (30 μm), and immunostained for DAB detection and for double immunofluorescence as previously described (Liu et al., 2008).
For the quantitative analysis of ER chaperones' expression in neurons accumulating αS abnormalities, fluorescence quantification of ER stress chaperones' signal was done using ImageJ software (National Institutes of Health). Mean values correspond to signal intensity of grp78/BIP or grp94 after subtraction of the nuclei fluorescence and normalized with the respective neuronal area. Values are expressed as the percentage of intensity of neurons in the same section that are syn303 or pS129-αS negative.
Immunoelectronic microscopy
Diseased A53T αS mice and nTg littermates were perfused with 4% PFA/0.1% glutaraldehyde. Brain and spinal cord (SpC) sections (50 μm) were stained with pS129-αS antibody as described above, labeled with 6 nm gold particle-conjugated secondary antibody, and embedded for electron microscopy. Samples were visualized using a Hitachi 7600 transmission electron microscope.
AAV2/6 plasmid construction, production, and titration
Human αS gene (GenBank accession no. NM_000345) carrying the A53T mutation was inserted in a pAAV-pgk-MCS-WPRE backbone modified from a pAAV-cmv-MCS (Stratagene), using standard cloning procedures. The noncoding pAAV-pgk-MCS-WPRE backbone was used to produce an empty control vector.
Recombinant pseudotyped AAV2/6 vectors were produced, purified, and titrated as described previously (Dusonchet et al., 2009). Briefly, we measured the integration of transcriptionally active transgene copies at 48 h in HEK293T cells and obtained the following titers: AAV2/6-pgk-αSyn-A53T-WPRE = 6.4 × 109 TU/ml, AAV2/6-pgk-MCS-WPRE = 1.5 × 1010 transducing units (TU)/ml.
AAV-rat model of A53TαS-dependent dopaminergic neurodegeneration
Female adult Sprague Dawley rats (Charles River Laboratories), weighing ∼200 g, were used in accordance with Swiss legislation and the European Community Council directive (86/609/EEC) for the care and use of laboratory animals. For stereotaxic injections, the animals were deeply anesthetized with a mixture of xylazine/ketamine and placed in a stereotaxic frame (David Kopf Instruments). Two microliters of viral preparation were injected in the right brain hemisphere using a 10 μl Hamilton syringe with a 34 gauge blunt tip needle connected to an automatic pump (CMA Microdialysis) at a speed of 0.2 μl/min. The needle was left in place for 7 additional minutes before being slowly withdrawn. The substantia nigra pars compacta (SNpc) was targeted at the following coordinates: anteroposterior, 5.2 mm; mediolateral, 2.0 mm relative to bregma; and dorsoventral, 7.8 mm relative to skull surface. For both the AAV2/6-pgk-αS-A53T-WPRE and AAV2/6-pgk-MCS-WPRE vectors, the total injected vector dose was 1.0 × 107 TUs.
Rats were treated with 1.5 mg/kg of Salubrinal (Alexis Biochemical) three times per week. The compound was solubilized in 5% DMSO mixed with chocolate milk. The rats were voluntarily drinking the Salubrinal solution from the tip of a 1 ml tuberculin syringe (Braun). Control animals were similarly treated with vehicle containing only DMSO.
Behavioral analysis of AAV-rat model
Cylinder test.
Spontaneous forelimb use during explorative behavior was assessed in the cylinder test (Schallert et al., 2000). Animals were placed in a transparent cylinder with a 20 cm diameter and movements were recorded using a video camera placed above the cylinder (5–15 min, depending on the level of animal activity). Paw touches on the cylinder wall were scored for each forelimb. Data are expressed as the percentage of left forelimb touches versus the total number of paw touches.
Drug-induced rotametry.
Apomorphine-induced rotational behavior was monitored for 40 min using automated rotameter bowls (Rotoscan, Rotometer v5.06; Omnitech) following subcutaneous injection of apomorphine-HCl (0.1 mg/kg) (Sigma). Animal rotational activity is expressed as the net number of counterclockwise turns per minute.
Quantitative neuropathological analysis of AAV-rat model.
Rats were killed and tissue processed for quantitative neuropathological analysis as described previously (Dusonchet et al., 2009).
Quantification of neurons displaying Golgi fragmentation
Coronal 40-μm-thick sections of the midbrain, immunostained for TH, αS, and GM130, were analyzed using a laser scanning confocal microscope (Zeiss LSM700). Golgi morphology was evaluated in the entire population of nigral TH-positive neurons expressing αS, across eight sections per animal, in a blind fashion (three animals per group). The Golgi apparatus was considered as “fragmented” when a completely scattered and punctuated GM130 signal was observed. When tubular structures were still present, the Golgi was considered to be in an intermediate stage of fragmentation and the cell was not attributed to any of the “normal” or “fragmented” populations.
Statistical analysis
All values are expressed as the mean ± SEM. Differences between means were analyzed by Student's t test and one- or two-way ANOVA with the Bonferroni's post test (Prism; Graph Pad Software). For survival curve log-rank (Mantel–Cox) test was used (Prism; Graph Pad Software).
For the rat AAV model, data are expressed as mean ± SEM. Statistical analysis of behavioral data were performed using one-way ANOVA (Statistica; StatSoft) followed by a Newman–Keuls post hoc test. Statistical analysis of striatal dopaminergic (DAergic) innervation and stereological quantification of the nigral TH-positive neurons was performed using a one-way ANOVA (Statistica; StatSoft) followed by an LSD Fisher post hoc test. Statistical analysis of Golgi fragmentation was performed using one-way ANOVA followed by a Tukey's post hoc test (Statistica; StatSoft).
Results
Increase in ER chaperones with disease in A53TαS Tg mice is not accompanied by activation of phospho-eIF2α
Cell culture studies implicate ERS in αS cell toxicity (Smith et al., 2005; Cooper et al., 2006). While PD is associated with signs of UPR (Hoozemans et al., 2007), a variety of antemortem and postmortem factors can activate UPR. Thus, it is not known if there are direct links between αS-dependent neurodegeneration and ERS in vivo. To address this issue, we analyzed an A53TαS Tg mouse model of α-synucleinopathy (Lee et al., 2002) for the presence of ERS/UPR activation.
First, we examined if A53TαS Tg mouse model exhibited increase in the expression of ER chaperones as grp94, grp78, and PDI. These markers are widely used indicators of ERS/UPR activation (Ron and Walter, 2007). Quantitative immunoblot analysis of pathologically affected regions [SpC and brainstem (BrSt)] show increased levels of grp78, grp94, and PDI with the progression of α-synucleinopathy (Fig. 1A,B). In SpC, increases in the ER chaperone levels were coincident with the onset of neurological abnormalities in the early symptomatic mice, which are characterized by a moderate wobbling gate (Martin et al., 2006). In addition, parallel analysis of BrSt from an end-stage A53TαS Tg mouse (Fig. 1C) shows significant increase in both grp94 (174 ± 4.1% of nTg, p < 0.01) and grp78 (165.3 ± 5.5% of nTg, p < 0.05) levels. The levels of ER chaperones in the cortex (Fig. 1C), a region with high levels of mutant αS expression without severe neuropathology, were comparable between the groups of mice. Consistent with the increased expression of ER chaperones, spinal cords of clinically affected mice show activation of the X-box-binding protein 1 (xbp1), a transcription factor involved in transcriptional induction of the ER chaperones at the early stage of disease process (Fig. 2A).
Figure 1.

Accumulation of ER chaperones with the onset of disease in A53TαS Tg mice. A, SpC lysates from A53TαS Tg mice at different disease stages and littermate nTg mice were immunoblotted for grp78, grp94, and PDI. The levels of ER chaperones in symptomatic Tg mice (Early and End stages) are clearly higher compared with the levels in the presymptomatic (PreS) or nTg animals. B, Quantitative analysis of various chaperone levels in Tg and nTg mice. The chaperone levels were normalized to the mean of nTg mice. All values represent mean ± SEM with n = 6 per group. ***p < 0.001, **p < 0.01, *p < 0.05 compared with the littermate nTg mice, Student's t test. C, Lysates from Brainstem, another area that develops pathology, and Cortex, a pathology-free region, from end-stage Tg and nTg mice were analyzed for grp78 and grp94. There is significant increase in the ER chaperones in the Brainstem (grp94, 174.07 ± 4.08% of nTg, p < 0.01; grp78, 165.31 ± 5.54 of nTg, p < 0.05; n = 6 per group) but not in Cortex. D, Levels of ER chaperones in the Spinal Cord of Tg mice (WT and A30PαS, 14–16 months old) that overexpress αS without pathology are not different from age-matched nTg mice. Also shown are α-tubulin (DM1A, α-tub) and Ponceau S staining (Pons) for controls. E–L, Immunohistochemical analysis of grp78 expression in nTg (E) and end-stage A53TαS Tg mice (F–L). Areas are SpC (E–G), Cbl (H, I), BrSt (J, K), and Ctx (L). Neurons exhibiting intense grp78 expression (denoted by asterisks) show abnormal morphology. Surrounding normal neurons show low basal levels of grp78 staining (arrowheads). Sections were counterstained with H&E. Scale bars, 10 μm.
Figure 2.

Alternative splicing of XBP-1 mRNA with the lack of phospho-eIF2α activation with α-synucleinopathy in the A53TαS Tg mice. A, Splicing of xbp1 mRNA mediated by the inositol-requiring enzyme, IRE1, leads to xbp1 mRNA variant encoding the active protein. RT-PCR analysis for xbp1 mRNA shows the presence of alternatively spliced variant (S) in the SpC from early-stage G2–3(A53T) mice but only the unspliced variant (U) is present in SpC from nTg mice. Also shown are control (C) and tunicamycin-treated (T) N2A cells. B, Western blot analysis for phospho-eIF2α (p-eIF2α) and total eIF2α (eIF2α) expression in SpC from mice at various disease stages. Also shown are the α-tub (DM1A) blots used for controls. The numbers below show the ratios of p-eIF2α/total eIF2α, as a percentage of nTg levels. Values are mean ± SEM (n = 6). C, Western blot analysis for phospho-eIF2α (p-eIF2α), total eIF2α (eIF2α), ATF4, and CHOP in A53TαS Tg and G37R-SOD1 Tg mice. Both mice were at end stage (paralyzed).
To further establish that induction of ER chaperones and UPR activation occurs with the presence of αS-associated neuropathology rather than simple interaction between aging and/or nonpathologic αS overexpression, we examined the expression of the ER chaperones in the αS overexpressing Tg mouse lines that do not develop neuropathology [line O2(A30P) and line I2–2(WT) (Lee et al., 2002)]. The ER chaperone levels in SpC of aged (14 months) A30P mice and WT αS Tg mice were not different from the nTg littermates (Fig. 1D). Combined with the fact that the ER chaperone levels in the cortex of end-stage A53TαS Tg mice, these results show that the onset of α-synucleinopathy and neurological abnormalities are intimately linked to the presence of ERS in brain. While the studies of using simpler systems (Cooper et al., 2006; Gitler et al., 2008) predicted that high levels of αS expression alone would be sufficient to cause ERS response, in mammalian brain, overt α-synucleinopathy and/or neurodegeneration appears to be a prerequisite for the induction of ERS.
In addition to the transcriptional induction of ER chaperones, UPR also involves general inhibition of protein translation during ER stress to reduce demand on the cell-folding machinery where the phosphorylation of the translation initiation factor, eIF2α, is thought to arrest general protein translation (Ron and Walter, 2007). Studies indicate that in cultured cells, phosphorylation of eIF2α is important for maintaining cell viability during chronic ER stress conditions (Boyce et al., 2005). Analysis of the A53TαS Tg mice for the phosphorylated eIF2α (p-eIF2α) show that α-synucleinopathy was associated with increased levels of phospho-eIF2α. However, because the total eIF2α was also elevated with α-synucleinopathy, the relative levels of p-eIF2α do not change with the progression of α-synucleinopathy (Fig. 2B). Consistent with the lack of increased p-eIF2α levels, analysis for CHOP and ATF4, which are selectively translated by p-eIF2α (Ron and Walter, 2007), show that these components are not induced with the onset of α-synucleinopathy (Fig. 2C). Absence of p-eIF2α activation is not due to limitations in documenting the activation of this pathway in an in vivo model system since we are able to clearly show the increases in the levels of p-eIF2α, ATF4, and CHOP in the end-stage G37R mutant superoxide dismutase-1 (SOD1) Tg mice (Fig. 2C), where the disease is associated with activation of UPR (Saxena et al., 2009). Since the phosphorylation of eIF2α is thought to protect cells from cell death induced by ERS (Boyce et al., 2005), our results indicate that the conditions in the A53TαS Tg mice are favorable for the activation of ERS-induced cell death cascade.
ER chaperones are induced in neurons with αS pathology in A53T mice
Upon establishing the specificity of ER stress with disease, we examined the cellular localization of the grp78 expression in brains of A53TαS Tg mice in relation to α-synucleinopathy. In nTg mice (Fig. 1E) and in cortical neurons (Fig. 1L), neuronal grp78 staining is sparsely distributed with punctuate perinuclear staining. In the end-stage A53TαS Tg mice, a subset of neurons in the areas affected by α-synucleinopathy, including deep cerebellar nuclei (Cbl), BrSt, and SpC, were highly reactive for grp78 (Fig. 1F–K). Moreover, the neurons with increased grp78 expression showed abnormal morphology (such as hypertrophy, atrophy, and/or dystrophic neurites) while the neighboring neurons with a normal morphology exhibited lower levels of grp78 immunoreactivity (Fig. 1F). These results indicate that UPR occurs in neurons that are pathologically affected.
To further link α-synucleinopathy with the presence of ERS, we asked whether the ER chaperone induction in the A53TαS Tg mice occurs in neurons with αS pathology. The αS abnormalities were documented using either syn303 or the anti-pS129-αS antibody and the ER chaperone expression was documented using antibodies to grp78 or grp94 (Ron and Walter, 2007). Confocal double-immunofluorescence microscopy reveals neurons in the affected areas and A53TαS Tg mice exhibiting the abnormal perikaryal and neuritic reactivity to syn303 or anti-pS129-αS (Fig. 3). While all neurons expressed ER chaperones as expected, neurons with abnormal αS generally exhibited stronger grp78/94 immunoreactivity (Fig. 3A,B,D, asterisks). To confirm our qualitative observations, the intensity of grp78- or grp94-associated immunofluorescence was quantified in cells with and without abnormal αS immunoreactivity. The results show that compared with ER chaperone levels in normal neurons within the same sections, neurons with abnormal αS (either syn303+ or pS129-αS+) exhibited higher levels of ER chaperones (Fig. 3F).
Figure 3.
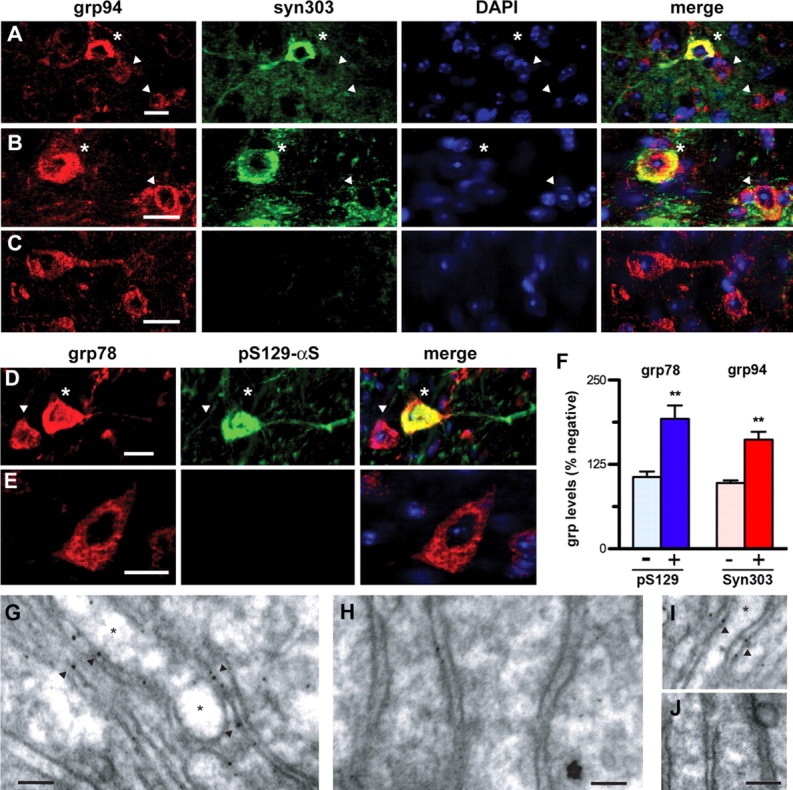
Induction of ER chaperones occurs in neurons with αS pathology. A–E, Confocal immunofluorescence colocalization of grp78 or grp94 with abnormal αS. SpC (A, D), BrSt (B, C), and Ctx (E) sections from end-stage A53TαS Tg mice (A, B, D, E) and nTg (C) mice were double-immunostained for grp94/syn303 (A–C) or for grp78/grp94 (KDEL)/pS129-αS (D, E). Nuclei were stained with DAPI. Neurons showing abnormal accumulation of syn303 or anti-pS129-αS are also associated with increased immunoreactivity for grp78/grp94 (asterisks). Neighboring neurons lacking abnormal αS show lower basal grp78/grp94 immunoreactivity (arrowheads). F, Quantitative analysis obtained using ImageJ software (National Institutes of Health) of neurons with αS abnormalities (stained for syn303 or pS129-αS) in BrSt and SpC of end-stage A53TαS Tg mice shows increased ER stress chaperones (grp78 for pS129-αS, grp94 for syn303). Values are mean ± SEM; n = 13 each for pS129-αS- and syn303-positive neurons and n = 25–31 for negative neurons. **p < 0.001, Student's t test. G–H, Immunogold labeling of αS aggregates shows αS aggregate association with the ER membrane in the diseased A53TαS mice. SpC sections from end-stage A53T (G, I) and nTg (H, J) were immunostained for pS129-αS and processed for electron microscopy. In neurons the pS129-αS-associated gold particles were associated with the ER membranes (arrowheads). No labeling was seen in nTg samples (H, J). The ER cisternae in diseased A53TαS Tg mice were often severely dilated (asterisks) while the ER in nTg controls showed a normal morphology. Scale bar, 50 nm.
Finally, immunoelectron microscopic analysis of diseased A53TαS Tg mice shows that a subset of pS129-αS reactivity localizes on the ER membranes (Fig. 3G,I). More important, the ER morphologies in these neurons were highly abnormal with severely dilated ER cisternae, an ultrastructural indication of ER dysfunction in the A53TαS Tg mice (Fig. 3G–J). Collectively, these results show that the ERS/UPR activation in A53TαS Tg mice is selective for neurons exhibiting αS pathology and the ER membranes show abnormal morphology in these neurons.
αS and αS aggregate are associated with ER/M fraction
Studies indicate that αS can functionally impact multiple organelles (Gosavi et al., 2002; Smith et al., 2005; Cooper et al., 2006; Martinez-Vicente et al., 2008; Winslow et al., 2010). Given the colocalization of α-synucleinopathy with ER chaperone activation and abnormal ER morphology (Fig. 3), ERS could be caused by direct effects of αS or αS aggregates on ER. As an initial test of this hypothesis, we examined whether αS can biochemically cofractionate with the microsomes. We found that αS and αS aggregates indeed copurify with the microsomes (Fig. 4). Significantly, microsomal αS was found in both Tg and nTg mice (Fig. 4B,C), as well as in human brain (Fig. 4C), indicating that αS associates with ER under normal conditions. Association of αS with ER is highly selective and is not related to the simple membrane-binding properties of synucleins since βS, which also interacts with membranes (Narayanan and Scarlata, 2001), is not associated with the ER/M fractions (Fig. 4B,C). Lack of βS in the ER/M fractions from Tg mice is not because of competition by high levels of αS, since βS does not associate with the microsomes in nTg mice and when overexpressed in SH-SY5Y cells (Fig. 4C). We also performed PK protection assay to determine whether microsomal αS is bound to the membrane surface or translocates into the microsomes. Our studies show that the bulk of microsomal αS are resistant to PK (Fig. 4D). This indicates that αS is located within the lumen of microsomes and not merely attached to the membrane surface.
Figure 4.

Accumulation and aggregation of αS in the ER. A, Schematic representation of the protocol used for subcellular fractionation. B, Various subcellular fractions, as shown in (A), from presymptomatic A53TαS mice were analyzed for organelle markers: calnexin (CLN) for ER, synaptotagmin (ST) for synaptic vesicles, syntaxin 6 (STX) for Golgi, cathepsin D (CTS D) for lysosomes, VDAC for mitochondria, and p38 for cytosol. Parallel analysis for αS and βS shows ER-associated αS but not βS. C, αS, but not βS, accumulates within the microsome fraction. ER/M (P100) and other intermediate fractions from nTg mice, human pons, and SH-SY5Y cell lines stably transfected with αS or βS were analyzed for αS/βS and organelle markers. In all cases, αS, but not βS is associated with ER/M fraction. Note that even with the overexpression of βS in the SH-SY5Y cell lines, βS does not associate with ER/M fractions (P100). D, ER-associated αS monomers in nTg, A53TαS Tg, and WTαS Tg mice are protected from proteolysis by PK, indicating that αS is in the ER lumen. In the presence of 1% TX-100, both αS and the ER-resident grp78 are completely proteolyzed by PK. Identical results were obtained with A30PαS mice. E, Accumulation of αS aggregates with ER-enriched microsomes fractions. One microgram of pure organelle fractions were also evaluated using organelle-specific markers: calnexin (CLN) for ER, synaptotagmin (ST) for synaptic vesicles, syntaxin 6 (STX) for Golgi, NeuN for nuclei (N), cytochrome c (CytoC) for mitochondria (M), βS for cytosol (C). Total SDS-soluble (T) and initial ER/M (P100) fractions are also shown. The gradient-enriched ER fraction is free from other organelle markers but is enriched for ER markers. The ER fraction also shows significant enrichments in αS monomer and the presence of SDS-stable high molecular weight αS aggregates (arrowheads). Quantitative data show that αS monomer is significantly enriched in the ER fractions, compared with M and N. All organelles were fractionated from SpCs of end-stage A53TαS Tg mice. Values are mean ± SEM (n = 3–4). *p < 0.05, **p < 0.01, one-way ANOVA. F, Microsomes from end-stage A53TαS Tg mice show accumulation of pS129 αS and oligomeric αS (arrowheads). Equal amounts of microsomes obtained from SpC of nTg and A53TαS Tg mice were immunoblotted for pS129 αS and total αS. Asymptomatic, (PreS); end stage, (Sick). G, Membrane floatation analysis of microsomes and detergent-insoluble αS. Microsome-associated αS from end-stage A53TαS Tg mice (Sick) and age-matched A30PαS Tg mice shows that αS floats with membranes. Lanes are input microsomes (P100), membrane fractions (M), and free (F) fractions from the floatation gradient. The detergent-insoluble αS aggregates (P-Tx) do not float with the M fractions. Arrowheads are high molecular weight aggregates of αS.
Subcellular fractionation of symptomatic A53TαS Tg mice reveals that higher molecular weight αS and pSer129-αS are enriched in ER/M fraction (Fig. 4E,F), indicating that ER could be directly affected by αS pathology. However, because αS aggregates can be pelleted by centrifugation, cofractionation of αS with ER/M could represent a fortuitous cosedimentation. To control for this possibility, we used the membrane floatation assay (Ding et al., 2002) to determine whether the αS aggregates “float” with the ER/M membranes on a density gradient. Analysis of the “membrane” and the “free” fractions obtained following the gradient centrifugation of the ER/M preparations from SpC show that both monomer and aggregated αS were recovered with the membranes along with the ER marker, calnexin (Fig. 4G). Microsomal αS monomers from A30P (Fig. 4G) and nTg mice (data not shown) are also recovered with the membranes in this assay. In contrast, total Triton X-100-insoluble αS aggregates (TX-Ptot) isolated from SpC of end-stage A53T mice are recovered in the free fraction and not in the membrane fraction (Fig. 4G). Thus, intact membranes are required for αS aggregates to float on the density gradient. Collectively, our results confirm that significant fraction of αS aggregates are actively bound to the microsomes.
αS accumulates in ER with disease, binds to ER chaperones, and αS overexpression sensitizes neuronal cells to ERS-induced toxicity
Since both αS and αS aggregates associate with ER/M, we asked whether quantitative changes in the microsomal αS levels correlate with the development of disease in the brain. In presymptomatic Tg mice, the level of ER/M αS was directly proportional to the total level of αS expression. Thus, mice from all Tg lines showed increased levels of ER/M αS compared with nTg mice (Fig. 5A). However, in the A53TαS Tg mice, the relative abundance of microsomal αS increases with disease progression in the pathologically affected areas (SpC, BrSt) (Fig. 5A,B). In particular, while the BrSt/SpC express lower levels of total αS than the cortex (Ctx), the microsomal αS is significantly higher in BrSt/SpC than the Ctx (Fig. 5B). Thus, there is a selective ER/M accumulation of αS in the areas that are vulnerable to α-synucleinopathy. Significantly, the analysis of ER/M fractions prepared from human PD cases also show that the level of microsomal αS is significantly higher in the PD cases compared with the non-PD controls (Fig. 5C).
Figure 5.

ER/M αS increase with the progression of α-synucleinopathy sensitizes neural cells to ERS-induced toxicity. A, Immunoblot analysis of αS levels in microsomes (ER/M) and total SDS-soluble lysates (Tot) obtained from different mouse lines. The samples were obtained from spinal cord. The graph shows the quantitative analysis of the immunoblots where the relative levels of microsomal αS normalized to total αS (mean ± SD, n = 3–4), *p <0.05, one-way ANOVA. B, Immunoblot analysis of αS levels in microsomes (ER/M) and total SDS-soluble lysates (Tot) obtained from different brain regions of end-stage A53TαS Tg mice. The graph shows the relative levels of microsomal αS normalized to total αS (mean ± SD, n = 3–4), **p < 0.01 (Ctx vs SpC), one-way ANOVA. C, Immunoblot analysis of αS levels in microsomes (ER/M) and total SDS-soluble lysates (Tot) obtained from pons of human PD cases and controls. The graph shows the levels of microsomal αS, normalized to the levels in the controls (C, 100%). n = 5, **p < 0.001, Student's t test. D, αS interacts with ER luminal chaperones grp78 and grp94. Immunoprecipitation with syn-1 antibody (IP: αS) or anti-grp78 antibody (IP:grp78) was performed on purified ER/M from nTg, asymptomatic A53TαS Tg (PreS), and end-stage A53TαS Tg (End) mice. Immunoprecipitates' pellets were probed with antibody for grp78 and grp94 (KDEL) and NAC-1 for αS. E, Doxycyclin (Dox)-dependent induction of αS expression in BE(2)-M17 cell lines. Lanes, LacZ (LZ); WTαS (WT); and A53TαS (AT). F, Immunoblot analysis of ER stress markers in M17 cells treated with tunicamycin (Tn) 12.5 ng/ml or thapsigargin (Tg) 5 nm for 24 h. Cells were induced for transgene expression for 3 d before treatment. C, control. G, Inducible αS BE(2)-M17 cells lines were induced for 3 d before treatment with increasing concentration of Tn (12.5 or 50 ng/ml) or Tg (1.25 or 5 nm) for 24 h. Cell survival was tested with XTT-based colorimetric method. The values are normalized to the control untreated cells and are mean ± SD (n = 4). **p < 0.001, two-way ANOVA.
Given the presence of αS and αS aggregates in the ER lumen, we asked whether αS promotes activation of ERS via competing for binding to ER chaperones. We used the ER/M fractions to determine whether αS can coimmunoprecipitate grp78 and grp94. Our results show that immunoprecipitation of microsomal αS also recovers grp78 and grp94 (Fig. 5D). Similarly, immunoprecipitation of grp78 leads to recovery of αS (Fig. 5D). Moreover, interaction of αS with ER chaperones occurs in both asymptomatic and symptomatic Tg mice indicating that ER chaperones normally interact with αS monomers (Fig. 5D). However, the interaction between αS and ER chaperones could be favored with an increase in the microsomal αS levels, as we were not able to consistently demonstrate interaction of endogenous mouse αS with ER chaperones (Fig. 5D).
If increased microsomal αS leads to increased interactions between αS and ER chaperones, increased αS expression may also sensitize neuronal cells to ERS-induced toxicity. Such would be significant for age-related vulnerability to α-synucleinopathy as increased ERS is likely associated with aging and/or environmental toxin exposure. Thus, we examined if increased expression of αS could affect the vulnerability of neuronal cells to inducers of ERS. We used M17 cell lines that show doxycycline-inducible expression of WT HuαS, A53T HuαS, or LacZ (Fig. 5E). Following the transgene induction, the cells were exposed to increasing concentration of ER stressors (tunicamycin or thapsigargin) for 24 h. Analysis of the cells for ERS markers in the M17 cell lines show that, even without exogenous ER stressors, overexpression of αS is sufficient to cause modest basal activation of grp78. With the exogenous ER stressors, αS expressing cells show higher induction of grp78 compared with LacZ controls (Fig. 5F). Significantly, analysis of phospho-eIF2α shows that the basal αS-dependent induction of grp78 is not associated with increased phospho-eIF2α. Moreover, despite the greater induction of grp78 following ER stressor to HuαS-expressing cells, the levels of phospho-eIF2α following ERS was comparable in all cells (Fig. 5F). Analysis of cell survival shows that increased expression of either WT or A53T HuαS leads to increased vulnerability of the M17 cells to ER stressors (Fig. 5G). In addition, we observe that human catecholaminergic neuroblastoma lines (SH-SY5Y) stably expressing HuαS (Li and Lee, 2005) are also more sensitive to cell death caused by ER stressors (data not shown). These results show that in cultured cell lines, overexpression of either WT or mutant αS can reliably cause modest levels of ERS and sensitizes cells to ER stress. Combined with the induction of αS pathology, as with the expression of A53T mutant, in vivo, ER/M-associated αS likely contributes to neurodegeneration.
Activation of ER caspase and ER accumulation of polyubiquitin in A53TαS Tg mice
The above results indicate that α-synucleinopathy in A53TαS Tg mice is associated with chronic ERS and overexpression of αS sensitizes neural cells to ER stressors. Combined with the presence of abnormal ER morphology and lack of increase in phospho-eIF2α, the conditions in the mice could promote the activation of cell death pathways. Thus, we examined whether the activation of ERS-associated caspase activation, such as cleavage/activation of caspase-12 in rodents (Zhang and Kaufman, 2006), occurs in the diseased A53TαS Tg mice. Our analysis shows that α-synucleinopathy is associated with the increased cleavage of caspase-12 and other downstream caspases (Fig. 6A). The activation of caspase-12 is selective for α-synucleinopathy since analyses of presymptomatic and pathology-free region (cortex) do not show accumulation of caspase-12 (Fig. 6B,C).
Figure 6.

ER accumulation of polyubiquitin, and ER caspase activation with α-synucleinopathy in the A53TαS Tg mice. A, Immunoblot analysis of total SpC lysates from the end-stage A53TαS Tg mice and nTg littermates. Neuropathology in the A53TαS Tg mice is clearly associated with cleavage of ER-dependent caspase-12 and its downstream effectors, caspase- 9 and -3 (pro, pro-peptide; Δ, cleaved). Both pro-peptide and cleaved peptide were detected on the same blot but cropped to save space. The bar graph is the quantitative analysis of blots shown. The amount of cleaved products was normalized to the full-length protein and represents mean ± SEM (n = 4). ***p < 0.001, **p < 0.01, Student's t test, Tg versus nTg littermates. B, C, Caspase-12 activation is selective for α-synucleinopathy. (B) Caspase-12 immunoblot showing cleavage (Δ) of caspase-12 propeptide (pro) in end-stage (End) Tg mice compared with presymptomatic (PreS) mice. (C) Caspase-12 analysis of affected areas (SpC) compared with disease-free regions (Ctx) from end-stage Tg and nTg mice. Note that the cleaved caspase is associated with the presence of end-stage disease. D, Ubiquitin immunoblot of total SDS-soluble fraction (Tot), Triton X-100-soluble (S-Tx), and Triton X-100-insoluble pellets (P-Tx) from SpC of end-stage A53TαS Tg mice and nTg littermates. The graph shows quantitative analysis. Mean ± SEM (n = 6), *p < 0.05, Student's t test. E, Ubiquitin immunoblot of microsome fractions from nTg and A53TαS Tg mice at various stages: 6 months old, 10 months old, presymptomatic stage (PreS), and symptomatic stages (Early, End). Densitometry analysis of immunoblots reveals the dramatic ER accumulation (30- to 40-fold increase) of polyubiquitin with α-synucleinopathy. In contrast, total tissue increase in polyubiquitin is only 2- to 3-fold (D). The plots are mean ± SEM of percentage of nTg average (n = 6). ***p < 0.001, **p < 0.01, one-way ANOVA. Both Early- and End-stage Tg mice are also different from PreS (p < 0.05 and p <0.001).
Previous studies indicate that overexpression of αS can cause ubiquitin–proteasome system (UPS) stress (Smith et al., 2005), and proteasome inhibition can cause abnormal UPR characterized by attenuated PERK-dependent phosphorylation of eIF2α (Nawrocki et al., 2005). Thus, we asked if α-synucleinopathy in mice was associated with signs of UPS stress (i.e., accumulation of polyubiquitinated proteins) to the ER. Analyses of unfractionated SpC extracts show that the disease in the symptomatic A53TαS Tg mice is associated with moderate (∼2- to 3-fold) increase in the levels of polyubiquitin in various extracts (Fig. 6D). However, when the ER/M fractions were analyzed for the polyubiquitin levels, ER/M from the symptomatic A53TαS Tg mice showed a more dramatic increase (∼20- to 40-fold) in the polyubiquitin levels (Fig. 6E). Moreover, parallel analyses of ER/M from A53TαS Tg mice at different disease stages show a progressive increase in polyubiquitin levels with the disease progression (Fig. 6E). These results suggest dysfunctional ER and abnormal ER-associated protein degradation (ERAD) with α-synucleinopathy. While additional studies are required to fully evaluate ERAD and UPS stress in α-synucleinopathy, our results suggest that α-synucleinopathy is associated with multiple markers of ER dysfunction.
Anti-ERS compound, Salubrinal, extends life span and reduces ER accumulation of αS in A53TαS Tg mouse model of α-synucleinopathy
The above studies show there are temporal and spatial associations between αS abnormalities, chronic UPR, and neurodegeneration. However, it will be important to show if the aspects of α-synucleinopathy-linked chronic ERS documented here are mechanistically linked to the onset and/or progression of α-synucleinopathy in vivo.
To determine whether chronic ERS-related toxicity is mechanistically linked to the onset and/or progression of disease in vivo, we treated cohorts of A53TαS Tg mice (line G2–3) with Salubrinal, a compound known to protect cells from chronic ER stress by inhibiting dephosphorylation of eIF2α (Boyce et al., 2005). Salubrinal has been shown to partially attenuate PC12 cells from A53T αS-dependent toxicity (Smith et al., 2005) and to extend the life span of G93A-SOD1 Tg mouse model of motor neuron disease by ∼20 d (∼20%) (Saxena et al., 2009). Thus, if chronic ER stress is an active and necessary participant in α-synucleinopathy, Salubrinal could attenuate the disease manifestation in mice. Moreover, the lack of p-eIF2α induction in the A53TαS Tg mouse model provides further rationale for using Salubrinal. Since the A53TαS Tg mouse model used here is one of the few models of fatal α-synucleinopathy, as it would be in humans, the life span was used as the primary outcome measure for the potential therapeutic effects of Salubrinal on α-synucleinopathy. To minimize any adaptive effects of treatment, Salubrinal treatment was initiated at 12 months of age. At this age, ∼20% of A53TαS Tg mice cohorts in various groups had developed the disease at the same rate (Fig. 7A). However, following the initiation of Salubrinal treatment, the rate of disease onset in the Salubrinal group was obviously slower than the control cohort (Fig. 7A).
Figure 7.

Salubrinal delays the onset of motoric dysfunction in the A53TαS Tg mouse model of α-synucleinopathy. A, Survival curve of A53TαS mice (line G2–3) treated with Salubrinal (Sal, n = 11) or vehicle (Veh, n = 10) and historical controls (hC, n = 23) show that Salubrinal delays the disease manifestation in the A53TαS mice (p < 0.01, Sal vs Veh; p < 0.05, Sal vs hC; not significant, hC vs Veh; log-rank test). The hC represents the life span of all A53TαS mice followed in the lab within the 1 year before treatment. The survival fractions between the hC and Veh mice were comparable, indicating that mouse handling did not influence their life span. The bar indicates the duration of the Salubrinal or vehicle treatments. B, C, Immunoblot analysis of total SpC lysates from the end-stage A53TαS mice treated with the vehicle (Sal−) or Salubrinal (Sal+). Among the four Salubrinal-treated mice analyzed, one mice showed induction of p-eIF2α (B). In other mice, Salubrinal treatment was only associated with the accumulation of CHOP, a downstream reporter for p-eIF2α (C). The densitometric analysis of immunoblots shows induction of CHOP. Values (mean ± SEM) are the percentage of average Sal− levels (n = 3). *p < 0.05, Student's t test. D, αS immunoblot of microsomal proteins (ER/M) and the total SDS-soluble proteins from SpC of vehicle (Sal−)- and Salubrinal (Sal+)-treated A53TαS mice. Arrowheads are high molecular weight aggregates of αS. The graph shows the quantitative analysis of the immunoblots. The ER/M fractions from Salubrinal (Sal)-treated A53TαS mice shows a significant decrease in αS while total αS is unchanged. Values are mean ± SEM (n = 3), *p < 0.05, Student's t test.
Analysis of brain extracts from Salubrinal- and vehicle-treated mice shows that while Salubrinal treatment did not consistently lead to increase in p-eIF2α levels (Fig. 7B,C), there was consistent and significant increase in the levels of CHOP expression (Fig. 7B,C), a downstream reporter of p-eIF2α (Ron and Walter, 2007).
To determine whether the Salubrinal treatment directly impacts αS expression or development of αS abnormalities, the brain lysates were analyzed for αS levels. The results show that the levels of total SDS-soluble αS were not affected by the Salubrinal treatment (Fig. 7D), confirming that Salubrinal did not simply reduce overall αS expression. However, Salubrinal treatment was associated with significantly reduced microsomal accumulation of monomeric and oligomeric αS (Fig. 7D). Moreover, our companion analysis for toxic αS oligomers shows that Salubrinal treatment attenuates the accumulation of toxic αS oligomers (Colla et al., 2012).
While Salubrinal treatment delayed the onset of motoric symptoms, Salubrinal treatment did not attenuate the progression of the disease following onset [16.7 ± 3.4 d (vehicle) compared with 17.4 ± 2.7 d (Salubrinal)]. Thus, immunocytochemical analysis of end-stage Tg mice for the accumulation of pSer129-αS or other neuropathology did not reveal obvious differences between the Salubrinal- and vehicle-treated mice. Thus, Salubrinal may provide protection from α-synucleinopathy by selectively decreasing the ER accumulation of αS and αS oligomers. This result also supports the view that microsomal αS oligomers are of pathologic importance. Moreover, our results suggest that anti-ER stress compounds, such as Salubrinal, should be developed as a therapy for α-synucleinopathy.
Salubrinal attenuates disease manifestation and decreases Golgi fragmentation in the AAV-rat model of A53TαS-dependent DAergic neurodegeneration
Because A53TαS Tg mice lack robust DAergic pathology, we used an AAV-transduced rat model to ask whether Salubrinal could also attenuate DAergic neurodegeneration following the overexpression of A53T HuαS in rat SNpc DA neurons. Unilateral injections of the AAV2/6-pgk-αS-A53T-WPRE vector in the rat SNpc achieve widespread expression of HuαS in DA neurons and a progressive degeneration of SNpc neurons (Kirik et al., 2002; Lo Bianco et al., 2002; Koprich et al., 2010) (Fig. 8A). To investigate whether Salubrinal protects neurons from A53TαS-induced neurodegeneration, the rats were administered either Salubrinal or vehicle starting at 1 week post AAV injection and evaluated at 12 weeks post AAV injection. Initial immunocytochemical analysis shows that Salubrinal treatment did not have an obvious, if any, effect on the expression of HuαS in SNpc (Fig. 8B). During the treatment, the animals were monitored for apomorphine-induced rotational behavior and spontaneous motor asymmetry (Fig. 8C,D).
Figure 8.

Salubrinal attenuates disease manifestation without protecting from neuronal loss in the A53TαS AAV model of DAergic neurodegeneration. A, AAV-pgk-αS-A53T stereotactic injection leads to overexpression of αSyn-A53T in the DAergic neurons of the SNpc. Shown are a wide-field and a higher magnification image of representative rat midbrain showing the overexpression of αS(A53T) across the SNpc (blue). TH-positive neurons are green. Scale bar, 10 μm. B, Salubrinal treatment does not change the level of αS overexpression in the SNpc of rats. Top, Representative low-magnification images of the injected rat SNpc showing αS overexpression in animals treated with either Salubrinal or vehicle (DMSO). Bottom, Representative high-magnification images of nigral neurons showing the overexpression of αS. Scale bars: 500 μm (top); 100 μm (bottom). C, D, Spontaneous motor behavior measured by forepaw touches in the cylinder test (C) and apomorphine-induced rotational behavior (D). n = 7 for all groups except the αSyn + DMSO group (n = 6); one-way ANOVA followed by Newman–Keuls post hoc test; *p < 0.05. ns, not significant. E, Representative sections of the SNpc (top), and striatum (bottom) immunostained for TH and illustrating nigrostriatal degeneration in all tested experimental conditions. F, Stereological analysis reveals that Salubrinal administration does not reduce the significant loss of TH-positive neurons (SN). Similarly, densitometric analysis does not reveal any change in the loss of TH immunoreactivity in the striatum (STR) induced by αS-A53T. Results are expressed as the percentage of loss of SNpc TH-positive neurons or TH immunoreactivity, when compared with the noninjected side, 12 weeks after injection. n = 7 for all groups except the αSyn + DMSO group (n = 6); one-way ANOVA followed by Fisher LSD post hoc test, *p < 0.05, ***p < 0.001.
The A53TαS vector-injected rats progressively developed signs of asymmetric motor behavior. In the cylinder test, the left paw contralateral to the injected SNpc was persistently impaired at both 6 (23.7 ± 2.4% of total touches) and 12 weeks (18.7 ± 7.1%) post injection (Fig. 8C). Salubrinal administration significantly attenuated the progression of the motor deficit, particularly at 6 weeks (35.6% ± 3.7% of total, p < 0.05) following injection. Measurement of apomorphine-induced rotations at 12 weeks post injection revealed a similar attenuation of motor abnormalities by Salubrinal (Fig. 8D). Specifically, while the vehicle-treated, A53TαS vector-injected rats showed significant rotational bias compared with the control rats, Salubrinal-treated rats were not significantly different from the controls (Fig. 8D). However, comparisons of Salubrinal- and vehicle-treated groups did not reach statistical significance.
While Salubrinal attenuated the progressive motor abnormalities, the behavioral amelioration by Salubrinal treatment is not reflected in the attenuation of DAergic neurodegeneration (Fig. 8E,F). This raises the possibility that Salubrinal treatment does not prevent the death of DA neurons, but allows remaining neurons to be more functional. To examine this issue, we evaluated the integrity of Golgi apparatus in DA neurons. Fragmentation of the Golgi apparatus is considered an early event preceding neuronal death in response to ER stress (Nakagomi et al., 2008) and has been reported to occur in vivo in conditions of αS expression (Gosavi et al., 2002). Thus, we hypothesized that Golgi fragmentation could provide a sensitive marker of A53TαS-induced ER-stress/toxicity in DA neurons, and may reveal the protective effects of Salubrinal treatment. We performed analysis of Golgi morphology in the DA neurons of the SNpc at 12 weeks post injection using the cis-Golgi matrix protein marker GM130 (Fig. 9A). Based on the morphology of GM130-positive structures, neurons were classified as “normal” or “fragmented” (Fig. 9A). In the animals injected with the control vector, very little Golgi fragmentation was seen (∼0.9%) with >97% being normal (Fig. 9B,C). The expression of A53TαS led to fragmented and normal Golgi in 16.5 ± 1.0% and 67.3 ± 4.1% of DA neurons, respectively (Fig. 9B,C). Remarkably, the Salubrinal treatment significantly reduced the proportion of DA neurons with a fragmented Golgi to 4.9 ± 0.6% and increased the number DA neurons with a normal Golgi to 85.9 ± 2.7% (Fig. 9B,C). Remaining neurons (∼16% in Vehicle group and ∼10% in Salubrinal group) were at intermediate states of Golgi morphology and were not classified.
Figure 9.

Salubrinal decreases Golgi fragmentation in the A53TαS AAV model DAergic neurodegeneration. A, Confocal images of Golgi apparatus (GM130 staining in green) in αS overexpressing neurons (αS staining in blue) in the rat SNpc. A well organized network of fine tubules around the nucleus was considered as “normal,” while a fine punctuated distribution of the GM130 staining was defined as “fragmented.” Other neurons belong to various intermediate stages of fragmentation. B, C, Analysis of Golgi morphology in DA neurons 12 weeks following AAV injection. Salubrinal administration significantly reduces fraction of neurons with fragmented Golgi (B) and increases DA neurons with normal Golgi apparatus (C). Thus, Salubrinal protects the Golgi apparatus in nigral DAergic neurons from αSyn-A53T-induced fragmentation. n = 3 per group, >400 cells/animal; one-way ANOVA followed by Tukey's post hoc test, **p < 0.01, ***p < 0.001. Scale bar, 5 μm.
These results indicate that A53TαS toxicity involves disruption of Golgi morphology in the surviving DA neurons at 12 weeks post AAV injection, and the Salubrinal treatment attenuates the Golgi fragmentation in surviving DA neurons. However, Salubrinal cannot prevent the initial loss of DA neurons caused by A53TαS. This latter fact is not surprising as A53TαS, when expressed at sufficient levels, can activate multiple cell death pathways (Smith et al., 2005; Martin et al., 2006).
Discussion
Our results provide the first direct evidence for pathological involvement of chronic ERS in a vertebrate model of α-synucleinopathy. Moreover, we show that UPR associated with α-synucleinopathy in brain is abnormal since the induction of ER chaperones is not accompanied by the increase in p-eIF2α. The onset of ERS and disease in the A53TαS Tg mice coincides with the accumulation of aggregated αS with ER microsomes and ERAD defect. More important, activation of ER-associated caspases and attenuation of disease manifestations by ER stress-protective compound, Salubrinal, demonstrate that chronic ERS is an active participant in onset/progression of α-synucleinopathy. Our results indicate that reducing the chronic ERS may represent an important disease-modifying therapeutic approach for PD and other α-synucleinopathies.
Based on the current results together with our companion report showing the genesis and evolution of toxic αS oligomers within the ER, we propose a model (Fig. 10) where a small fraction of αS normally locates to the lumen of the ER/M compartment. With aging and other conditions, αS oligomer forms and matures into insoluble aggregates with the disease progression. Accumulation and maturation of αS oligomer is favored by the lack of βS in the ER as well as sequestration of ER chaperones by increasing amounts of αS. Initially, soluble αS monomer and oligomer are not exposed to the cytosol, but the insoluble aggregates become exposed to the cytosol likely by destabilizing the membranes. Together with the fact that therapeutic effects of Salubrinal treatment seem to be associated with reduced αS oligomers in the ER/M, we hypothesize that the αS-related abnormalities contribute to chronic ERS and neurodegeneration.
Figure 10.

A model showing the evolution of αS abnormalities in the ER. The model is derived from considering the findings of this report and the companion report (Colla et al., 2012). The red arrows indicating “αS flux” are meant to show increased ER accumulation of αS with disease, but the mechanism of αS accumulation in the ER is unknown. Similarly, while our results show Salubrinal inhibits accumulation of αS and reduces ERS, the detailed mechanism is undefined.
Normal presence of αS in the lumen of ER/M seems consistent with prior data from cell culture showing that αS can be secreted by neurons by transiting through the normal ER–Golgi secretory compartments (Lee et al., 2005). More recently, Desplats et al. (2009) showed that secreted αS is transmitted from neuron to neuron, seeding the formation of “aggregates” in the neighbor “accepting” neurons (Desplats et al., 2009). Moreover, recent studies show that secreted αS can be toxic to neuronal cells (Emmanouilidou et al., 2010). Thus, the presence of toxic αS species, like the ER-associated αS aggregates, could be responsible for accelerated inclusion formation in neighboring neurons or neurotoxicity. While the biology behind ER-associated αS needs further studies, the lack of βS in ER/M is a strong indication that ER-associated αS is not a nonspecific byproduct, lipid-binding, or contamination of subcellular fractionation. Regardless, the fact is that increases in the levels of ER-associated αS are a common feature of α-synucleinopathy in mice and in human support the pathological link between ER-associated αS and neurodegeneration. Significantly, in addition to αS monomers, significant amounts of αS aggregates were selectively associated with microsomes, and our companion report shows that toxic αS oligomers initially form within the ER/M lumen (Colla et al., 2012). Since the ERS response is activated by accumulation of misfolded proteins in the ER (Kozutsumi et al., 1988; Bertolotti et al., 2000) toxic αS oligomers could induce ERS directly by interaction with ER chaperones and/or by affecting ER function (i.e., interfering with calcium metabolism). Even in the absence of αS aggregate, interactions between αS and ER chaperones are significant as they could reduce the amount of ER chaperones available to binding to other UPR transducers and ER clients, particularly during ERS conditions. For example, lack of grp78 binding to IP3Rs could lead to defective Ca2+ homeostasis and mitochondrial defect (Higo et al., 2010). Such conditions are consistent with our data showing that αS sensitizes cells to ERS toxicity. This finding is significant, as αS would increase the vulnerability of neurons to ERS caused by aging and/or environmental agents.
The fact that α-synucleinopathy in vivo is associated with chronic ERS is consistent with the studies from cellular and invertebrate models that implicate ER stress in αS toxicity (Smith et al., 2005; Cooper et al., 2006). While some of the studies, particularly derived from the yeast model of αS toxicity (Cooper et al., 2006), suggested that αS monomers could cause ER stress by affecting ER–Golgi membrane trafficking, the ER stress in the vertebrate brain seems mechanistically variable from the αS-induced ER stress in yeast. In yeast, the membrane-binding capacity of αS monomers seems important for toxicity (Volles and Lansbury, 2007). In brain of αS Tg mice, ER stress is most obvious with the overt αS pathology, including ER accumulation of αS oligomers. However, while αS toxicity in many viral models is independent of αS membrane-binding properties (Kirik et al., 2002; Lo Bianco et al., 2002; Koprich et al., 2010), toxicity in the rat AAV2/6 model is somewhat dependent on membrane-binding capacity of αS (i.e., A30P mutant is least toxic; M. Gaugler and B. Schneider et al., unpublished observation). These variations amplify the notion that multiple toxic pathways are activated by αS abnormalities, where the mode of toxicity may depend on the context and dynamics of αS expression. In particular, significant early αS pathology in synaptic terminals and axons do not colocalize with markers of ER (Fig. 3) and may be related to distinct toxic events (Volpicelli-Daley et al., 2011). Moreover, while we hypothesize that α-synucleinopathy causes abnormal ERS/UPR, which is mechanistically linked to neurodegeneration, we note that our documentation of ERS/UPR is not complete. In particular, due to technical limitations with reagents, we were not able to effectively evaluate the status of other UPR reporters such as ATF6 and PERK. Because induction of the ER chaperones and the xbp1 cleavage can occur via non-UPR mechanisms (Kim et al., 2008), observed lack of p-eIF2α induction in αS Tg mice could reflect activation of processes other than ERS/UPR. Despite this caveat, ER accumulations of both αS oligomers and polyubiquitin support our view that αS pathology causes ER dysfunction. We believe future targeted studies using models where UPR pathways are genetically altered will provide valuable insights in this area.
While there are variations in the mechanistic details, it is important that both overt α-synucleinopathy in the A53TαS Tg model and αS toxicity in the rat AAV2/6 model is associated with ERS response. The pathologic importance of ERS to neurodegeneration is supported by the recent studies showing that chronic ERS condition can lead to neurodegeneration (Higo et al., 2010), and studies implicate ERS in chronic neurodegenerative conditions (Sokka et al., 2007; Lin et al., 2008; Zhu et al., 2008; Saxena et al., 2009; Egawa et al., 2011; Wang et al., 2011). The pathological significance of chronic ER stress in α-synucleinopathy is also supported by our result that pharmacological inhibitor of ER stress can extend the life span of the A53TαS Tg mouse model and attenuate toxicity of αS in the AAV2/6-transduced rat model. Significantly, we show here (Fig. 7) and in the companion report (Colla et al., 2012) that Salubrinal is able to selectively reduce levels of ER-associated αS oligomers without affecting total αS levels. These results indicate that, consistent with the known action of Salubrinal on ER homeostasis, this compound selectively effects ER accumulation of αS in models of α-synucleinopathy. One intriguing possibility is that since p-eIF2α has been linked to induction of autophagy (Tallóczy et al., 2002; Py et al., 2009), it is possible that Salubrinal may have facilitated the removal of damaged ER via autophagy. It is also important to note that while Salubrinal is generally considered an anti-ERS compound that inhibits p-eIF2α dephosphorylation, the exact basis for neuroprotection here and in other studies (Sokka et al., 2007; Zhu et al., 2008; Saxena et al., 2009) is unknown. In the current study, despite the accumulation of CHOP (Fig. 7C), we were not able to consistently demonstrate Salubrinal-dependent increase in p-eIF2α levels (Fig. 7B,C). Thus, while we and others have used Salubrinal to affect the p-eIF2α levels in vivo (Kim et al., 2008), we cannot rule out the possibility that the neuroprotective effects of Salubrinal are independent of p-eIF2α or unrelated to ERS, such as inhibiting translation of protein(s) required for cell death. The possible attenuation of αS expression by Salubrinal seems unlikely based on our results and the prior studies showing that the attenuation of p-eIF2α dephosphorylation does not cause a general decrease in protein translation (Jiang et al., 2010). Overall, while our results provide the initial pathological links between α-synucleinopathy, ER stress, and αS oligomers, the mechanistic details will need further examination.
Salubrinal treatment clearly delays the disease onset in the A53TαS Tg mouse model and attenuates disease manifestation in the AAV2/6 model. However, the results from the AAV2/6 model show that Salubrinal does not increase the survival of DA neurons destined for cell death. Based on the motor tests and the analysis of Golgi morphology, it is clear that Salubrinal corrects αS-induced abnormalities in the surviving neurons. The rescue of Golgi morphology indicates positive effects of Salubrinal on the secretory function of DA neurons, which is further confirmed by improved motor activity in treated animals. Significantly, the therapeutic effects of Salubrinal are comparable to the therapeutic effects of Rab1A overexpression in this model (Coune et al., 2011). Therefore, the defects induced by αS in the surviving neurons are possibly related to impaired vesicle trafficking at the ER–Golgi level, thereby reducing neuronal secretory capacity. It is also possible that the dose of Salubrinal used was not sufficient to overcome the initial toxic effects of high levels of αS expression achieved in the AAV2/6 model. Thus, either higher Salubrinal dose and/or longer follow-up may have revealed overt neuroprotection. In particular, Golgi fragmentation is an early precursor to cell death and neuronal health (Nakagomi et al., 2008). Thus, given that Salubrinal reduces Golgi fragmentation in αS-expressing neurons, one may expect less ongoing neurodegeneration at later time points. The results are also consistent with the view that αS toxicity is associated with the activation of multiple cytotoxic pathways (Vila and Przedborski, 2004; Obeso et al., 2010). The presence of additional toxic processes is also consistent with the fact that while Salubrinal delayed the onset of disease in the A53TαS Tg mice, progression of the disease was not affected. Moreover, in the end-stage animals, we did not observe qualitative differences in neuropathology between Salubrinal- and vehicle-treated A53TαS Tg mice (data not shown). Another caveat is that the increase in CHOP expression resulting from Salubrinal may have antagonized the neuroprotective effects of Salubrinal as CHOP can promote cell death (Silva et al., 2005). However, the pathogenic importance of CHOP in rodent models of PD is unclear, as despite robust CHOP induction, the loss of CHOP does not protect from neurodegeneration in some cases (Silva et al., 2005).
In summary, combined with our companion report (Colla et al., 2012), our data show that pathogenic αS oligomers initially accumulate within ER/M fractions, likely causing ER dysfunction and chronic ER stress. Treating the αS Tg mouse model the rat AAV2/6 model of α-synucleinopathy with Salubrinal, a pharmacological inhibitor of ER-stress toxicity, dramatically delays the onset of motoric symptoms and decreases accumulation of αS oligomers in vivo. Thus, our results establish that toxic αS oligomers accumulate with α-synucleinopathy in brain and identify chronic ER stress as the manifestations of toxicity involved the disease progression. Further, our data indicate that modulation of ERS/UPR constitutes an important therapeutic target for PD and other α-synucleinopathies.
Footnotes
This work was supported by National Institutes of Health Grants NS038065, NS0380377, NS055776, and ES017384 (M.K.L.); and Swiss National Science Foundation, Grant 31003A_120653 (B.L.S.).We thank Drs. David Ron, Darren Moore, and Shaida Andrabi for helpful discussions. We also thank Dr. Virginia Lee for kindly providing the syn303 antibody.
The authors declare no competing financial interests.
References
- Bertolotti A, Zhang Y, Herdershot LM, Harding HP, Ron D. Dynamic interaction of BIP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe CG, Lee MK. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci, 2012;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coune P, Bensadoun JC, Aebischer P, Schnieder BL. Rab1A Over-expression prevents Golgi apparatus fragmentation and partially corrects motor deficits in an alpha-synuclein based rat model of Parkinson's disease. J Parkinson's Dis. 2011;1:373–387. doi: 10.3233/JPD-2011-11058. [DOI] [PubMed] [Google Scholar]
- Cox B, Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2006;1:1872–1878. doi: 10.1038/nprot.2006.273. [DOI] [PubMed] [Google Scholar]
- Croze EM, Morré DJ. Isolation of plasma membrane, Golgi apparatus, and endoplasmic reticulum fractions from single homogenates of mouse liver. J Cell Physiol. 1984;119:46–57. doi: 10.1002/jcp.1041190109. [DOI] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding TT, Lee SJ, Rochet JC, Lansbury PT., Jr Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM, Trojanowski JQ. Widespread nitration of pathological inclusions in neurodegenerative synucleinopathies. Am J Pathol. 2000;157:1439–1445. doi: 10.1016/S0002-9440(10)64781-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusonchet J, Bensadoun JC, Schneider BL, Aebischer P. Targeted overexpression of the parkin substrate Pael-R in the nigrostriatal system of adult rats to model Parkinson's disease. Neurobiol Dis. 2009;35:32–41. doi: 10.1016/j.nbd.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. 2011;286:7947–7957. doi: 10.1074/jbc.M110.156430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Bevis BJ, Shorter J, Strathearn KE, Hamamichi S, Su LJ, Caldwell KA, Caldwell GA, Rochet JC, McCaffery JM, Barlowe C, Lindquist S. The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–150. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, Nakamura T, Mikoshiba K. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron. 2010;68:865–878. doi: 10.1016/j.neuron.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–711. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Jiang Z, Belforte JE, Lu Y, Yabe Y, Pickel J, Smith CB, Je HS, Lu B, Nakazawa K. eIF2alpha Phosphorylation-dependent translation in CA1 pyramidal cells impairs hippocampal memory consolidation without affecting general translation. J Neurosci. 2010;30:2582–2594. doi: 10.1523/JNEUROSCI.3971-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Björklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koprich JB, Johnston TH, Reyes MG, Sun X, Brotchie JM. Expression of human A53T alpha-synuclein in the rat substantia nigra using a novel AAV1/2 vector produces a rapidly evolving pathology with protein aggregation, dystrophic neurite architecture and nigrostriatal degeneration with potential to model the pathology of Parkinson's disease. Mol Neurodegener. 2010;5:43. doi: 10.1186/1750-1326-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozutsumi Y, Segal M, Normington K, Gething MJ, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Stirling W, Xu Y, Xu X, Qui D, Mandir AS, Dawson TM, Copeland NG, Jenkins NA, Price DL. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc Natl Acad Sci U S A. 2002;99:8968–8973. doi: 10.1073/pnas.132197599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Lee MK. Antiapoptotic property of human alpha-synuclein in neuronal cell lines is associated with the inhibition of caspase-3 but not caspase-9 activity. J Neurochem. 2005;93:1542–1550. doi: 10.1111/j.1471-4159.2005.03146.x. [DOI] [PubMed] [Google Scholar]
- Li W, Hoffman PN, Stirling W, Price DL, Lee MK. Axonal transport of human alpha-synuclein slows with aging but is not affected by familial Parkinson's disease-linked mutations. J Neurochem. 2004a;88:401–410. doi: 10.1046/j.1471-4159.2003.02166.x. [DOI] [PubMed] [Google Scholar]
- Li W, Lesuisse C, Xu Y, Troncoso JC, Price DL, Lee MK. Stabilization of alpha-synuclein protein with aging and familial Parkinson's disease-linked A53T mutation. J Neurosci. 2004b;24:7400–7409. doi: 10.1523/JNEUROSCI.1370-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jäkälä P, Hartmann T, Price DL, Lee MK. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci U S A. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Yoo MJ, Savonenko A, Stirling W, Price DL, Borchelt DR, Mamounas L, Lyons WE, Blue ME, Lee MK. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2008;28:13805–13814. doi: 10.1523/JNEUROSCI.4218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci U S A. 2002;99:10813–10818. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson's disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, Dauer W, Przedborski S, Ischiropoulos H, Lansbury PT, Sulzer D, Cuervo AM. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest. 2008;118:777–788. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi S, Barsoum MJ, Bossy-Wetzel E, Sütterlin C, Malhotra V, Lipton SA. A Golgi fragmentation pathway in neurodegeneration. Neurobiol Dis. 2008;29:221–231. doi: 10.1016/j.nbd.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan V, Scarlata S. Membrane binding and self-association of alpha-synucleins. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Carew JS, Dunner K, Jr, Boise LH, Chiao PJ, Huang P, Abbruzzese JL, McConkey DJ. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- Obeso JA, Rodriguez-Oroz MC, Goetz CG, Marin C, Kordower JH, Rodriguez M, Hirsch EC, Farrer M, Schapira AH, Halliday G. Missing pieces in the Parkinson's disease puzzle. Nat Med. 2010;16:653–661. doi: 10.1038/nm.2165. [DOI] [PubMed] [Google Scholar]
- Pletnikova O, West N, Lee MK, Rudow GL, Skolasky RL, Dawson TM, Marsh L, Troncoso JC. Abeta deposition is associated with enhanced cortical alpha-synuclein lesions in Lewy body diseases. Neurobiol Aging. 2005;26:1183–1192. doi: 10.1016/j.neurobiolaging.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5:393–396. doi: 10.4161/auto.5.3.7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. 2005;95:974–986. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet. 2005;14:3801–3811. doi: 10.1093/hmg/ddi396. [DOI] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallóczy Z, Jiang W, Virgin HW, 4th, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;10(Suppl):S58–S62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- Volles MJ, Lansbury PT., Jr Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J Mol Biol. 2007;366:1510–1522. doi: 10.1016/j.jmb.2006.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Martin E, Gonzales V, Borchelt DR, Lee MK. Differential regulation of small heat shock proteins in transgenic mouse models of neurodegenerative diseases. Neurobiol Aging. 2008;29:586–597. doi: 10.1016/j.neurobiolaging.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow AR, Chen CW, Corrochano S, Acevedo-Arozena A, Gordon DE, Peden AA, Lichtenberg M, Menzies FM, Ravikumar B, Imarisio S, Brown S, O'Kane CJ, Rubinsztein DC. alpha-Synuclein impairs macroautophagy: implications for Parkinson's disease. J Cell Biol. 2010;190:1023–1037. doi: 10.1083/jcb.201003122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Fenik P, Zhan G, Sanfillipo-Cohn B, Naidoo N, Veasey SC. Eif-2a protects brainstem motoneurons in a murine model of sleep apnea. J Neurosci. 2008;28:2168–2178. doi: 10.1523/JNEUROSCI.5232-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]