

Abstract
Iron overload is associated with increased diabetes risk. We therefore investigated the effect of iron on adiponectin, an insulin-sensitizing adipokine that is decreased in diabetic patients. In humans, normal-range serum ferritin levels were inversely associated with adiponectin, independent of inflammation. Ferritin was increased and adiponectin was decreased in type 2 diabetic and in obese diabetic subjects compared with those in equally obese individuals without metabolic syndrome. Mice fed a high-iron diet and cultured adipocytes treated with iron exhibited decreased adiponectin mRNA and protein. We found that iron negatively regulated adiponectin transcription via FOXO1-mediated repression. Further, loss of the adipocyte iron export channel, ferroportin, in mice resulted in adipocyte iron loading, decreased adiponectin, and insulin resistance. Conversely, organismal iron overload and increased adipocyte ferroportin expression because of hemochromatosis are associated with decreased adipocyte iron, increased adiponectin, improved glucose tolerance, and increased insulin sensitivity. Phlebotomy of humans with impaired glucose tolerance and ferritin values in the highest quartile of normal increased adiponectin and improved glucose tolerance. These findings demonstrate a causal role for iron as a risk factor for metabolic syndrome and a role for adipocytes in modulating metabolism through adiponectin in response to iron stores.
Introduction
Increased iron stores are associated with increased risk of type 2 diabetes (1–4), gestational diabetes (5), prediabetes (6), metabolic syndrome (MetS) (7), central adiposity (8), and cardiovascular disease (9, 10). The mechanisms underlying these associations are poorly understood. The commonly used marker for total body iron stores, serum ferritin, is also responsive to inflammatory stress (11, 12), so increased ferritin in diabetes could simply reflect the inflammatory component of that disease (13). On the other hand, phlebotomy improves glycemia and MetS traits (14–17), arguing that iron may play a causal role in diabetes.
The possible mediators of the association between iron and diabetes risk are not known. Decreases in both insulin secretion and sensitivity have been linked to iron. Excess iron impairs pancreatic β cell function and causes β cell apoptosis (18–21). Recent studies have also found a negative correlation between serum ferritin and the insulin-sensitizing adipokine, adiponectin (3, 22–24). The hypothesis that adiponectin links iron and insulin resistance is appealing, as decreased adiponectin levels are associated with obesity and type 2 diabetes (25) and are causally linked with insulin resistance (26).
We therefore investigated the mechanisms underlying the relationships among serum ferritin, adiponectin, and MetS in mice and humans. We demonstrate in humans that the association between serum ferritin and adiponectin is independent of inflammation and that serum ferritin, even within its normal ranges, is among the best predictors of serum adiponectin. Studies in cell culture, mouse models, and humans demonstrate that iron plays a direct and causal role in determining adiponectin levels and diabetes risk. The adipocyte expresses specialized proteins related to iron metabolism that make it well suited to perform as an iron sensor, allowing it to integrate iron availability into its broader nutrient-sensing function.
Results
Human ferritin levels are inversely associated with serum adiponectin independently of inflammation.
We studied 110 individuals with (n = 49) and without (n = 61) diabetes recruited for an independent study of metabolic flexibility (27). Serum ferritin was negatively associated with serum adiponectin (r = –0.294, P = 0.0017). To mitigate the effects of inflammation and/or extreme iron overload and anemia, we next restricted the analysis to individuals with normal serum ferritin (men, >30 ng/ml and <300 ng/ml; women, <200 ng/ml) (28, 29). Excluding 28 individuals outside this range strengthened the association between ferritin and adiponectin (Figure 1A, r = –0.385, P = 0.0003). Serum levels of the inflammatory marker C-reactive protein (CRP) were elevated in diabetic subjects compared with those in nondiabetics (log[CRP] 3.40 ± 0.07 vs. 3.01 ± 0.08, P = 0.0005). Nonetheless, multivariate analysis, including CRP, BMI, and diabetes status, had little effect on the ferritin-adiponectin association (Table 1). There was no association of IL-6 or TNF-α levels with serum ferritin (r = –0.13, P = 0.29 and r = –0.010, P = 0.94, respectively) or adiponectin (r = 0.085, P = 0.51 and r = 0.13, P = 0.35, respectively).
Figure 1. Serum ferritin levels are inversely associated with serum adiponectin ferritin levels and predict presence or absence of MetS.

(A) Serum adiponectin and ferritin levels were measured and correlated in a cohort of subjects with normal ferritin. P = 0.0003. (B) Ferritin levels in the same subjects subdivided by diabetes status. *P < 0.0004. (C) Adiponectin levels in the same subjects subdivided by diabetes status. *P < 0.0012. (D) Ferritin levels in the obese subjects without MetS (white bars) or with MetS, without or with diabetes (gray and black bars, respectively). *P < 0.03 for the diabetic subgroup compared with either other group. (E) Adiponectin levels in the subset of the subjects described D in whom adiponectin levels were determined. *P < 0.03 for either MetS subgroup compared with the non-MetS group.
Table 1.
Multiple regression models for the relationship of adiponectin to ferritin
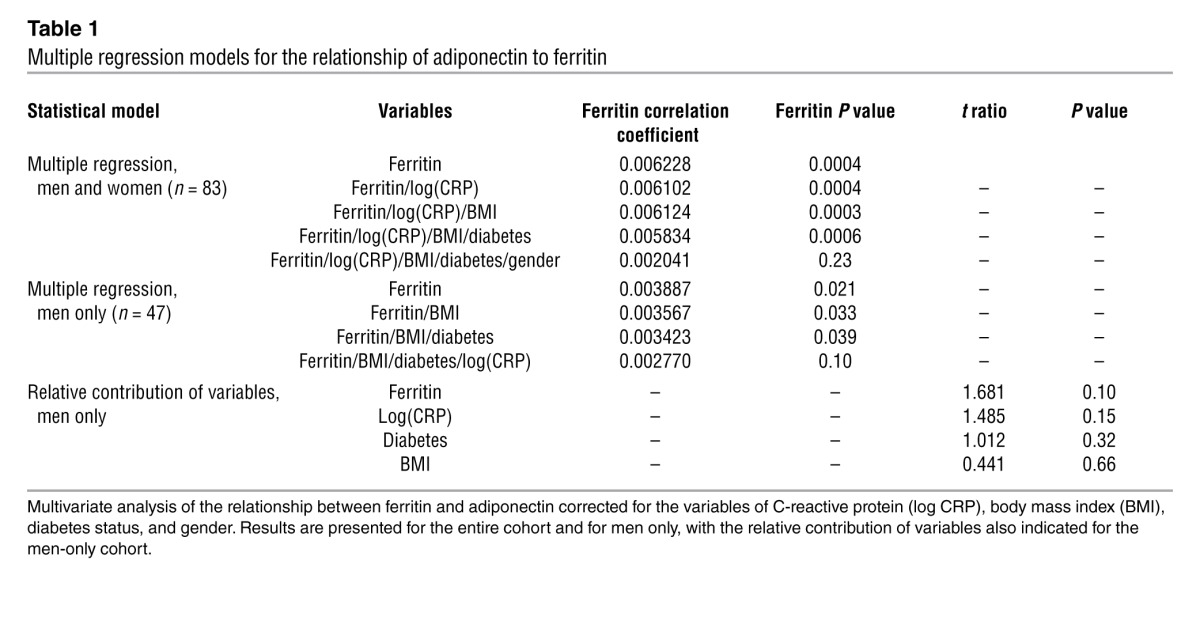
Inclusion of gender in the analysis affected the serum ferritin and adiponectin association (Table 1). The relationship was weaker in women, likely because the range of ferritin values was significantly narrower and lower compared with that in men (average ferritin, 75.3 ± 10.5 ng/ml in women and 183.6 ± 20.9 ng/ml in men, P < 0.0001) (30). Overall, however, and consistent with previous reports (31), these lower ferritin levels were accompanied by higher adiponectin levels in women (3.42 ± 0.26 μg/ml vs. 2.39 ± 0.14 μg/ml in men, P = 0.0006). In men, after adjusting for BMI and diabetes status, serum ferritin remained significantly associated with adiponectin (Table 1, P = 0.04) but was reduced to a trend after adjusting for CRP (Table 1, P = 0.10). Serum ferritin levels in men, however, remained more predictive of adiponectin levels than BMI, CRP, and diabetes status (Table 1, see Relative contribution of variables). In women, none of the variables were significantly associated with adiponectin in the multivariate analysis.
To further explore the physiological significance of the correlation between iron and adiponectin, we subdivided the cohort by diabetes status. In the subjects with diabetes, serum ferritin levels were 2-fold higher (Figure 1B, P = 0.0004) and adiponectin levels were 24% lower (Figure 1C, P = 0.012). We also determined that, in the entire cohort with normal ferritin levels, insulin sensitivity, as determined for the original study (27) by the glucose clamp technique, was inversely correlated with ferritin (r = 0.365, P = 0.0003, n = 93) and directly correlated with adiponectin levels (r = 0.354, P = 0.0004, n = 95).
Ferritin predicts adiponectin and the presence of MetS in obese men.
To extend and validate these findings in a separate cohort, we measured serum adiponectin and ferritin levels of obese men, with or without MetS (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI44421DS1). BMI was similar between the groups (39.6 ± 0.7 kg/m2 vs. 39.7 ± 0.6 kg/m2). The MetS group had all 5 features of the syndrome: obesity, hypertension (>130/85 mmHg), HDL cholesterol <40 mg/dl, serum triglycerides >150 mg/dl, and either fasting glucose >100 mg/dl or diabetes (32). Those without MetS had no more than 1 factor in addition to obesity, and none had diabetes. Serum ferritin was measured in all subjects (n = 125), and adiponectin was measured in a randomly selected subset (n = 38). Overall, the MetS group had significantly higher ferritin (260 ± 23 ng/ml vs. 185 ± 21 ng/ml, P < 0.01) and lower adiponectin (11.5 ± 1.0 μg/ml vs. 18.9 ± 1.9 μg/ml, P < 0.005) than the non-MetS group. Higher ferritin values were seen in the MetS subset with diabetes compared with those in either the non-MetS group or the MetS subgroup without diabetes (Figure 1D, P < 0.03). Adiponectin was higher in the non-MetS group compared with that in either MetS subgroup (Figure 1E, P < 0.03). Consistent with the data in Figure 1A, ferritin was inversely correlated with adiponectin in this cohort, although in this smaller group the relationship did not quite reach the level of statistical significance (r = 0.304, n = 38, P = 0.06, data not shown). Insulin resistance estimated from the homeostasis model (HOMA-IR) was correlated positively with ferritin (r = 0.264, n = 125, P < 0.01) and negatively with adiponectin (r = 0.48, n = 38, P < 0.005).
Adipocyte iron increases and adiponectin mRNA and serum protein levels decrease in dietary iron overload.
Several studies have identified effects of iron on adipocyte metabolism (e.g., refs. 33, 34). To explore the regulation of adipocyte iron levels, we first demonstrated that adipocyte iron levels respond to dietary iron content in WT C57BL6/J mice by measuring mRNA levels of the transferrin receptor (Tfrc). Tfrc mRNA contains iron response elements in its 3′ untranslated region that result in decreased Tfrc mRNA levels as cellular iron levels increase (35). We observed a 40% decrease in Tfrc mRNA in adipocytes from mice fed a high-iron diet (20 g/kg iron) compared with that in mice fed normal chow (330 mg/kg iron) (Figure 2A, P < 0.05). Technical difficulties precluded the direct assay of cytosolic iron in isolated adipocytes. To investigate a possible direct and causal role of iron in the regulation of adiponectin, we studied the effects of dietary iron overload on serum adiponectin levels in mice. We fed 129SvEvTac male mice high- (20,000 mg/kg carbonyl iron), normal (330 mg/kg), or low- (7 mg/kg) iron diets for 2 months. Body weights were significantly lower in mice fed high-iron diets and significantly higher in those fed low-iron diets compared with those of mice fed normal chow (low iron, 34.6 ± 1.1 g; normal chow, 29.0 ± 0.5 g; high iron, 26.7 ± 0.6 g, P < 0.001 between groups by ANOVA, n = 8–12/group). Despite decreased body weight, serum adiponectin levels were 29% lower in iron-overloaded mice (Figure 2B, P = 0.0002). Conversely, dietary iron restriction increased serum adiponectin levels by 31% despite increased body weight (Figure 2B, P = 0.0036). A similar 29% decrease in serum adiponectin was also seen in a different strain, namely male C57BL6/J mice fed normal chow and high-iron diets (6.46 ± 0.15 μg/ml vs. 4.55 ± 0.16 μg/ml, respectively, P < 0.0001). Adiponectin mRNA levels in isolated epididymal adipocytes were 30% lower in iron-overloaded mice (Figure 2C, P = 0.07), mirroring the changes in serum adiponectin levels. With a smaller cohort of high-iron diet– and normal chow–fed mice (n = 4/group), we determined body composition by magnetic resonance imaging, and the high-iron diet caused not only a decrease in weight but an even larger relative effect on fat mass because of a parallel increase in lean body mass (Figure 2D). Both food intake and oxygen consumption rates were higher in the lower-weight, high-iron group (Figure 2D). The differences in weight seen with the high-iron diet were not observed in mice with genetic deletion of adiponectin (Figure 2E). The expected inverse linear relationship between weight and adiponectin was observed in mice fed normal chow (r = 0.48, n = 22, P = 0.02, not shown), but this relationship was lost in mice fed the high-iron diet, in fact even trending toward a positive relationship (r = 0.17, n = 17, P = 0.51).
Figure 2. Serum adiponectin and adipocyte mRNA levels decrease with dietary iron overload and with iron treatment in 3T3-L1 cells.

(A) Tfrc mRNA quantified by RT-PCR and normalized to cyclophilin in collagenased adipocytes from epididymal fat pads of mice fed normal chow (NC) or a high-iron diet (HI) for 8 weeks. *P < 0.05. (B) Serum adiponectin levels were measured in 7-month-old 129/SvEvTac background mice following 2 months of being fed low-iron (7 mg/kg carbonyl iron), normal chow (330 mg/kg), or high-iron (20 g/kg) diets. *P = 0.004, low iron vs. normal chow; ‡P = 0.0002, high iron vs. normal chow. (C) Adiponectin mRNA levels in isolated epididymal adipocytes from mice fed high-iron diet or normal chow. P = 0.07. (D) Body weights were determined in C57BL6/J mice after 8 weeks on normal chow or high-iron diets, and body composition was determined by magnetic resonance imaging (n = 6/group or 8/group, *P < 0.05, ‡P < 0.01). (E) Body weights were determined in mice with knockout of the adiponectin gene compared with those of controls after 8 weeks on normal chow or high-iron diets (n = 5–12/group, ‡P < 0.001). (F) Euglycemic hyperinsulinemic clamps were performed on WT mice on normal or high-iron diets. Glucose infusion rates (GIRs) trended lower when normalized to total body weight (P = 0.22) but differed significantly when normalized to lean body mass (‡P < 0.0005).
Finally, to demonstrate that the change in adiponectin was accompanied by changes in glucose metabolism, we performed hyperinsulinemic clamp studies on WT mice on normal chow and those that had been on the high-iron diet for 8 weeks. There was a trend toward lower glucose disposal, normalized to total body weight, in the mice fed high-iron diet (Figure 2F, P = 0.22). However, because most glucose uptake at hyperinsulinemia is into skeletal muscle, we also normalized to lean mass based on the magnetic resonance imaging findings, and the mice on high-iron diet had a significant decrease in their maximal glucose disposal rate per gram of lean tissue (P < 0.001).
Iron decreases adiponectin transcription.
To demonstrate further that the decreased adiponectin mRNA levels are due to decreased transcription and that iron regulates adiponectin directly, we examined the effects of iron on adiponectin in a cell culture model. Treatment of 3T3-L1 adipocytes with iron sulfate decreased media adiponectin protein levels in a dose-dependent manner (Figure 3A, P < 0.0001). Adiponectin mRNA levels also decreased 30% with iron treatment (Figure 3B, P = 0.02). We measured luciferase activity driven by the proximal 1,460 bp of the murine adiponectin promoter, which contains most of the previously identified sites that regulate adiponectin transcription (36). Iron decreased promoter activity by 28% (Figure 3C, P = 0.0025). Iron did not decrease the half-life of the endogenous mRNA or the reporter construct measured after actinomycin C or cycloheximide D treatment of cells (data not shown). Most physiologic regulation of adiponectin gene transcription is attributable to the factors FOXO1 and PPARγ (36, 37). To explore the mechanism of regulation of adiponectin by iron, we first examined posttranslational modification of FOXO1. Iron caused decreased acetylation of FOXO1 without changing its level of phosphorylation or total protein (Figure 3, D and E). In agreement with the lack of change of FOXO1 phosphorylation, we also detected no differences in basal or insulin-stimulated phosphorylation of AKT in iron-treated cells (Figure 3F).
Figure 3. Transcriptional regulation of adiponectin by iron.

(A) Media adiponectin levels in 3T3-L1 cells 12 hours following 12-hour pretreatment. P < 0.0001. (B) RT-PCR quantification of adiponectin mRNA levels in 3T3-L1 adipocytes treated with no iron or 100 μM FeSO4 for 24 hours, normalized to cyclophilin A. *P = 0.02. (C) Adiponectin promoter-driven luciferase activity in the presence or absence of 100 μM FeSO4. ‡P = 0.0025. (D) Western blot for acetylated FOXO1 (Ac-FOXO1), phosphorylated FOXO1 (P-FOXO1), total FOXO1 (Tot-FOXO1), and β-actin in 3T3-L1 adipocytes treated with no iron or 100 μM FeSO4 for 8 hours. (E) Quantitation of Western blots (total n = 6 independent determinations) normalized to β-actin. *P < 0.05. (F) Quantitation of Western blots for phosphorylated AKT in 3T3-L1 adipocytes treated with no iron or 100 μM FeSO4 for 8 hours and insulin (10 nM) for 1 hour. (G) ChIP showing FOXO1 occupancy of adiponectin promoter FOXO1 sites and PPRE and PPARγ occupancy of PPRE in 3T3-L1 adipocytes (n = 3 experiments each assayed in duplicate, *P < 0.05). (H) Immunoprecipitation of 3T3-L1 adipocyte extracts, treated overnight in the presence or absence of 100 μM FeSO4, by antibodies to FOXO1, followed by immunoblotting for FOXO1 (t-FOXO1) and C/EBPα (0.58 ± 0.15 density units for control, 0.61 ± 0. 26 density units for iron-treated extracts, P = 0.93).
Contrary to the observed effects of iron on adiponectin transcription, deacetylation of FOXO1 is generally associated with increased adiponectin transcription (36, 38). We therefore measured FOXO1 occupancy at its 2 known sites of transcriptional activation using ChIP. As predicted by FOXO1 acetylation status, cells treated with iron exhibited a 3.1-fold increase in occupancy by FOXO1 at the sites reported to stimulate adiponectin transcription (P < 0.01, Figure 3G). FOXO1, however, has also been reported to transrepress adiponectin transcription when associated with the PPARγ response element (PPRE) of the adiponectin promoter (39). Iron treatment resulted in a 3.5-fold enhancement of FOXO1 binding to the PPRE (P = 0.01, Figure 3G). There was no significant increase in PPARγ binding to the PPRE (1.6 fold, P = 0.07, Figure 3G). Because the interaction of C/EBPα with FOXO1 has also been implicated in regulation of adiponectin transcription (36), we also measured the association of C/EBPα with FOXO1 by coimmunoprecipitation but saw no effect of iron on this association (P = 0.93, Figure 3H).
Adipocytes express the specialized iron channel ferroportin.
Adipocytes express genes that are generally restricted to iron-sensing tissues (40, 41). We therefore sought to obtain evidence that adipocytes might have a specialized iron-sensing capacity by assessing adipocyte expression of the iron export channel ferroportin, whose significant tissue expression has been reported to be limited to gut enterocytes, placenta, and reticuloendothelial cells, including macrophages (42, 43). Ferroportin mRNA and protein were detectable by quantitative RT-PCR and Western blot in differentiated 3T3-L1 adipocytes. Treatment with iron sulfate increased ferroportin mRNA in a dose-dependent manner (Figure 4A, P < 0.0001). Protein levels were responsive both to iron and to treatment by hepcidin, a ferroportin ligand that results in ferroportin downregulation (Figure 4B and ref. 44).
Figure 4. Functional expression of ferroportin in adipocytes.

(A) Fpn1 mRNA levels quantified by RT-PCR in 3T3-L1 adipocytes exposed to different concentrations of iron (FeSO4) in the culture medium. *P < 0.05 compared with 0.1 mM, ‡P < 0.01, P < 0.0001 for overall trend. (B) FPN1 protein levels in 3T3-L1 adipocytes, as detected by immunoblotting in adipocytes treated with no iron, 100 μM FeSO4, or 1 μg/ml hepcidin for 8 hours. (C) Fpn1 mRNA in adipose tissue and spleen from WT (Fpn1fl/fl) and Fpn1–/– (AP2 Cre ferroportin knockout) mice. ‡P < 0.001. (D) Tfrc mRNA quantified by RT-PCR and normalized to cyclophilin in collagenased adipocytes from epididymal fat pads of WT and Fpn1–/– mice (n = 10–14/group, ‡P < 0.001). (E) Adiponectin mRNA in the adipocytes used in E. ‡P < 0.01. (F) Serum adiponectin in WT and Fpn1–/– mice (n = 9–12/group, *P < 0.01). (G) High molecular weight (HMW) adiponectin determined as a percentage of total in the same group depicted in F. (H) Glucose tolerance testing of WT and Fpn1–/– mice (n = 5–6/group, *P < 0.05 for individual glucose values). (I) Body composition by magnetic resonance imaging in WT and Fpn1–/– mice on normal chow or high-iron diets (n = 11–20/group, *P < 0.05).
Deletion of adipocyte ferroportin results in increased adipocyte iron levels, decreased serum adiponectin, and increased insulin resistance.
To demonstrate that ferroportin serves as a functional iron channel and exporter in adipocytes, we generated mice lacking the ferroportin gene in adipocytes (Fpn1–/– mice). An Fpn1fl/fl mouse (45), provided by Nancy C. Andrews (Duke University, Durham, North Carolina, USA), was crossed to a mouse expressing Cre recombinase under control of the 5.4-kB AP2 promoter. The Ap2-Cre:Fpn1fl/fl mice were subsequently backcrossed onto the 129 strain for at least 5 generations. Ferroportin mRNA was undetectable in adipocytes purified by collagenase digestion from Fpn1–/– mice (Figure 4C). Because macrophages can also express the Ap2 gene, we examined ferroportin expression in splenocytes, wherein the only cell expressing significant ferroportin is the macrophage. There was no decrease in ferroportin mRNA in splenocytes from the Ap2-Cre:Fpn1fl/fl mice (Figure 4C).
To demonstrate a role for ferroportin in modulating adipocyte iron, we measured Tfrc mRNA in isolated adipocytes. Compared with WT Fpn1fl/fl adipocytes, Ap2-Cre:Fpn1fl/fl adipocytes exhibited a 44% decrease in Tfrc (Figure 4D, P < 0.001), consistent with increased cytosolic iron, and functionality of the ferroportin channel is in adipocytes.
The increased levels of adipocyte iron in the Fpn1–/– mice resulted in a 58% decrease in levels of adiponectin mRNA in adipocytes (Figure 4E, P < 0.01), and this was reflected in decreased serum adiponectin (Figure 4F). Because of the heterogeneity in the weights of the mice and the effect of weight on adiponectin, serum adiponectin was determined in a cohort of mice all weighing less than 30 g. In control (Fpn1fl/fl) mice, the high-iron diet resulted in a 12% decrease in serum adiponectin (Figure 4F, P < 0.05). Serum adiponectin was also lower (13%, P < 0.05) in the Ap2-Cre:Fpn1fl/fl mice on normal chow compared with controls and did not decrease further in mice on the high-iron diet. No changes were noted in the distribution of adiponectin molecular weight isoforms, as analyzed by an adiponectin assay that detects both total and high molecular weight isoforms (Figure 4G) and as analyzed by native SDS-PAGE (data not shown).
To determine whether the change in adiponectin was physiologically significant, we performed glucose tolerance testing in WT and Fpn1–/– mice. Fpn1–/– mice had significantly higher glucose excursions at 30 and 60 minutes after challenge (Figure 4H, P < 0.05), and the areas under the glucose curve differed significantly between the groups (16,372 mg-min/dl in WT mice and 20,272 mg-min/dl in Fpn1–/– mice, 24% increase, P < 0.001, data not shown). Fasting glucose and insulin levels were also determined in WT and Fpn1–/– mice on different levels of dietary iron, and insulin resistance, as determined by homeostasis model assessment (HOMA-IR), was increased in the Fpn1–/– mice (P < 0.01, data not shown). The effects of high-iron diet on body weight and body composition were also lost in the Fpn1–/– mice. Body weights of the Fpn1–/– mice on normal chow compared with those of mice on high-iron diets did not differ (normal chow, 32.1 ± 1.5 g, high iron 31.5 ± 1.3 g, P = 0.68), and the changes in body composition induced by high-iron diet (Figure 2D) and replicated in a cohort of control Fpn1fl/fl mice (Figure 4I, P < 0.05 for both percentage of lean and fat mass) were also not seen in the Fpn1–/– mice (Figure 4I, P = 0.37 and P = 0.56 for percentage of lean and fat mass, respectively).
Lower adipocyte iron, higher serum adiponectin, and increased insulin sensitivity in hereditary hemochromatosis.
The effects of iron on adiponectin levels present a paradox. Namely, adiponectin decreases with dietary iron overload, and yet serum adiponectin levels are increased in a mouse model of genetic iron overload, wherein the gene most commonly mutated in human hereditary hemochromatosis (HH) has been deleted (Hfe–/– mice) (46). It has been shown that the relative lack of hepcidin in HH results in failure to downregulate ferroportin, so that cells that express significant amounts of the iron channel are paradoxically less loaded with iron in HH (47, 48). We therefore sought to determine whether the same were true of adipocytes. Tfrc mRNA levels, which inversely reflect cytosolic iron, were increased by 67% in adipocytes from Hfe–/– mice compared with those from WT mice (Figure 5A, P = 0.05). Thus, the previously reported increased adiponectin in a mouse model of HH (46) is consistent with lower adipocyte iron levels.
Figure 5. Adiponectin in mouse and human hemochromatosis.

(A) Tfrc mRNA levels in isolated adipocytes from WT and Hfe–/– mice on normal chow, normalized to cyclophilin A (n = 5/group, *P = 0.05). (B) Serum adiponectin levels in male (*P = 0.04) and female (P = 0.06) subjects with HH compared with non-HH sibling controls. (C) Serum adiponectin levels plotted as a function of BMI for subjects with HH (black circles) and non-HH sibling controls (white circles). Sexes were combined for linear regression analysis of HH (P = 0.87) and control subjects (P = 0.03). (D) Fasting glucose levels in WT, Hfe–/–, and Hfe–/–:APN–/– double-knockout mice (n = 5–7/group, *P = 0.006 by ANOVA).
Because humans with HH also trend toward increased insulin sensitivity prior to the onset of clinical diabetes (20), we hypothesized that serum adiponectin levels would likewise be increased in human HH. Serum adiponectin levels were increased by 89% in male subjects with HH, compared with non-HH, male sibling controls (Figure 5B, P = 0.04). In women, serum adiponectin levels also trended higher (136%) in patients with HH (Figure 5B, 3.3 μg/ml vs. 1.4 μg/ml, P = 0.06). In non-HH sibling controls of the subjects with HH, serum adiponectin and BMI were closely and inversely associated (Figure 5C, r = 0.77, P = 0.03), consistent with previous reports (31, 49). However, the association between BMI and adiponectin was lost in patients with HH (Figure 5C, r = 0.09, P = 0.87). Serum ferritin, which largely reflects hepatic iron stores, was not associated with serum adiponectin levels in patients with HH (data not shown).
To determine whether the increased adiponectin levels in HH mice were functionally significant, we generated mice with deletion of the adiponectin gene (APN–/–, provided by Phillip Scherer, ref. 50) on the C57BL6/J-HH (Hfe–/–) background. Because the effects of adiponectin deletion on glucose tolerance are more manifest in obese mice or mice exposed to a high-fat diet (26, 50), the mice were fed a high-fat diet for 8 weeks. The Hfe–/– mice exhibited a 19% decrease in fasting glucose compared with APN–/– mice, an improvement that was completely lost in the APN–/–:Hfe–/– double-knockout mice (Figure 5D, P = 0.006 by ANOVA). A similar trend was seen in animals on normal chow but was not significant because of the relatively smaller effects of both the Hfe–/– and APN–/– genotypes on glucose in mice on normal chow (fasting glucose levels, 126 ± 7 mg/dl in WT, 115 ± 4 mg/dl in Hfe–/–, and 129 ± 11 mg/dl in APN–/–:Hfe–/–, P = 0.16, data not shown).
Phlebotomy increases adiponectin and improves glucose tolerance in humans with high-normal serum ferritin.
We next sought to determine whether iron plays a causal role in determining adiponectin levels and the risk of MetS in humans. We studied humans with impaired glucose tolerance (IGT) whose serum ferritin levels were in the highest quartile of normal (221 ± 42 ng/ml, see Supplemental Table 2). Individuals with chronic inflammatory states, such as hepatitis, arthritides, or infections, were excluded, as were individuals with HH. Subjects received oral and frequently sampled intravenous glucose tolerance tests (OGTTs and FSIVGTTs) before and approximately 6 months after phlebotomy, which was sufficient to result in a fall in serum ferritin to the lowest quartile of normal (33 ± 12 ng/ml, P < 0.02 compared with before phlebotomy). The average blood donation was 3.7 units.
After phlebotomy, all subjects improved in the area under the glucose curve during OGTT, and the difference in the groups before and after phlebotomy was significant (Figure 6A, P = 0.03). All subjects had both IGT and impaired fasting glucose (IFG) prior to phlebotomy. After phlebotomy, one exhibited correction of both parameters, one exhibited correction of IGT only, and one exhibited correction of IFG only. There was a nonsignificant trend toward improvement in the FSIVGTT parameters of insulin secretory capacity (acute insulin response to glucose [AIRg], ~2.5 fold, Figure 6B) and insulin sensitivity (Si, ~3 fold, Figure 6C). Similar trends toward improvement were seen in the homeostasis model indices of β cell function and insulin resistance (data not shown). The disposition index, the product of Si and AIRg and the better predictor of overall diabetes risk (51), improved significantly (Figure 6D, P < 0.05). After phlebotomy, all subjects showed an increase in serum adiponectin (range of increase 9%–55%, see Supplemental Table 2, P < 0.02 by paired t test). Similar to the results seen in mice on a low-iron diet (Figure 2A), adiponectin increased despite an average weight gain of 2.6 kg, with weight gain occurring in two-thirds of the subjects.
Figure 6. Phlebotomy improves glucose tolerance.

Human men with impaired glucose tolerance and serum ferritin levels in the highest quartile of normal underwent phlebotomy to decrease their ferritin values to the lowest quartile of normal. (A) Results of oral glucose tolerance testing before (open symbols) and approximately 6 months after initiation of phlebotomy (closed symbols). The integrated area under the glucose curve for the 120-minute test is shown. Squares represent individual values, and circles represent the means. (*P < 0.03 by paired t test). (B) Insulin secretory capacity (AIRg) and (C) Si were determined from FSIVGTT performed before and after phlebotomy. (D) The disposition index was calculated from the data in B and C. *P < 0.05 by paired t test.
Discussion
Increased serum ferritin is associated with insulin resistance and increased risk for diabetes (1–3). Recent studies (3, 22, 23) have also noted an association between serum ferritin and adiponectin, the adipocyte-specific, insulin-sensitizing hormone. We have verified that serum ferritin levels, reflecting tissue iron stores, are more tightly associated with adiponectin than its more common predictor, obesity. More importantly, the relationship is causal, reflecting regulation of adiponectin transcription by iron. We have demonstrated this in cultured cells, by manipulation of iron stores and adipocyte iron levels in rodents, and in humans. Adiponectin is causally linked to insulin sensitivity (26), and, consistent with this, the changes in adiponectin in response to iron are accompanied by changes in glucose tolerance and insulin sensitivity.
The fact that adipocytes use iron levels to regulate adiponectin suggests a role for adipocytes in coordinating organism-wide metabolic responses to iron availability, as they do for responses to overall macronutrient status. There is other evidence for cross-talk between iron and adipocyte metabolism. Insulin treatment, for example, increases iron uptake by increasing cell surface expression of transferrin receptor 1 in 3T3-L1 and rat adipocytes (52, 53). Iron induces lipolysis in cultured adipocytes and modulates the lipolytic response to norepinephrine (34, 54). Close coregulation of iron levels and metabolic parameters, such as fuel preference, is conserved from yeast (55, 56) to mammals (46). The need for this coregulation is consistent with the necessity of iron for electron transport and other redox reactions combined with its dangers as a potent oxidant.
Adipocytes are well suited for their iron-sensing role. They express not only common regulators of iron homeostasis, such as ferritin and iron regulatory proteins (57), but also iron-related proteins with restricted tissue expression, including transferrin receptor 2, HFE, hepcidin (40, 41), and, as shown herein, ferroportin. Ferroportin is the only known cellular iron export protein and is critical in the regulation of tissue iron stores (42, 43, 45). Hepcidin, a peptide secreted by the liver in response to serum iron, binds ferroportin, resulting in ferroportin internalization and degradation (44). Under conditions of high iron, hepcidin-induced downregulation of ferroportin in duodenal enterocytes prevents dietary iron from entering the circulation. Significant expression of ferroportin has been heretofore reported to be limited to duodenal enterocytes, hepatocytes, and macrophages (42, 43). Expression of ferroportin in adipocytes allows them to serve an iron-sensing function with greater dynamic range and sensitivity, as evidenced by the effect of tissue-specific ferroportin deletion.
Ferroportin expression dictates that adipocytes will respond to hepcidin by downregulating ferroportin and sequestering iron. Of added significance, hepcidin is not only regulated by iron but is also induced in inflammation (58). Thus, adipocytes will regulate adiponectin not only in response to the availability of iron but also to the inflammatory state of the organism. Such integration should be adaptive, given the dangers posed by the potentially additive dangers of oxidative stress, resulting from high rates of fuel oxidation, high tissue iron levels, and inflammation. We have previously shown, for example, that high iron itself activates fat oxidation (59), and we speculate that in situations of high-iron stores, dampening fatty acid oxidation by decreasing adiponectin (60) might be protective to tissues because of these additive oxidative stresses.
Most physiologic regulation of adiponectin levels is attributable to changes in transcription mediated by FOXO1 and PPARγ (36, 37). We demonstrate that iron causes decreased acetylation of FOXO1, without changing its level of phosphorylation. Deacetylation of FOXO1, however, is generally associated with increased adiponectin transcription (61). The resolution of this paradox appears to be that deacetylation is accompanied by increased binding of FOXO1, not only to activator sites in the adiponectin promoter, but also to a PPRE, which has been reported to repress transcription when co-occupied by FOXO1 and PPARγ (39). These results differ somewhat from those of Qiao et al. who reported that FOXO1 interaction with C/EBPα was enhanced by SIRT1, leading to increased adiponectin transcription in 3T3-L1 cells (36). Although we did see increased SIRT1 activity associated with increased binding of FOXO1 to the adiponectin promoter, we saw no enhancement of C/EBPα binding to FOXO1. Furthermore, the effect of SIRT1 activation was not sufficient to increase adiponectin transcription, so we can only speculate that the effect of occupancy of the PPARγ site that is stimulated by iron treatment is dominant, as has been reported for that site (39). Interestingly, fatty acid–induced reactive oxygen species have been shown to decrease adiponectin transcription by inducing FOXO1 phosphorylation and nuclear exclusion (62). We did not, however, observe any change in FOXO1 phosphorylation with iron, suggesting that inflammation is not the direct mediatory of adiponectin regulation by iron. FOXO1 also regulates adipocyte differentiation, size, and energy expenditure (63, 64), and the FOXO family of transcription factors is involved in nutrient sensing and adaptation of metabolism to available nutrients (65). Our results add iron to the list of factors that are integrated by FOXO1 in determining its regulation of adipocyte metabolism and adiponectin secretion.
The regulation of adiponectin by iron sheds light on glucose homeostasis in HH, a condition associated with a high prevalence of diabetes (20, 66). In HH, failure to induce hepcidin in the liver results in low hepcidin levels. This results in failure to downregulate ferroportin in enterocytes, leading to unregulated iron absorption from the gut. Iron overload of islets leads to diabetes, largely on the basis of decreased insulin secretion rather than insulin resistance (20, 67). In fact, a mouse model of HH and nondiabetic humans with HH exhibit increased insulin sensitivity (46), although they may also develop secondary insulin resistance from independent conditions such as obesity (20). This presents a paradox in that dietary iron overload is associated with typical type 2 diabetes, a condition of insulin resistance (1–4). The paradox is resolved by our finding of ferroportin in adipocytes. In the limited tissues expressing significant levels of ferroportin, such as macrophages, decreased hepcidin in HH results in increased ferroportin expression and therefore decreased iron levels despite total body iron overload (48, 68). As reported herein, adipocytes follow this pattern of decreased iron content in HH, explaining our previously reported increased levels of adiponectin in mouse HH (46) and the parallel findings in humans reported here. Adiponectin is the mediator of the effects of low adipocyte iron on insulin sensitivity, as demonstrated by the lack of improved glucose tolerance in Hfe–/– mice that also have deletion of the adiponectin gene. Decreased adipocyte iron may also explain our previous demonstration of increased size of Hfe–/– adipocytes, in that iron-deficient adipocytes exhibit decreased rates of lipolysis (34).
Within the range of iron used in the current studies the inverse relationship between iron and adiponectin is a continuum. It is possible, however, that adiponectin levels might also drop under conditions of severe iron deficiency. A modal relationship between iron and adiponectin would be adaptive, in that an organism might also seek to limit fat oxidation in situations of very low iron, wherein metallation of mitochondrial proteins might be limited. Consistent with adiponectin levels falling with very low adipocyte iron, we have reported that insulin sensitivity decreases in HH subjects following phlebotomy (19), accompanied by decreased serum adiponectin levels (data not shown).
Proof of the effects of ferroportin in adipocytes, and of adiponectin as one of the mediators of the metabolic effects of iron, is provided by the phenotype of mice with adipocyte-specific deletion of ferroportin. These mice exhibit increased adipocyte iron, decreased adiponectin, insulin resistance, and decreased glucose tolerance. Thus, iron overload restricted to adipocytes is sufficient to affect glucose tolerance, although iron also exerts effects in other cells (e.g., muscle and β cells) that add to this diabetes risk (18, 19, 21, 59). Of note, in our tissue-specific deletion of ferroportin, we used a mouse expressing Cre recombinase under control of the AP2 promoter. Although certain AP2-driven constructs may be expressed in macrophages, we observed no change in macrophage ferroportin expression in the Fpn1–/– mice. Thus, the metabolic effects induced by changes in ferroportin expression in the knockout model can be confidently ascribed to the adipocyte.
The studies of the non-HH human populations also reveal close and causal relationships among serum ferritin, adiponectin, and glucose tolerance status. In men, ferritin predicted adiponectin approximately 4 times better than BMI. In the cohort of obese men, with or without MetS (Figure 1, D and E), ferritin was more strongly associated with diabetes than MetS. Although an association of MetS with ferritin has been reported (7), the stronger relationship with diabetes may be explained by the adverse effects of iron on both insulin secretion and adiponectin. The fact that MetS subjects without diabetes only trended toward higher ferritin but still had lower adiponectin demonstrates that other factors such as obesity modulate adiponectin independently of iron.
The relationship of ferritin to adiponectin was weaker in women, likely because the range of ferritin values was significantly narrower and lower in women than in men. On average, however, lower ferritin levels are accompanied by higher adiponectin levels in women, consistent with our hypothesis. The data suggest that ferritin may be a major determinant of the higher adiponectin levels that exist in women (31). The results may also speak to the differences between men and women in diabetes risk, with women having a decreased incidence of diabetes prior to menopause (69).
Insulin resistance, obesity, and diabetes are associated both with iron and with chronic inflammation (2, 3, 7, 8, 57), and ferritin is a known acute-phase reactant to inflammatory stimuli (11). We believe, however, that the association between serum ferritin and adiponectin reflects tissue iron rather than systemic inflammation for three reasons. First, while we found an association between serum ferritin and adiponectin in the human population as a whole, restricting the analysis to individuals with normal serum ferritin strengthened rather than weakened the association. In healthy adults, serum ferritin levels within this normal range are closely associated with mobilizable iron and reliably predict total body iron stores (70). Second, inclusion in multivariate analysis of CRP, another marker of the acute-phase response, had little effect on the correlation coefficient of serum ferritin with adiponectin, as was the case in other studies (3, 6, 7). In this study, the relationship between ferritin and adiponectin was also independent of IL-6 and TNF-α. Third, our in vitro and in vivo studies reveal that decreasing tissue iron stores is sufficient to decrease ferritin and increase adiponectin. Our studies do not eliminate the possibilities, however, that systemic inflammation is making an additional contribution to increased serum ferritin levels or that adiponectin transcription might also be regulated independently by signals that reflect oxidant stress and/or inflammation.
Because of the association of iron both with insulin resistance and insulin deficiency (18, 20, 21, 67, 71, 72), we reasoned that phlebotomy might improve both, leading to additive effects in decreasing diabetes risk. Phlebotomy in humans with IGT led to significant improvements in glucose tolerance and the glucose disposition index, a reflection of insulin secretion and sensitivity. The fact that phlebotomy decreased ferritin also confirms that the initially high-normal ferritin values were not due to inflammation. These results are consistent with our recent demonstration that iron restriction affords significant protection from diabetes to the Lep–/– (ob/ob) mouse, improving both insulin sensitivity and insulin secretion (21). The metabolic effects of iron, even within the “normal” range, therefore likely explain the many epidemiologic observations that have been made concerning iron and diabetes risk, such as the fact that periodic blood donation is associated with increased insulin sensitivity (73).
It should be emphasized that this small clinical study was designed as a proof of concept and does not serve as an adequate clinical trial to guide medical practice. To minimize variance, for example, we only studied men and, to maximize the effect size, we examined only those with IGT. The weight gain observed in 2 out of the 3 subjects is also of potential concern, although it also suggests that the improvements seen were not related to coincident changes in lifestyle. Iron deficiency is associated with obesity (74), although the phlebotomy was controlled such that none of these subjects became iron deficient. The mechanism underlying weight gain in iron deficiency is unknown, although we have demonstrated that decreased iron results in decreased capacity for fatty acid oxidation (59). The results suggest that larger clinical trials with more diverse populations are clearly indicated. In addition, because of the interrelationships between iron and oxygen sensing (75), and because this study was performed in subjects living at 4,000–5,000 feet altitude, any future trials should include subjects living at sea level to allow generalization of the results.
In sum, we have presented evidence that adipocyte iron levels regulate adiponectin transcription and serum protein levels. These data further highlight the role of the adipocyte as a key regulator of metabolism in all tissues, based on integrated sensing of nutritional stores and iron availability. Understanding the mechanisms by which adipocytes sense and respond to intracellular iron levels is fundamental to understanding this role and merits further investigation. In addition to regulating adiponectin, the fact that iron is also a factor in determining insulin secretory capacity (19, 21) demonstrates a rational basis for the observed associations among iron, glucose tolerance, and the MetS. These data also support the need for further clinical trials on the effects of iron reduction on diabetes and the MetS.
Methods
Experimental animals.
Dietary iron manipulations were accomplished with diets containing 7 mg/kg, 330 mg/kg, 500 mg/kg, or 20 g/kg carbonyl iron (Harlan Teklad) for a period of 2 months before phenotyping. High-fat diet (Research diets D12451) contained 45% calories from fat. Targeted mutagenesis produced a knockout of the Hfe gene (76). This and the mutations listed below were bred onto either the 129/SvEvTac or C57BL6/J genetic backgrounds for at least 5 generations. Adipocyte-specific ferroportin knockout mice were generated by breeding mice with LoxP sites flanking exon 6 and 7 of the ferroportin gene (45), provided by Nancy C. Andrews (Duke University), with mice containing an AP2-promoter–driven Cre recombinase (The Jackson Laboratory). Mice with deletion of the adiponectin gene (50) were provided by Phillip Scherer (University of Texas Southwestern, Dallas, Texas, USA). Age- and sex-matched WT littermates were used as controls. Body composition analysis was performed using magnetic resonance imaging.
Euglycemic clamp procedure.
The jugular vein was catheterized under avertin anesthesia, using Micro-Renathane tubing (Braintree Scientific Inc., MRE 025). After a 48-hour recovery, mice were fasted overnight. A dual infusion pump (Harvard Apparatus, Pump 33) was used to infuse insulin at a constant flow rate and 50% dextrose at a variable rate to maintain glucose at 100–150 mg/dl. Glucose was measured at 10-minute intervals as described above for glucose tolerance testing. After steady state was reached for 3 successive glucometer readings, the glucose infusion rate was determined.
Human subjects.
Subjects with HH have been described elsewhere (19). Studies with these subjects were approved by the University of Utah IRB. The subjects depicted in Figure 1, A–C, were part of a larger study and were described previously (27). Diabetes status was confirmed by OGTT. Diabetic subjects were on dietary therapy only, with fasting plasma glucose between 125 mg/dl and 175 mg/dl. The study protocol was approved by Pennington Biomedical Research Center’s IRB. All sera were obtained after a 12-hour overnight fast. Insulin sensitivity was assessed by a 1-step, high-dose (120 mU × m–2 × min–1) hyperinsulinemic-euglycemic clamp (27).
The obese men (BMI ≥35, with or without MetS) whose data are presented in Figure 1, D and E, were selected from subjects recruited from the general population as members of pedigrees for genetics studies or as subjects in a gastric bypass study (77), under a protocol approved by the University of Utah IRB.
For the phlebotomy study, informed consent was obtained, and OGTT, serum ferritin, and blood counts were measured to determine eligibility for phlebotomy. Subjects were excluded who had any significant chronic or inflammatory diseases, including cancer, hepatitis, renal failure (serum creatinine >1.4), arthritis, or autoimmune diseases. HFE genotyping using allele-specific PCR primers (20) was performed to rule out HH. OGTT and FSIVGTT (78) were performed in the CSC, each after a 12-hour fast. Glucose tolerance status was defined according to World Health Organization criteria (79). FSIVGTT results were analyzed by MINMOD software to determine the AIRg and Si (80). Subjects then donated blood at a Red Cross center (1 unit per month). After 2 to 3 units were donated by each subject, hematocrit, hemoglobin, and iron indices were determined, and phlebotomy continued until ferritin was in the lowest quartile of normal (goal = 50 μg/dl). In no case did blood counts decrease by more than 10%. Two to three months after reaching the ferritin goal, subjects were retested with OGTT and FSIVGTT.
Reagents and assays.
Reagents were purchased from Sigma-Aldrich unless otherwise noted. Serum CRP and TNF-α were measured by ELISA (R&D Systems), IL-6 was measured by ELISA (Abnova), ferritin was measured by ARUP Laboratories, insulin was measured by radioimmunoassay (Diagnostic Products), and adiponectin was measured by ELISA (ALPCO).
3T3-L1 adipocyte culture and differentiation.
3T3-L1 adipocytes (ATCC) were maintained in high-glucose DMEM (HG-DMEM) supplemented with 10% bovine serum and penicillin/streptomycin (Invitrogen). For differentiation (81), cells were incubated in HG-DMEM with 10% FBS (Thermo Scientific) for 48 hours after confluence. Cells were then cultured in differentiation media I (HG-DMEM, 10% FBS, 1 μg/ml insulin, 0.25 μg/ml dexamethasone, 0.5 mM IBMX) for 4 days, followed by differentiation media II (HG-DMEM, 10% FBS, 1 μg/ml insulin) for 48 hours. Prior to experiments, cells were cultured overnight in low-glucose DMEM (LG-DMEM, Invitrogen) with 10% FBS. All experiments were performed in LG-DMEM.
Plasmids, nucleofection, and luciferase assay.
The proximal 1,460 bp of the murine adiponectin promoter was amplified from genomic epididymal fat pad DNA and inserted into the pGL4.10 vector (Promega). Plasmids were nucleofected into differentiated 3T3-L1 adipocytes using an Amaxa Nucleofector with Kit L (Lonza). Luciferase and renilla activity were quantified using the Dual Luciferase Reporter Assay System (Promega) and a 96-well plate luminometer.
Isolation of primary adipocytes.
Epididymal and retroperitoneal fat pads were removed from male and female mice, respectively, and incubated in HBSS with 1% BSA and 20 mg collagenase, type I, for 30 minutes at 37°C, rotating at 180 rpm. Adipocytes were filtered through 100-μm nylon mesh and rinsed with HBSS–1% BSA. Following centrifugation at 500 g, the fat cake was transferred to a clean tube, rinsed, and centrifuged. Isolated adipocytes were either used immediately or flash frozen in liquid nitrogen.
Quantification of transcript.
Quantitative RT-PCR was performed as described previously (46). Briefly, mRNA was extracted from primary and 3T3-L1 adipocytes using TRIzol (Invitrogen), purified using an RNeasy column (QIAgen), and synthesized into cDNA using a First-Strand cDNA Synthesis Kit (Invitrogen). Real-time PCR was performed with a LightCycler (Roche Diagnostics). cDNA products were quantified using the LightCycler software (ΔΔCT method). mRNA levels of specific genes were normalized to cyclophilin A.
Western blotting.
Antibodies used included pFOXO1 (Ser 256), acFOXO1, total FOXO1, and pAKT from Santa Cruz Biotechnologies Inc. Rabbit α-mouse ferroportin antibody was provided by Jerry Kaplan (University of Utah).
ChIP.
ChIP studies were performed as described previously (82). Chromatin was extracted from 3T3-L1 adipocytes on day 9 after differentiation using the SimpleChIP Kit (Cell Signaling Technology). Cells were treated with FeSO4 or 0.1 N HCl control for 24 hours prior to cross-linking for 10 minutes with 1% formaldehyde. Cells were then lysed and sonicated 3 times for 20 seconds using a sonic dismembrator (Fisher Scientific). Lysates were precleared with protein A agarose beads (Millipore). FKHR or PPARγ antibody (Santa Cruz Biotechnology Inc.) was applied. DNA was released from protein-DNA complexes by proteinase K digestion and then subjected to quantitative real-time PCR for the APN response elements for PPARγ (PPRE) (83) and FOXO1 using the Power SYBR Green Kit (Applied Biosystem). ChIP-qPCR data were normalized to input samples for the amount of chromatin and for immunoprecipitation efficiency by normal IgG controls.
Statistics.
Descriptive statistics in the text and figures are represented as average ± SEM. The Pearson’s correlation coefficient was calculated to test for correlation between 2 parameters. Difference between 2 slopes was calculated by analysis of covariance. Multivariate logistic regression analysis was sequentially performed to investigate the role of CRP, BMI, diabetes, and gender on the ferritin-adiponectin association. An unpaired 2-tailed Student’s t test was used to determine significance between controls and individual experimental groups, whereas a paired t test was used to compare values in the same subjects before and after phlebotomy. One-way ANOVA was used to compare series of data. P < 0.05 was considered significant for all tests. All statistical analyses were performed with GraphPad PRISM 5.0d for Macintosh. In the studies depicted in Figure 1, adiponectin values (n = 2) that were more than 3 SD from the mean were excluded from the analysis.
Study approval.
Animal studies were approved by the Institutional Animal Care and Use Committee of the University of Utah, under IACUC approval A3031-01. The human studies were approved by the IRB of the University of Utah, protocols 0880 and 20094. Studies were also reviewed and approved by the Advisory Committee to the Clinical Services Core (CSC) of the Center for Clinical and Translational Science. Additional previously unpublished data was used from human studies completed at the Pennington Biomedical Research Center and at the University of Utah. Both studies were approved by the IRBs of the respective institutions, as previously published (27, 77).
Supplementary Material
Acknowledgments
This work was supported by the Research Service of the Veterans Administration and NIH grants DK59512 and 5-T32-DK-007115 and the University of Utah Clinical and Translational Science Award funded by the National Center for Advancing Translational Sciences (UL1RR025764). We would like to thank Tom Greene (UL1 RR025764) for help with the statistical analysis of our data and also to acknowledge the assistance of the Clinical Services, Technology, and Biostatistics and Study Design Cores of the CTSA. Mice used in the study were supplied by Nancy Andrews (Duke University) and Phillip Scherer (University of Texas Southwestern).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3529–3540. doi:10.1172/JCI44421.
References
- 1.Fernandez-Real JM, Lopez-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. 2002;51(8):2348–2354. doi: 10.2337/diabetes.51.8.2348. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Cogswell ME. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care. 1999;22(12):1978–1983. doi: 10.2337/diacare.22.12.1978. [DOI] [PubMed] [Google Scholar]
- 3.Forouhi NG, et al. Elevated serum ferritin levels predict new-onset type 2 diabetes: results from the EPIC-Norfolk prospective study. Diabetologia. 2007;50(5):949–956. doi: 10.1007/s00125-007-0604-5. [DOI] [PubMed] [Google Scholar]
- 4.Jiang R, Manson JE, Meigs JB, Ma J, Rifai N, Hu FB. Body iron stores in relation to risk of type 2 diabetes in apparently healthy women. JAMA. 2004;291(6):711–717. doi: 10.1001/jama.291.6.711. [DOI] [PubMed] [Google Scholar]
- 5.Afkhami-Ardekani M, Rashidi M. Iron status in women with and without gestational diabetes mellitus. J Diabetes Complications. 2009;23(3):194–198. doi: 10.1016/j.jdiacomp.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 6.Sharifi F, Nasab NM, Zadeh HJ. Elevated serum ferritin concentrations in prediabetic subjects. Diab Vasc Dis Res. 2008;5(1):15–18. doi: 10.3132/dvdr.2008.003. [DOI] [PubMed] [Google Scholar]
- 7.Jehn M, Clark JM, Guallar E. Serum ferritin and risk of the metabolic syndrome in U.S. adults. Diabetes Care. 2004;27(10):2422–2428. doi: 10.2337/diacare.27.10.2422. [DOI] [PubMed] [Google Scholar]
- 8.Gillum RF. Association of serum ferritin and indices of body fat distribution and obesity in Mexican American men—the Third National Health and Nutrition Examination Survey. Int J Obes Relat Metab Disord. 2001;25(5):639–645. doi: 10.1038/sj.ijo.0801561. [DOI] [PubMed] [Google Scholar]
- 9.Iwasaki T, et al. Serum ferritin is associated with visceral fat area and subcutaneous fat area. Diabetes Care. 2005;28(10):2486–2491. doi: 10.2337/diacare.28.10.2486. [DOI] [PubMed] [Google Scholar]
- 10.Qi L, van Dam RM, Rexrode K, Hu FB. Heme iron from diet as a risk factor for coronary heart disease in women with type 2 diabetes. Diabetes Care. 2007;30(1):101–106. doi: 10.2337/dc06-1686. [DOI] [PubMed] [Google Scholar]
- 11.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340(6):448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 12.De Domenico I, McVey Ward D, Kaplan J. Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol. 2008;9(1):72–81. doi: 10.1038/nrm2295. [DOI] [PubMed] [Google Scholar]
- 13.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 14.Bofill C, et al. Response to repeated phlebotomies in patients with non-insulin-dependent diabetes mellitus. Metabolism. 1994;43(5):614–620. doi: 10.1016/0026-0495(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Lopez-Bermejo A, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on vascular reactivity. Diabetes Care. 2002;25(12):2249–2255. doi: 10.2337/diacare.25.12.2249. [DOI] [PubMed] [Google Scholar]
- 16.Facchini FS, Hua NW, Stoohs RA. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology. 2002;122(4):931–939. doi: 10.1053/gast.2002.32403. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Real JM, Penarroja G, Castro A, Garcia-Bragado F, Hernandez-Aguado I, Ricart W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and beta-cell function. Diabetes. 2002;51(4):1000–1004. doi: 10.2337/diabetes.51.4.1000. [DOI] [PubMed] [Google Scholar]
- 18.Jouihan HA CP. Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol Med. 2008;14(3–4):98–108. doi: 10.2119/2007-00114.Jouihan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abraham D, Rogers J, Gault P, Kushner J, McClain D. Increased insulin secretory capacity but decreased insulin sensitivity after correction of iron overload by phlebotomy in hereditary haemochromatosis. Diabetologia. 2006;49(11):2546–2551. doi: 10.1007/s00125-006-0445-7. [DOI] [PubMed] [Google Scholar]
- 20.McClain D, et al. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia. 2006;49(7):1661–1669. doi: 10.1007/s00125-006-0200-0. [DOI] [PubMed] [Google Scholar]
- 21.Cooksey RC, et al. Dietary iron restriction or iron chelation protects from diabetes and loss of beta-cell function in the obese (ob/ob lep–/–) mouse. . Am J Physiol Endocrinol Metab. 2010;298(6):E1236–1243. doi: 10.1152/ajpendo.00022.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fargnoli JL, Fung TT, Olenczuk DM, Chamberland JP, Hu FB, Mantzoros CS. Adherence to healthy eating patterns is associated with higher circulating total and high-molecular-weight adiponectin and lower resistin concentrations in women from the Nurses’ Health Study. Am J Clin Nutr. 2008;88(5):1213–1224. doi: 10.3945/ajcn.2008.26480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mojiminiyi OA, Marouf R, Abdella NA. Body iron stores in relation to the metabolic syndrome, glycemic control and complications in female patients with type 2 diabetes. Nutr Metab Cardiovasc Dis. 2008;18(8):559–566. doi: 10.1016/j.numecd.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Ku B-J, Kim S-Y, Lee T-Y, Park K-S. Serum ferritin is inversely correlated with serum adiponectin level: population-based cross-sectional study. Dis Markers. 2009;27(6):303–310. doi: 10.3233/DMA-2009-0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weyer C, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86(5):1930–1935. doi: 10.1210/jc.86.5.1930. [DOI] [PubMed] [Google Scholar]
- 26.Kubota N, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277(29):25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 27.Stull AJ, Galgani JE, Johnson WD, Cefalu WT. The contribution of race and diabetes status to metabolic flexibility in humans. Metabolism. 2010;59(9):1358–1364. doi: 10.1016/j.metabol.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fleming DJ, et al. Iron status of the free-living, elderly Framingham Heart Study cohort: an iron-replete population with a high prevalence of elevated iron stores. Am J Clin Nutr. 2001;73(3):638–646. doi: 10.1093/ajcn/73.3.638. [DOI] [PubMed] [Google Scholar]
- 29.Nelson R, Chawla M, Connolly P, LaPorte J. Ferritin as an index of bone marrow iron stores. South Med J. 1978;71(12):1482–1484. doi: 10.1097/00007611-197812000-00011. [DOI] [PubMed] [Google Scholar]
- 30.Cook JD, Lipschitz DA, Miles LEM, Finch CA. Serum ferritin as a measure of iron stores in normal subjects. Am J Clin Nutr. 1974;27(7):681–687. doi: 10.1093/ajcn/27.7.681. [DOI] [PubMed] [Google Scholar]
- 31.Arita Y, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 32.[No authors listed] Third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. [PubMed] [Google Scholar]
- 33.Green A, Basile R, Rumberger JM. Transferrin and iron induce insulin resistance of glucose transport in adipocytes. Metabolism. 2006;55(8):1042–1045. doi: 10.1016/j.metabol.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 34.Rumberger JM, Peters T, Jr, Burrington C, Green A. Transferrin and iron contribute to the lipolytic effect of serum in isolated adipocytes. Diabetes. 2004;53(10):2535–2541. doi: 10.2337/diabetes.53.10.2535. [DOI] [PubMed] [Google Scholar]
- 35.Casey JL, et al. Iron-responsive elements: regulatory RNA sequences that control mRNA levels and translation. Science. 1988;240(4854):924–928. doi: 10.1126/science.2452485. [DOI] [PubMed] [Google Scholar]
- 36.Qiao L, Shao J. SIRT1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein alpha transcriptional complex. J Biol Chem. 2006;281(52):39915–39924. doi: 10.1074/jbc.M607215200. [DOI] [PubMed] [Google Scholar]
- 37.Liu M, Liu F. Transcriptional and post-translational regulation of adiponectin. Biochem J. 2010;425(1):41–52. doi: 10.1042/BJ20091045. [DOI] [PubMed] [Google Scholar]
- 38.van der Heide LP, Smidt MP. Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci. 2005;30(2):81–86. doi: 10.1016/j.tibs.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 39.Fan W, et al. FOXO1 transrepresses peroxisome proliferator-activated receptor gamma transactivation, coordinating an insulin-induced feed-forward response in adipocytes. J Biol Chem. 2009;284(18):12188–12197. doi: 10.1074/jbc.M808915200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farahani P, et al. Obesity in BSB mice is correlated with expression of genes for iron homeostasis and leptin. Obes Res. 2004;12(2):191–204. doi: 10.1038/oby.2004.26. [DOI] [PubMed] [Google Scholar]
- 41.Bekri S, et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology. 2006;131(3):788–796. doi: 10.1053/j.gastro.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–19912. doi: 10.1074/jbc.M000713200. [DOI] [PubMed] [Google Scholar]
- 43.Donovan A, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403(6771):776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 44.Nemeth E, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 45.Donovan A, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. doi: 10.1016/j.cmet.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Huang J, et al. Increased glucose disposal and AMP-dependent kinase signaling in a mouse model of hemochromatosis. J Biol Chem. 2007;282(52):37501–37507. doi: 10.1074/jbc.M703625200. [DOI] [PubMed] [Google Scholar]
- 47.Ross CE, Muir WA, Alan BP, Graham RC, Kellermeyer RW. Hemochromatosis. Pathophysiologic and genetic considerations. Am J Clin Pathol. 1975;63(2):179–191. doi: 10.1093/ajcp/63.2.179. [DOI] [PubMed] [Google Scholar]
- 48.Cairo G, Recalcati S, Montosi G, Castrusini E, Conte D, Pietrangelo A. Inappropriately high iron regulatory protein activity in monocytes of patients with genetic hemochromatosis. Blood. 1997;89(7):2546–2553. [PubMed] [Google Scholar]
- 49.Furukawa S, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114(12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nawrocki AR, et al. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem. 2006;281(5):2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- 51.Bergman RN, Finegood DT, Kahn SE. The evolution of beta-cell dysfunction and insulin resistance in type 2 diabetes. Eur J Clin Invest. 2002;3(1):35–45. doi: 10.1111/j.1365-2362.1973.tb00326.x. [DOI] [PubMed] [Google Scholar]
- 52.Tanner LI, Lienhard GE. Insulin elicits a redistribution of transferrin receptors in 3T3-L1 adipocytes through an increase in the rate constant for receptor externalization. J Biol Chem. 1987;262(19):8975–8980. [PubMed] [Google Scholar]
- 53.Davis RJ, Corvera S, Czech MP. Insulin stimulates cellular iron uptake and causes the redistribution of intracellular transferrin receptors to the plasma membrane. J Biol Chem. 1986;261(19):8708–8711. [PubMed] [Google Scholar]
- 54.Yamagishi H, Okazaki H, Shimizu M, Izawa T, Komabayashi T. Relationships among serum triacylglycerol, fat pad weight, and lipolysis in iron-deficient rats. J Nutr Biochem. 2000;11(9):455–460. doi: 10.1016/S0955-2863(00)00113-3. [DOI] [PubMed] [Google Scholar]
- 55.Haurie V, Boucherie H, Sagliocco F. The Snf1 protein kinase controls the induction of genes of the iron uptake pathway at the diauxic shift in Saccharomyces cerevisiae. J Biol Chem. 2003;278(46):45391–45396. doi: 10.1074/jbc.M307447200. [DOI] [PubMed] [Google Scholar]
- 56.Shakoury-Elizeh M, et al. Metabolic response to iron deficiency in Saccharomyces cerevisiae. J Biol Chem. 2010;285(19):14823–14833. doi: 10.1074/jbc.M109.091710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Festa M, Ricciardelli G, Mele G, Pietropaolo C, Ruffo A, Colonna A. Overexpression of H ferritin and up-regulation of iron regulatory protein genes during differentiation of 3T3-L1 pre-adipocytes. J Biol Chem. 2000;275(47):36708–36712. doi: 10.1074/jbc.M004988200. [DOI] [PubMed] [Google Scholar]
- 58.Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci U S A. 2005;102(6):1906–1910. doi: 10.1073/pnas.0409808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang J, et al. Iron overload and diabetes risk: a shift from glucose to fatty acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes. 2011;60(1):80–87. doi: 10.2337/db10-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamauchi T, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 61.Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. 2010;285(35):27396–27401. doi: 10.1074/jbc.M110.140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Subauste AR, Burant CF. Role of FoxO1 in FFA-induced oxidative stress in adipocytes. Am J Physiol Endocrinol Metab. 2007;293(1):E159–E164. doi: 10.1152/ajpendo.00629.2006. [DOI] [PubMed] [Google Scholar]
- 63.Nakae J, Kitamura T, Kitamura Y, Biggs WH, 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell. 2003;4(1):119–129. doi: 10.1016/S1534-5807(02)00401-X. [DOI] [PubMed] [Google Scholar]
- 64.Nakae J, et al. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes. 2008;57(3):563–576. doi: 10.2337/db07-0698. [DOI] [PubMed] [Google Scholar]
- 65.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27(16):2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 66.Hatunic M, Finucane FM, Brennan AM, Norris S, Pacini G, Nolan JJ. Effect of iron overload on glucose metabolism in patients with hereditary hemochromatosis. Metabolism. 2010;59(3):380–384. doi: 10.1016/j.metabol.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 67.Cooksey RC, et al. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology. 2004;145(11):5305–5312. doi: 10.1210/en.2004-0392. [DOI] [PubMed] [Google Scholar]
- 68.Brink B, Disler P, Lynch S, Jacobs P, Charlton R, Bothwell T. Patterns of iron storage in dietary iron overload and idiopathic hemochromatosis. J Lab Clin Med. 1976;88(5):725–731. [PubMed] [Google Scholar]
- 69.Szmuilowicz ED, Stuenkel CA, Seely EW. Influence of menopause on diabetes and diabetes risk. Nat Rev Endocrinol. 2009;5(10):553–558. doi: 10.1038/nrendo.2009.166. [DOI] [PubMed] [Google Scholar]
- 70.Walters GO, Miller FM, Worwood M. Serum ferritin concentration and iron stores in normal subjects. J Clin Pathol. 1973;26(10):770–772. doi: 10.1136/jcp.26.10.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mangiagli A, Italia S, Campisi S. Glucose tolerance and beta-cell secretion in patients with thalassaemia major. J Pediatr Endocrinol Metab. 1998;11:985–986. [PubMed] [Google Scholar]
- 72.Cheng K, et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010;120(6):2171–2183. doi: 10.1172/JCI35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez-Real JM, Lopez-Bermejo A, Ricart W. Iron stores, blood donation, and insulin sensitivity and secretion. Clin Chem. 2005;51(7):1201–1205. doi: 10.1373/clinchem.2004.046847. [DOI] [PubMed] [Google Scholar]
- 74.McClung J, Karl J. Iron deficiency and obesity: the contribution of inflammation and diminished iron absorption. Nutr Rev. 2009;67(2):100–104. doi: 10.1111/j.1753-4887.2008.00145.x. [DOI] [PubMed] [Google Scholar]
- 75.Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114(10):2015–2019. doi: 10.1182/blood-2009-05-189985. [DOI] [PubMed] [Google Scholar]
- 76.Zhou XY, et al. HFE gene knockout produces mouse model of hereditary hemochromatosis. Proc Natl Acad Sci U S A. 1998;95(5):2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adams TD, et al. Health outcomes of gastric bypass patients compared to nonsurgical, nonintervened severely obese. Obesity (Silver Spring). 2010;18(1):121–130. doi: 10.1038/oby.2009.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saad MF, et al. Method of insulin administration has no effect on insulin sensitivity estimates from the insulin-modified minimal model protocol. Diabetes. 1997;46(12):2044–2048. doi: 10.2337/diabetes.46.12.2044. [DOI] [PubMed] [Google Scholar]
- 79.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part. 1998;15(7):539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 80.Boston RC, Stefanovski D, Moate PJ, Sumner AE, Watanabe RM, Bergman RN. MINMOD Millennium: a computer program to calculate glucose effectiveness and insulin sensitivity from the frequently sampled intravenous glucose tolerance test. Diabetes Technol Ther. 2003;5(6):1003–1015. doi: 10.1089/152091503322641060. [DOI] [PubMed] [Google Scholar]
- 81.Chen Z, Torrens JI, Anand A, Spiegelman BM, Friedman JM. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1(2):93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 82.Chakrabarti P, Kandror KV. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem. 2009;284(20):13296–13300. doi: 10.1074/jbc.C800241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Seo JB, et al. Adipocyte determination- and differentiation-dependent factor 1/sterol regulatory element-binding protein 1c regulates mouse adiponectin expression. J Biol Chem. 2004;279(21):22108–22117. doi: 10.1074/jbc.M400238200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.