

Abstract
Peripheral T cell lymphomas (PTCLs) are heterogeneous neoplasms and represent about 12% of all lymphoid malignancies. They are often regarded as “orphan diseases,” a designation that does not reflect their real incidence but rather signifies the difficulties encountered in their classification, diagnosis, and treatment. Here we revise the current understanding of the pathobiological characteristics of the most common nodal PTCLs by focusing on the contribution given by high-throughput technologies and the identification of potential therapeutic targets proposed by translational studies.
Introduction
Tumors from peripheral T and NK cells (PTCLs) constitute about 12% of all lymphoid neoplasms (see WHO classification 2008 and ref. 1). They are often regarded as “orphan diseases,” reflecting the difficulties encountered in their classification, diagnosis, and treatment. On histogenetic grounds, PTCLs derive from distinct subpopulations of cells with different functions and molecular profiles. These include NK cells and γδ T lymphocytes, elements of the innate immune system, and αβ T cells that correspond to naive, central memory, effector, and specialized subsets, which contribute to the adaptive immune response (2). PTCLs can be detected worldwide, although some varieties display an endemic distribution; for example, adult T cell lymphoma/leukemia (ATLL) is most prevalent in Japan and the Caribbean basin, EBV-related extranodal NK/T cell lymphoma of the nasal type (ENKTCL/NT) most prevalent in Hong Kong and Central America, and enteropathy-associated T cell lymphoma (EATCL) most prevalent in the United Kingdom and Scandinavian countries (1). PTCLs are morphologically heterogeneous and include several distinct and provisional entities. Of these, angioimmunoblastic T cell lymphoma (AITL), PTCL not otherwise specified (PTCL-NOS), and anaplastic large cell lymphoma (ALCL) ALK+ or ALK– (anaplastic lymphoma kinase–positive or –negative, respectively) (Table 1 and ref. 1). Phenotypically, PTCLs typically lack one or more of the T cell–associated antigens (3, 4), and some of these antigens are the target of humanized mAbs administered in clinics (e.g., CD52) (5), emphasizing the utility of determining the targeted antigen before therapy initiation in order to maximize efficacy and avoid unwanted toxicities. Furthermore, PTCLs can express distinctive, diagnostically relevant markers, for example, markers of follicular T helper lymphocytes (FTHs) in AITL; CD30, EMA, and perforin in ALCL; and CD25 and FOXP3 in ATLL (1). PCR reveals clonal rearrangement of TCR genes in 98% of cases when the BIOMED2 β and γ probes are applied on fresh and/or frozen tissues (6). This protocol has been developed in order to provide an efficient and standardized tool for the investigation of clonality in lymphoproliferative processes and is currently applied in routine diagnostics (6). Conventional cytogenetics, comparative genomic hybridization, and SNP array studies have revealed complex karyotypes (Figure 1 and ref. 7) but only a few recurrent genetic aberrations, including t(2;5)(p23;q35) and variants in ALK+ ALCL and iso7q in hepatosplenic (HS) T cell lymphoma (TCL) (1). Other abnormalities, such as the t(6;7)(p25;q32.3) recently shown in ALK– ALCLs by massive parallel sequencing, need confirmation (8).
Table 1.
Clinico-pathological features of the most common nodal PTCL subtypes
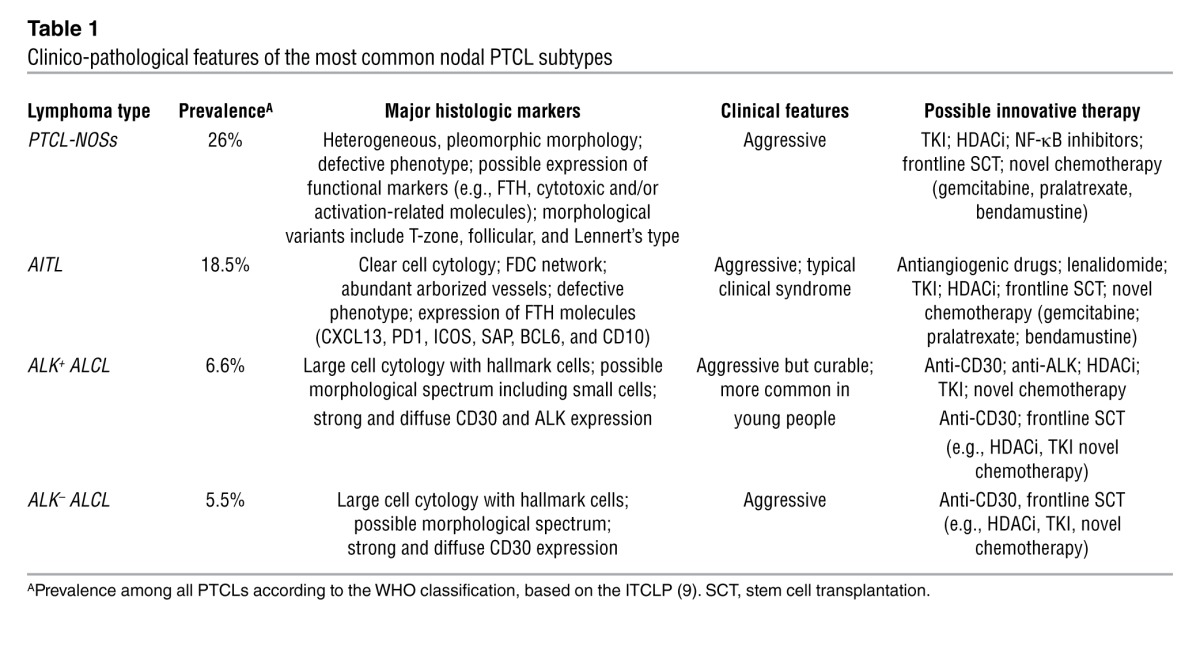
Figure 1. Prevalence of genomic imbalances in terms of copy number variants in PTCL-NOS, based on SNP array karyotyping.

Created with data from ref. 7.

On clinical grounds, according to the results of the International Peripheral T Cell Lymphoma Project (ITCLP), which analyzed more than 1,300 cases worldwide, with a few exceptions, PTCLs run a very aggressive clinical course (9). In most instances, the 5-year overall survival rate ranges from 20% to 30%. This is at least in part due to the lack of sensitivity to alkylating agents and anthracyclines, as shown retrospectively by the ITCLP and experimentally by our group (9, 10). Thus, there is a cogent need to better understand the pathobiology of PTCLs and to develop novel and more effective therapeutic strategies. Here we review the most recent developments in this field, with special attention to the most common subtypes, the contribution given by high-throughput technologies, and the identification of potential targets proposed by translational studies.
Histogenesis
The origin of the different PTCLs was initially investigated by immunohistochemical analysis and has thus far been the object of a limited number of studies. In 2004 Tsuchiya and coworkers tried to subdivide PTCLs into Th1- and Th2-derived neoplasms, based on the expression of markers CXCR3, CCR6, and ST2(L) (11). Additionally, these authors assessed OX40 (also known as CD134) expression as an indicator of TCR activation. AITL and ALCL samples were both positive for OX40 but differed in the expression of CXCR3 (detected in the former) and ST2(L) (recorded in the latter), findings that suggested these cancers derived from activated Th1 and Th2 cells, respectively. PTCL-NOSs appeared much more heterogeneous, with variable staining patterns for ST2(L), CCR6, and CXCR3. Notably, about 50% of these tumors could not be assigned to either cell type because they lacked expression of all markers tested (11). In 2006, Geissinger et al. reported that AITL and ALCL share a common effector cell phenotype (CD45RA–CD45R0+CD27–) but differ in the expression of activation and cytotoxic markers (characteristically observed in the ALCL setting) (12). On the other hand, five of eight PTCL-NOSs tested by these authors expressed CD4 and carried a central memory profile (CD45RA–CD45R0+CD27+), while the remaining three could not be assigned to any normal counterpart because of the lack of both CD4 and CD8 expression (12). More recently, antibodies have been developed that allow the distinction of α/β from γ/δ T lymphocytes in formalin-fixed, paraffin-embedded (FFPE) tissue samples. These may assist in the lineage definition of the neoplastic cells, along with markers of activation (e.g., CD25 and CD30), Treg elements (FOXP3), or specialized categories of T lymphocytes such as FTH (see below). In addition, antibodies against the γ chain or γ/δ dimer of TCR identify a morphologically heterogeneous group of neoplasms with putative common derivation that span from most HS TCLs to mucocutaneous TCLs, T-LGLs (T cell large granular lymphocyte leukemias), some ENKTCLs/NTs, type II EATCL (13), and a small minority of PTCL-NOSs, all marked by an aggressive clinical course (14). However, immunohistochemistry remains an imprecise tool for the histogenetic definition of PTCLs, in part because these tumors are characterized by the loss of one or more of the T cell–associated markers as well as by the aberrancy of the global phenotypic profile (3, 10).
Much more relevant is the contribution expected from gene expression profiling. Unfortunately, most of these studies (15–19) provided very little information on the histogenesis of PTCLs, although they focused on several categories, from mycosis fungoides (MF) to cutaneous TCLs, AITL, PTCL-NOSs, and ALK+ and ALK– ALCL. This was related to limitations in the study design and/or technologies employed. Specifically, the normal match usually consisted of a pool of T lymphocytes, with no distinction between different subsets (the aim of the analyses being to identify the differences of the gene signature among PTCL histotypes), and/or the expression chip used contained a limited number of genes. In 2007 our group profiled a series of PTCL-NOSs, AITLs, and ALCLs mostly consisting of neoplastic cells and various samples of purified normal T lymphocytes (10). We found that on the whole, the PTCL-NOSs expression profile was closer to that of activated T lymphocytes and most PTCL-NOSs were related to CD4/helper T cells, with only a minority being more similar to the CD8/cytotoxic compartment. Interestingly, the comparison between the gene expression patterns (CD4/helper and CD8/cytotoxic) and the immunohistochemical search for CD4 and CD8 antigens showed a partial dissociation of these two parameters. Such results have been recently expanded by our group on a larger series of FFPE cases by adopting the Illumina DASL technology. In addition to confirming previous observations, this approach has shed light on the relationships between neoplastic cells and different functional stages of T lymphocytes, such as Th1, Th2, follicular T helper (FTH), central memory T, and effector T cell memory (2). Distinct groups of cases were clearly identified, related to the different counterparts. These findings are in keeping with the observations of other groups. De Leval et al. found a small subset of PTCL-NOSs with an expression signature related to FTH, suggesting similarity to AITL (see below) (20), while Iqbal et al. reported PTCL-NOSs cases showing an expression profile that suggested an origin either in FTH or in helper or cytotoxic T lymphocytes (21). The latter possessed more aggressive clinical behavior and in a separate study (22) were shown to correspond at least in part to the so-called Lennert’s lymphoma.
More recently, Wang et al. developed a mouse model in which SNF5, a chromatin-remodeling complex subunit with suppressor activity, could be conditionally inactivated in immature T cells (23). This led to onset of PTCLs via the activation of a MYC-driven signaling network, a stem cell transcriptional program, and TCR signaling engagement. The tumors had a CD44hiCD122loCD8+ phenotype, resembling a subset of memory T cells (23).
In 2007, our group (24) and others (20) reported that AITL is related to FTH cells, a finding that is in keeping with the hyperplasia of follicular dendritic cells (FDC) and the presence of EBV-infected B-lymphocytes characteristic of the tumor. These results—also confirmed by Iqbal et al. (21)—are supported by immunohistochemical analysis of a series of markers (CD10, BCL6, PD1, ICOS, SAP, CXCL13, c-MAF, and CCR5) that are expressed by the FTH compartment (25). Importantly, under neoplastic conditions at least three of these markers should be simultaneously expressed to designate a given population as FTH. Indeed, one can aberrantly be observed in a different setting because of cell plasticity (25). In addition, BCL6 and SAP are carried by FOXP3+ follicular Tregs (26), which are usually depleted in AITL (27). The availability of these gene expression profiling surrogates has allowed for the analysis of large series of FFPE PTCLs by confirming that the FTH phenotype is always carried by AITL and by showing that tumors assigned to other categories can share the same profile (28). This holds true for a certain number of PTCL-NOSs, at times corresponding to the so-called follicular variant, which are characterized by small- to medium-sized neoplastic cells with slightly irregular nuclear contours and a wide rim of clear cytoplasm in the absence of FDC and high-endothelial venules (28). An FTH origin has also been postulated for the primary cutaneous CD4+ small-medium TCL (29).
Additional histogenetic observations provided by gene expression profiling pertain to ALCL and ENKTCL/NT. According to Huang et al. (30), the latter are more similar to activated than to resting NK cells. Notably, in the same analysis, both tumors of NK and T derivation as well as a γ/δ TCL clustered together, showing high levels of granzyme H transcripts. In ALCL, both ALK+ and ALK– forms cluster together, their distinction being based on the different ALK/STAT3 signatures as well as on the expression of a series of genes (BCL6, PTPN12, C/EBPβ and serpinA1 for ALK+ ALCL, and CCR7, CNTFR, IL22, and IL21 for ALK– ALCL) (31). The close histogenetic relationship between the two forms of ALCL is underlined by the fact that the 16 genes that distinguish ALK– ALCL from PTCL-NOSs and AITL are similarly expressed in ALK+ ALCL (32). These findings provide a rationale for the decision taken in the 2008 WHO classification to maintain ALK– ALCL as a provisional entity distinct from PTCL-NOSs (1).
Molecular pathogenesis
In addition to histogenetic information, gene expression profiling studies provided relevant insights into the functional alterations of different PTCL subtypes.
First, a careful comparison of PTCL-NOSs with the closest normal cellular counterparts revealed the extensive deregulation of genes that control functions typically damaged in malignant cells, such as matrix remodeling, cell adhesion, transcription regulation, proliferation, and apoptosis. In particular, an analysis of ours (10) might explain the dissemination pattern of PTCL-NOSs, which involves frequent extranodal sites and bone marrow and spreads to peripheral blood, because it demonstrates the upregulation of the FN1, LAMB1, COL1A2, COL3A1, COL4A1, COL4A2, and COL12A1 genes, which promote local invasion and metastasis in different types of human cancers (33–35). In addition, our analysis revealed the deregulation of genes involved in apoptosis (e.g., MOAP1, ING3, GADD45A and GADD45B) (36–42) and chemo-resistance (e.g., CYR61 and NNMT) (33–35, 43–53), which may be responsible for the poor response to conventional chemotherapy.
Second, some studies based on gene expression profiling and/or immunohistochemistry suggested frequent deregulation of the NF-κB pathway in PTCL-NOSs (15, 16, 54, 55). Such findings, however, have not been confirmed by ourselves or others (10, 56) (see below).
Third, multiple groups have characterized aberrant tyrosine kinase (TK) signaling in PTCLs (10, 30, 32, 57). In particular, we showed that PTCLs/NOS constantly over-express PDGFRA and present with constitutive phosphorylation (i.e., activation) of the encoded protein (10, 57). Our group also found that the activation of PDGFRA is sustained by an autocrine stimulation, a mechanism subsequently found also in T-LGL (ref. 58 and Pileri, personal communication). Notably, PDGFR overexpression was later demonstrated in other T/NK-derived tumors, further strengthening its potential therapeutic relevance (see below) (30). In addition, it might play an important role in promoting and/or sustaining the neoplastic growth by counterbalancing the frequent alterations of the NF-κB pathway (see below) (ref. 59 and Figure 2). Along the same lines, in ALK+ ALCL the oncogenic TK nucleophosmin (NPM)-ALK activates STAT3, a major contributor to the tumor molecular signature (32), and sustains the expression of the growth promoter ICOS (60). In a subset of PTCL-NOSs, Streubel et al. detected the t(5;9)(q33; q22) translocation, which leads to the fusion transcript ITK-SYK with constitutive kinase activity (61) and possible pathogenic significance. Indeed, inhibition of SYK, a TCR downstream molecule, was found to be effective ex vivo by inhibiting cell proliferation and inducing cell death (62). More recently, somatic mutations of isocitrate dehydrogenase 2 (IDH2) have been specifically detected in AITL (63), but their role has not yet been established.
Figure 2. Schematic representation of the PDGF/PDGFR signaling pathway (Generated using MetaCore software; GeneGo Inc.).

Notably, different key points of PDGFR signaling can be the object of pharmacological inhibition. First, PDGFRs can be directly blocked by TKIs. Second, the immediate downstream JAK/STAT pathway, SRC kinase and PI3K can be selectively disrupted. Finally, in cases retaining NF-κB activity (around 30% of PTCL-NOSs), different molecules might be targeted. Adapted with permission from Expert Review of Hematology (59).
Fourth, the role of micro-RNAs (miRNAs) has so far been investigated mainly in ALCL. In particular, Merkel et al. (64) reported that five members of the miR17–92 cluster were expressed more highly in ALK+ ALCL, whereas expression levels of miR155 were more than 10-fold higher in ALK– ALCL. Moreover, miR101 is downregulated in all types of ALCL, a fact of potential therapeutic interest because mTOR is the target of this miRNA. Additional data have recently been reported regarding the potential pathogenetic effect of miR19a and miR135b in ALK+ ALCL (65, 66). It was shown that ALK+ ALCLs express a low level of miR29a, and that this downmodulation requires an active NPM-ALK kinase. Importantly, it was also reported that low expression of miR29a, probably achieved through methylation-mediated repression, appeared to play an important regulatory role in MCL-1 overexpression, promoting tumor cell survival by inhibiting apoptosis. Consistently, forced miR29a expression was found to modulate apoptosis through inhibition of MCL-1 expression with concomitant tumor growth reduction in vivo (64).
Finally, novel insights into PTCL pathogenesis will be likely be provided by massive parallel sequencing in the near future. In this vein, a novel recurrent t(6;7)(p25.3;q32.3) translocation has been just discovered in ALK– ALCL (8). This lesion, which disrupts the DUSP22 phosphatase gene on 6p25.3 and adjoins the FRA7H fragile site on 7q32.3, was found to be associated with downregulation of DUSP22 and upregulation of miR29, mapped on 7q32.3 (8).
Prognosis
As previously mentioned, with a few exceptions, PTCLs usually are characterized by an aggressive clinical course and dismal prognosis (1, 9). Among the latter, ALK+ ALCL has been a matter of controversy. In fact, although it is obvious that it is less aggressive than the ALK– form, its course is nevertheless quite variable, being influenced by several factors: the age of the patient, the International Prognostic Index (IPI; a clinical tool developed to aid in predicting the prognosis of patients with aggressive non-Hodgkin lymphoma, and based on age, disease stage, serum lactic dehydrogenase (LDH), performance status, and presence of extranodal disease) and the state of the bone marrow. In addition, the prognosis of ALK+ ALCL may soon be improved by the recent introduction of ALK inhibitors and SGN-35 in the clinical practice (67, 68).
Several attempts have been made to define the optimal prognostic approach for PTCL-NOSs, but none of the proposed systems has completely achieved such a goal. In 2004, Gallamini et al. (69) proposed the Prognostic Index for PTCL-Unspecified (PIT), including age >60, Eastern Cooperative Oncology Group (ECOG) ≥1, elevated LDH levels, and bone marrow involvement. Two years later, our group refined this scoring system by substituting Ki67 expression for bone marrow involvement (3), which allowed for the identification of three distinct subgroups of patients with clearly different clinical behavior. Remarkably, the proposed score considered for the first time a biological parameter (the proliferative index), consistent with the observation made later by Cuadros et al. that a gene signature related to proliferation heralds a poor prognosis (70). Interestingly, we also found in a univariate analysis that EBV positivity and cytotoxic marker expression were adverse prognostic indicators (3). These findings are in line with the results of two studies carried out by the Groupe d’Etude des Lymphomes de l’Adulte (GELA) (71) and by Iqbal et al. (21), highlighting the negative impact of neoplastic cell EBV infection and of a cytotoxic gene signature, respectively. Finally, Martinez-Delgado et al. (15) and Briones et al. (55) reported that the activation of the NF-κB pathway, assessed by expression profiling or BCL10/p65 immunohistochemistry, identifies PTCL-NOSs cases with a more favorable outcome.
These results were not reproduced by our group, which observed downregulation or deregulation of the genes involved in the NF-κB cascade in about two-thirds of our PTCL-NOS patients with no impact on the prognosis, as shown by expression profiling and immunohistochemical evaluation of both the canonical and alternative NF-κB pathway components (10, 56, 59). Interestingly, our results were confirmed in an independent SNP array–based study by Hartmann et al., who found genomic gains of the REL locus and nuclear localization of the REL protein in about 25% of their cases (7). These discrepancies may be due to the different cellular composition of the analyzed cases in each study; in particular, it is conceivable that many of the cases analyzed by Martinez-Delgado et al. and Briones et al. contained significant numbers of reactive cells, which in turn might have influenced the NF-κB genes and related protein expression (15, 54, 55).
The development of other prognostic indices has mainly focused on ENKTCL/NT. Once it had been demonstrated that the extranasal forms of the tumor run a more aggressive clinical course, Lee et al. proposed a prognostic model based on B-symptoms, stage, LDH values, and regional lymph node involvement, which showed a superior predictive value as compared with the IPI by allowing the identification of four risk categories (72). These findings have been confirmed by the ITCLP, which however found that IPI also identifies three groups of patients with significantly different clinical courses (63, 72).
It has also recently been shown that the derivation from γ/δ T lymphocytes might represent a further prognostic indicator, the tumors stemming from this compartment being highly aggressive irrespective of the histological subtype (14). Thus, the IPI is currently the only tool that can be applied to different PTCL categories, though it is far from perfect and lacks relevance in the setting of ATLL, ENKTCL/NT, and EATCL (9).
Targeted therapy
TK inhibitors.
Based on the evidence of TK deregulation in different subtypes of PTCLs, the application of TK inhibitors (TKIs) has been tested ex vivo (10, 30). In particular, our group designed experiments aiming to test the sensitivity of PTCL-NOS cells to different TKIs (10). We found a remarkable reduction of cell proliferation and viability. Conversely, TKIs exerted a limited effect on normal lymphocytes. Huang et al. treated T/NK tumor–derived cell lines with imatinib and obtained analogous results (30). Following these observations, clinical trials were initiated and the effectiveness of TK inhibition was demonstrated in vivo in PTCL-NOS patients. In this regard, a phase I/II trial designed to evaluate the safety and clinical activity of dasatinib in relapsed/refractory non-Hodgkin lymphomas indicated a remarkable activity of the compound in PTCL, inducing complete remission in both enrolled cases (73). In ALK+ALCL, Chiarle et al. clearly showed that targeting ALK and its downstream target STAT3 represents an effective strategy (74, 75). In addition, PDGFR inhibition was shown to be effective in the same setting, inducing clinical responses in vivo (76).
Histone deacetylase inhibitors.
Interestingly, expression profiling demonstrated widespread silencing of genes, possibly regulated by epigenetic mechanisms such as histone deacetylation (10). Our group found that histone deacetylase inhibitor (HDACi) compounds induced a dramatic reduction in cell viability in PTCL-NOS primary cells and cell lines, with G0-G1 cell cycle arrest and apoptosis at therapeutic concentrations (10), suggesting a possible role for this class of drugs in PTCL-NOS therapy, and indeed, this idea was supported by clinical observations (77). Notably, the combination of TK inhibition and HDAC inhibition produced a remarkable effect on cell viability and might represent a promising option for future therapeutic applications (10). More recently, the effectiveness of associations of HDACis and demethylating agents was suggested by ex vivo studies (78).
Anti-angiogenetic therapy.
Increased angiogenesis is a major characteristic of AITL. However, the molecular mechanisms that underlie this have remained elusive. Recently, two studies documented the upregulation of the VEGF gene in this tumor (20, 24). Our group immunohistochemically analyzed a large number of cases on tissue microarrays and demonstrated that VEGF is mainly expressed by the neoplastic elements (24), and not only by the abundant vascular component, as was initially proposed (20). We further showed that AITL cells do also express a VEGF receptor, VEGFR2/KDR (24), suggesting the intriguing hypothesis that autocrine/paracrine stimulation also occurs in this setting. These findings also suggest that AITL might be sensitive to anti-angiogenetic drugs such as thalidomide and bevacizumab. Indeed, several reports documented their positive activity in AITL cases (79–84). More recently, a possible pathogenetic role of VEGF has been proposed for MF as well (85, 86). Indeed, VEGF deregulation may be an early event in this disease.
mAbs directed against surface antigens
Although PTCLs typically present with defectiveness in T cell–associated surface molecules, mAbs are promising agents in this setting. In this regard, alemtuzumab, a humanized anti-CD52 mAb, was successfully administered to PTCL patients in combination with other chemotherapeutics (CHOP therapy) (87, 88). The complete response rates ranged from 71% to 90%, with more than 50% of patients alive at two years (87, 88). However, in general, toxicity remains a persistent problem with this agent, although there may be strategies available to overcome this obstacle, allowing a more rational usage of the compound, including effective antimicrobial prophylaxis and alemtuzumab dose reduction (59). On the other hand, the overall effectiveness may be improved by patient selection based on CD52 positivity, as around half of patients present a consistent downregulation of this antigen (5, 89–92). Conversely, CD30 is theoretically an optimal target for therapy owing to its minimal expression on nonmalignant cells, although the unconjugated anti-CD30 agents SGN-30 and MDX-060 demonstrated minimal effects in Hodgkin lymphoma and ALCL (93, 94). However, more recently, in order to enhance the antitumor activity, the antitubulin agent monomethyl auristatin E (MMAE) was attached to a CD30-specific mAb by an enzyme-cleavable linker, producing the antibody-drug conjugate brentuximab vedotin (SGN-35) (95). Notably, this drug induced remarkable clinical responses in all relapsed/refractory CD30+ lymphomas with acceptable toxicity, leading to its rapid approval by FDA in this setting (95).
PTCLs are relatively rare tumors; however, they account for a substantial number of deaths worldwide. The most common subtype, PTCL-NOS, probably represents a “wastebasket” category that cannot be further classified, at present, based on morphology and phenotype. In addition, the distinction of AITL and ALK– ALCL from PTCL-NOSs is often challenging. Thus, novel molecular tools are warranted to better understand these tumors. Indeed, only a deeper biological knowledge of PTCLs, hopefully by both the characterization of primary samples and the creation of adequate preclinical experimental models, will lead to significant improvements in their clinical management. In this regard, the recent evidence of deregulation of specific pathways, including TK signaling, HDAC activity, and aurora kinase A (96), as well as the availability of remarkably active conjugated mAbs (e.g., brentuximab vedotin [SGN-35]) herald the advent of a new era for targeted therapy in this setting.
Note added in proof.
While the present paper was in press, three contributions appeared in the literature that should be mentioned. First, next generations sequencing showed a second mutation recurrent in AITL and affecting the TET2 coding sequence (97). Its incidence turned out to be slightly lower than the one of IDH2. Interestingly, it occurred also in 38% of PTCLs-NOS — almost all these cases were related to FTH lymphocytes.
Second, Agnelli et al. have undertaken a transcriptional profiling meta-analysis of 309 cases, including ALCL and other primary T-NHL samples. Pathway discovery and prediction analyses defined a minimum set of genes capable to recognize ALK– ALCL. Application of RT-qPCR in independent data sets from cryo-preserved and FFPE samples validated a three-gene model (TNFRSF8, BATF3, TMOD1) able to successfully separate ALK– ALCL from PTCL-NOS, with overall accuracy near 97% (98).
Finally, Rossi et al. profiled 40 PTCLs (28 NOS, 6 ALCLs, and 6 AITLs), four reactive lymph nodes, and 20 samples of normal T-lymphocytes, by showing significantly lower BCL10 expression in all tumors in comparison to normal samples, the lowest values being detected in ALCL. Immunohistochemistry on TMA revealed BCL10 positivity in only 10/52 PTCLs, NOS (19%) with no significant correlation with either expression of Ki-67 and the T-cell markers or NFκB activation, as well as with progression free survival and overall survival, although a favorable trend was recorded in BCL10+ cases (99).
Acknowledgments
This work was supported by the Centro Interdipartimentale per la Ricerca sul Cancro “G. Prodi”, BolognAIL, Associazione Italiana per la Ricerca sul Cancro (AIRC, grants IG10519 and 5xMille 10007 to S.A. Pileri), Ricerca Fondamentale Orientata (RFO) (to S.A. Pileri and P.P. Piccaluga), Fondazione Cassa di Risparmio in Bologna, Fondazione della Banca del Monte e Ravenna, and Progetto Strategico di Ateneo 2006 (to S.A. Pileri and P.P. Piccaluga). The authors thank Anna Gazzola and Fabio Fuligni for the assistance in manuscript and figure preparation.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3448–3455. doi:10.1172/JCI61205.
References
- 1.2008. pp. 158–166. Jaffe ES, Harris NL, Stein H, Campo E, Pileri SA, Swerdlow SH. Introduction and overview of the classification of the lymphoid neoplasms. In: Swerdlow, S., et al., eds. WHO classification of tumors of hematopoietic and lymphoid tissues. Lyon, France: International Agency For Research On Cancer; [Google Scholar]
- 2.Piccaluga PP, et al. Peripheral T-cell lymphoma classification: the matter of cellular derivation. Expert Rev Hematol. 2011;4(4):415–425. doi: 10.1586/ehm.11.37. [DOI] [PubMed] [Google Scholar]
- 3.Went P, et al. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J Clin Oncol. 2006;24(16):2472–2479. doi: 10.1200/JCO.2005.03.6327. [DOI] [PubMed] [Google Scholar]
- 4.2008. pp. 306–308. Pileri SA, et al. Peripheral T–cell lymphoma, not otherwise specified. In: Swerdlow, S., et al., eds. WHO Classification Of Tumors Of Hematopoietic And Lymphoid Tissues. Lyon, France: International Agency For Research On Cancer; [Google Scholar]
- 5.Piccaluga PP, Agostinelli C, Righi S, Zinzani PL, Pileri SA. Expression of CD52 in peripheral T-cell lymphoma. Haematologica. 2007;92(4):566–567. doi: 10.3324/haematol.10767. [DOI] [PubMed] [Google Scholar]
- 6.van Dongen JJ, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 7.Hartmann S, et al. High resolution SNP array genomic profiling of peripheral T cell lymphomas, not otherwise specified, identifies a subgroup with chromosomal aberrations affecting the REL locus. Br J Haematol. 2010;148(3):402–412. doi: 10.1111/j.1365-2141.2009.07956.x. [DOI] [PubMed] [Google Scholar]
- 8.Feldman AL, et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011;117(3):915–919. doi: 10.1182/blood-2010-08-303305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vose J, Armitage J, Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008;26(25):4124–4130. doi: 10.1200/JCO.2008.16.4558. [DOI] [PubMed] [Google Scholar]
- 10.Piccaluga PP, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117(3):823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohshima K, et al. Classification of distinct subtypes of peripheral T-cell lymphoma unspecified, identified by chemokine and chemokine receptor expression: Analysis of prognosis. Int J Oncol. 2004;25(3):605–613. [PubMed] [Google Scholar]
- 12.Geissinger E, et al. Nodal peripheral T-cell lymphomas correspond to distinct mature T-cell populations. J Pathol. 2006;210(2):172–180. doi: 10.1002/path.2046. [DOI] [PubMed] [Google Scholar]
- 13.Chan JK, et al. Type II enteropathy-associated T-cell lymphoma: a distinct aggressive lymphoma with frequent gammadelta T-cell receptor expression. Am J Surg Pathol. 2011;35(10):1557–1569. doi: 10.1097/PAS.0b013e318222dfcd. [DOI] [PubMed] [Google Scholar]
- 14.Tripodo C, et al. Gamma-delta T-cell lymphomas. Nat Rev Clin Oncol. 2009;6(12):707–717. doi: 10.1038/nrclinonc.2009.169. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Delgado B, et al. Expression profiling of T-cell lymphomas differentiates peripheral and lymphoblastic lymphomas and defines survival related genes. Clin Cancer Res. 2004;10(15):4971–4982. doi: 10.1158/1078-0432.CCR-04-0269. [DOI] [PubMed] [Google Scholar]
- 16.Ballester B, et al. Gene expression profiling identifies molecular subgroups among nodal peripheral T-cell lymphomas. Oncogene. 2006;25(10):1560–1570. doi: 10.1038/sj.onc.1209178. [DOI] [PubMed] [Google Scholar]
- 17.Tracey L, et al. Mycosis fungoides shows concurrent deregulation of multiple genes involved in the TNF signaling pathway: an expression profile study. Blood. 2003;102(3):1042–1050. doi: 10.1182/blood-2002-11-3574. [DOI] [PubMed] [Google Scholar]
- 18.Pise-Masison CA, et al. Gene expression profiling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood. 2009;113(17):4016–4026. doi: 10.1182/blood-2008-08-175901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Litvinov IV, Jones DA, Sasseville D, Kupper TS. Transcriptional profiles predict disease outcome in patients with cutaneous T-cell lymphoma. Clin Cancer Res. 2010;16(7):2106–2114. doi: 10.1158/1078-0432.CCR-09-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Leval L, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109(11):4952–4963. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- 21.Iqbal J, et al. Molecular signatures to improve diagnosis in peripheral T–cell lymphoma and prognostication in angioimmunoblastic T–cell lymphoma. Blood. 2010;115(5):1026–1036. doi: 10.1182/blood-2009-06-227579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartmann S, et al. Hartmann S, et al. Revising the historical collection of epithelioid cell-rich lymphomas of the Kiel Lymph Node Registry: what is Lennert’s lymphoma nowadays? Histopathology. 2011;59(6):1173–1182. doi: 10.1111/j.1365-2559.2011.04069.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, et al. TCR-dependent transformation of mature memory phenotype T cells in mice. J Clin Invest. 2011;121(10):3834–3845. doi: 10.1172/JCI37210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piccaluga PP, et al. Gene expression analysis of angioimmunoblastic lymphoma indicates derivation from T follicular helper cells and vascular endothelial growth factor deregulation. Cancer Res. 2007;67(22):10703–10710. doi: 10.1158/0008-5472.CAN-07-1708. [DOI] [PubMed] [Google Scholar]
- 25.Laurent C, Fazilleau N, Brousset P. A novel subset of T-helper cells: follicular T-helper cells and their markers. Haematologica. 2010;95(3):356–358. doi: 10.3324/haematol.2009.019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17(8):975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruneau J, et al. Regulatory T-cell depletion in angioimmunoblastic T-cell lymphoma. Am J Pathol. 2010;177(2):570–574. doi: 10.2353/ajpath.2010.100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agostinelli C, et al. Peripheral T cell lymphomas with follicular T helper phenotype: a new basket or a distinct entity? Revising Karl Lennert’s personal archive. Histopathology. 2011;59(4):679–691. doi: 10.1111/j.1365-2559.2011.03981.x. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez Pinilla SM, et al. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma expresses follicular T-cell markers. Am J Surg Pathol. 2009;33(1):81–90. doi: 10.1097/PAS.0b013e31818e52fe. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y, et al. Gene expression profiling identifies emerging oncogenic pathways operating in extranodal NK/T-cell lymphoma, nasal-type. Blood. 2010;115(6):1226–1237. doi: 10.1182/blood-2009-05-221275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamant L, et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007;109(5):2156–2164. doi: 10.1182/blood-2006-06-028969. [DOI] [PubMed] [Google Scholar]
- 32.Piva R, et al. Gene expression profiling uncovers molecular classifiers for the recognition of anaplastic large-cell lymphoma within peripheral T-cell neoplasms. J Clin Oncol. 2010;28(9):1583–1590. doi: 10.1200/JCO.2008.20.9759. [DOI] [PubMed] [Google Scholar]
- 33.Tapper J, Kettunen E, El-Rifai W, Seppala M, Andersson LC, Knuutila S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet Cytogenet. 2001;128(1):1–6. doi: 10.1016/S0165-4608(01)00386-7. [DOI] [PubMed] [Google Scholar]
- 34.Sado Y, et al. Organization and expression of basement membrane collagen IV genes and their roles in human disorders. J Biochem. 1998;123(5):767–776. doi: 10.1093/oxfordjournals.jbchem.a022003. [DOI] [PubMed] [Google Scholar]
- 35.van den Boom J, et al. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163(3):1033–1043. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin S, et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene. 2002;21(57):8696–8704. doi: 10.1038/sj.onc.1206034. [DOI] [PubMed] [Google Scholar]
- 37.Papa S, et al. Gadd45 beta mediates the NF-kappa B suppression of JNK signalling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6(2):146–153. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 38.Chen F, et al. Opposite effect of NF-kappa B and c-Jun N-terminal kinase on p53-independent GADD45 induction by arsenite. J Biol Chem. 2001;276(14):11414–11419. doi: 10.1074/jbc.M011682200. [DOI] [PubMed] [Google Scholar]
- 39.Hirose T, et al. p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene. 2003;22(49):7762–7773. doi: 10.1038/sj.onc.1207091. [DOI] [PubMed] [Google Scholar]
- 40.Tan KO, et al. MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with Bax through its Bcl-2 homology domains. J Biol Chem. 2001;276(4):2802–2807. doi: 10.1074/jbc.M008955200. [DOI] [PubMed] [Google Scholar]
- 41.Nagashima M, et al. A novel PHD-finger motif protein, p47ING3, modulates p53-mediated transcription, cell cycle control, and apoptosis. Oncogene. 2003;22(3):343–350. doi: 10.1038/sj.onc.1206115. [DOI] [PubMed] [Google Scholar]
- 42.Gunduz M, et al. Allelic loss and reduced expression of the ING3, a candidate tumor suppressor gene at 7q31, in human head and neck cancers. Oncogene. 2002;21(28):4462–4470. doi: 10.1038/sj.onc.1205540. [DOI] [PubMed] [Google Scholar]
- 43.Lee MS, Hanspers K, Barker CS, Korn AP, McCune JM. Gene expression profiles during human CD4+ T cell differentiation. Int Immunol. 2004;16(8):1109–1124. doi: 10.1093/intimm/dxh112. [DOI] [PubMed] [Google Scholar]
- 44.Chtanova T, et al. Identification of T cell-restricted genes, and signatures for different T cell responses, using a comprehensive collection of microarray datasets. J Immunol. 2005;175(12):7837–7847. doi: 10.4049/jimmunol.175.12.7837. [DOI] [PubMed] [Google Scholar]
- 45.Chtanova T, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol. 2004;173(1):68–78. doi: 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- 46.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(10):R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han JS, Macarak E, Rosenbloom J, Chung KC, Chaqour B. Regulation of Cyr61/CCN1 gene expression through RhoA GTPase and p38MAPK signaling pathways. Eur J Biochem. 2003;270(16):3408–3421. doi: 10.1046/j.1432-1033.2003.03723.x. [DOI] [PubMed] [Google Scholar]
- 48.Leu SJ, Liu Y, Chen N, Chen CC, Lam SC, Lau LF. Identification of a novel integrin alpha 6 beta 1 binding site in the angiogenic inducer CCN1 (CYR61). J Biol Chem. 2003;278(36):33801–33808. doi: 10.1074/jbc.M305862200. [DOI] [PubMed] [Google Scholar]
- 49.Schober JM, Lau LF, Ugarova TP, Lam SC. Identification of a novel integrin alphaMbeta2 binding site in CCN1 (CYR61), a matricellular protein expressed in healing wounds and atherosclerotic lesions. J Biol Chem. 2003;278(28):25808–25815. doi: 10.1074/jbc.M301534200. [DOI] [PubMed] [Google Scholar]
- 50.Tsai MS, Bogart DF, Castaneda JM, Li P, Lupu R. Cyr61 promotes breast tumorigenesis and cancer progression. Oncogene. 2002;21(53):8178–8185. doi: 10.1038/sj.onc.1205682. [DOI] [PubMed] [Google Scholar]
- 51.Lin MT, et al. Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaB-dependent XIAP up-regulation. J Biol Chem. 2004;279(23):24015–24023. doi: 10.1074/jbc.M402305200. [DOI] [PubMed] [Google Scholar]
- 52.Kassem H, Sangar V, Cowan R, Clarke N, Margison GP. A potential role of heat shock proteins and nicotinamide N-methyl transferase in predicting response to radiation in bladder cancer. Int J Cancer. 2002;101(5):454–460. doi: 10.1002/ijc.10631. [DOI] [PubMed] [Google Scholar]
- 53.Xu J, Capezzone M, Xu X, Hershman JM. Activation of nicotinamide N-methyltransferase gene promoter by hepatocyte nuclear factor-1beta in human papillary thyroid cancer cells. Mol Endocrinol. 2005;19(2):527–539. doi: 10.1210/me.2004-0215. [DOI] [PubMed] [Google Scholar]
- 54.Martinez-Delgado B, et al. Differential expression of NF-kappaB pathway genes among peripheral T-cell lymphomas. Leukemia. 2005;19(12):2254–2263. doi: 10.1038/sj.leu.2403960. [DOI] [PubMed] [Google Scholar]
- 55.Briones J, et al. Bcl-10 protein highly correlates with the expression of phosphorylated p65 NF-kappaB in peripheral T-cell lymphomas and is associated with clinical outcome. Histopathology. 2009;54(4):478–485. doi: 10.1111/j.1365-2559.2009.03250.x. [DOI] [PubMed] [Google Scholar]
- 56.Agostinelli C, et al. Peripheral T cell lymphoma, not otherwise specified: the stuff of genes, dreams and therapies. J Clin Pathol. 2008;61(11):1160–1167. doi: 10.1136/jcp.2008.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piccaluga PP, et al. Expression of platelet-derived growth factor receptor alpha in peripheral T-cell lymphoma not otherwise specified. Lancet Oncol. 2005;6(6):440. doi: 10.1016/S1470-2045(05)70213-8. [DOI] [PubMed] [Google Scholar]
- 58.Yang J, et al. Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood. 2010;115(1):51–60. doi: 10.1182/blood-2009-06-223719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roncolato F, Gazzola A, Zinzani PL, Pileri SA, Piccaluga PP. Targeted molecular therapy in peripheral T-cell lymphomas. Expert Rev Hematol. 2011;4(5):551–562. doi: 10.1586/ehm.11.55. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Q, et al. Oncogenic tyrosine kinase NPM-ALK induces expression of the growth-promoting receptor ICOS. Blood. 2011;118(11):3062–3071. doi: 10.1182/blood-2011-01-332916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia. 2006;20(2):313–318. doi: 10.1038/sj.leu.2404045. [DOI] [PubMed] [Google Scholar]
- 62.Wilcox RA, Sun DX, Novak A, Dogan A, Ansell SM, Feldman AL. Inhibition of Syk protein tyrosine kinase induces apoptosis and blocks proliferation in T-cell non-Hodgkin’s lymphoma cell lines. Leukemia. 2010;24(1):229–232. doi: 10.1038/leu.2009.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cairns RA, et al. IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood. 2012;119(8):1901–1903. doi: 10.1182/blood-2011-11-391748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Merkel O, et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proc Natl Acad Sci U S A. 2010;107(37):16228–16233. doi: 10.1073/pnas.1009719107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Desjobert C, et al. MiR-29a down-regulation in ALK-positive anaplastic large cell lymphomas contributes to apoptosis blockade through MCL-1 overexpression. Blood. 2011;117(24):6627–6637. doi: 10.1182/blood-2010-09-301994. [DOI] [PubMed] [Google Scholar]
- 66.Matsuyama H, et al. miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood. 2011;118(26):6881–6892. doi: 10.1182/blood-2011-05-354654. [DOI] [PubMed] [Google Scholar]
- 67.Cheng M, et al. CEP-28122, a highly potent and selective orally active inhibitor of anaplastic lymphoma kinase with antitumor activity in experimental models of human cancers. Mol Cancer Ther. 2012;11(3):670–679. doi: 10.1158/1535-7163.MCT-11-0776. [DOI] [PubMed] [Google Scholar]
- 68.Younes A, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 69.Gallamini A, et al. Peripheral T-cell lymphoma unspecified (PTCL-U): a new prognostic model from a retrospective multicentric clinical study. Blood. 2004;103(7):2474–2479. doi: 10.1182/blood-2003-09-3080. [DOI] [PubMed] [Google Scholar]
- 70.Cuadros M, et al. Identification of a proliferation signature related to survival in nodal peripheral T-cell lymphomas. J Clin Oncol. 2007;25(22):3321–3329. doi: 10.1200/JCO.2006.09.4474. [DOI] [PubMed] [Google Scholar]
- 71.Dupuis J, et al. Prognostic significance of Epstein-Barr virus in nodal peripheral T-cell lymphoma, unspecified: A Groupe d’Etude des Lymphomes de l’Adulte (GELA) study. Blood. 2006;108(13):4163–4169. doi: 10.1182/blood-2006-04-017632. [DOI] [PubMed] [Google Scholar]
- 72.Lee J, et al. Extranodal natural killer T-cell lymphoma, nasal-type: a prognostic model from a retrospective multicenter study. J Clin Oncol. 2006;24(4):612–618. doi: 10.1200/JCO.2005.04.1384. [DOI] [PubMed] [Google Scholar]
- 73.William BM, et al. Phase I/II study of dasatinib in relapsed or refractory non-Hodgkin’s lymphoma (NHL). 53rd ASH Annual Meeting and Exposition. December 6, 2010. Abstract 288. [Google Scholar]
- 74.Chiarle R, et al. The anaplastic lymphoma kinase is an effective oncoantigen for lymphoma vaccination. Nat Med. 2008;14(6):676–680. doi: 10.1038/nm1769. [DOI] [PubMed] [Google Scholar]
- 75.Chiarle R, et al. Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11(6):623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- 76.Kenner L, et al. New and highly efficient therapy for treatment NPM-ALK associated lymphomas. 53rd ASH Annual Meeting and Exposition. December 10, 2010. Abstract 1659. [Google Scholar]
- 77.Piekarz RL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117(22):5827–5834. doi: 10.1182/blood-2010-10-312603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marchi E, et al. Combination of epigenetic agents synergistically reverse the malignant phenotype in models of T-cell lymphoma. ASH Annual Meeting Abstracts. 2011;118(21):2727. [Google Scholar]
- 79.Strupp C, Aivado M, Germing U, Gattermann N, Haas R. Angioimmunoblastic lymphadenopathy (AILD) may respond to thalidomide treatment: two case reports. Leuk Lymphoma. 2002;43(1):133–137. doi: 10.1080/10428190210190. [DOI] [PubMed] [Google Scholar]
- 80.Bruns I, et al. Complete remission in a patient with relapsed angioimmunoblastic T-cell lymphoma following treatment with bevacizumab. Leukemia. 2005;19(11):1993–1995. doi: 10.1038/sj.leu.2403936. [DOI] [PubMed] [Google Scholar]
- 81.Dogan A, Ngu LS, Ng SH, Cervi PL. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19(5):873–875. doi: 10.1038/sj.leu.2403710. [DOI] [PubMed] [Google Scholar]
- 82.Ramasamy K, Lim Z, Pagliuca A, Salisbury JR, Mufti GJ, Devereux S. Successful treatment of refractory angioimmunoblastic T-cell lymphoma with thalidomide and dexamethasone. Haematologica. 2006;91(8 suppl):ECR44. [PubMed] [Google Scholar]
- 83.Aguiar Bujanda D. Complete response of relapsed angioimmunoblastic T-cell lymphoma following therapy with bevacizumab. Ann Oncol. 2008;19(2):396–397. doi: 10.1093/annonc/mdm579. [DOI] [PubMed] [Google Scholar]
- 84.Gottardi M, et al. Complete remission induced by thalidomide in a case of angioimmunoblastic T-cell lymphoma refractory to autologous stem cell transplantation. Leuk Lymphoma. 2008;49(9):1836–1838. doi: 10.1080/10428190802233165. [DOI] [PubMed] [Google Scholar]
- 85.Shin J, et al. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood. 2007;110(8):3015–3027. doi: 10.1182/blood-2006-12-061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karpova MB, Fujii K, Jenni D, Dummer R, Urosevic-Maiwald M. Evaluation of lymphangiogenic markers in Sezary syndrome. Leuk Lymphoma. 2011;52(3):491–501. doi: 10.3109/10428194.2010.517877. [DOI] [PubMed] [Google Scholar]
- 87.Gallamini A, et al. Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood. 2007;110(7):2316–2323. doi: 10.1182/blood-2007-02-074641. [DOI] [PubMed] [Google Scholar]
- 88.Kluin-Nelemans HC, et al. Intensified alemtuzumab-CHOP therapy for peripheral T-cell lymphoma. Ann Oncol. 2011;22(7):1595–1600. doi: 10.1093/annonc/mdq635. [DOI] [PubMed] [Google Scholar]
- 89.Rodig SJ, et al. Heterogeneous CD52 expression among hematologic neoplasms: implications for the use of alemtuzumab (CAMPATH-1H). Clin Cancer Res. 2006;12(23):7174–7179. doi: 10.1158/1078-0432.CCR-06-1275. [DOI] [PubMed] [Google Scholar]
- 90.Chang ST, Lu CL, Chuang SS. CD52 expression in non-mycotic T- and NK/T-cell lymphomas. Leuk Lymphoma. 2007;48(1):117–121. doi: 10.1080/10428190601016167. [DOI] [PubMed] [Google Scholar]
- 91.Jiang L, et al. Variable CD52 expression in mature T cell and NK cell malignancies: implications for alemtuzumab therapy. Br J Haematol. 2009;145(2):173–179. doi: 10.1111/j.1365-2141.2009.07606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Geissinger E, Bonzheim I, Roth S, Rosenwald A, Muller-Hermelink HK, Rudiger T. CD52 expression in peripheral T-cell lymphomas determined by combined immunophenotyping using tumor cell specific T-cell receptor antibodies. Leuk Lymphoma. 2009;50(6):1010–1016. doi: 10.1080/10428190902926981. [DOI] [PubMed] [Google Scholar]
- 93.Ansell SM, et al. Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol. 2007;25(19):2764–2769. doi: 10.1200/JCO.2006.07.8972. [DOI] [PubMed] [Google Scholar]
- 94.Forero-Torres A, et al. A phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol. 2009;146(2):171–179. doi: 10.1111/j.1365-2141.2009.07740.x. [DOI] [PubMed] [Google Scholar]
- 95.Younes A, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2011;363(19):1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 96.Iqbal J, et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic gammadelta T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia. 2011;25(2):348–358. doi: 10.1038/leu.2010.255. [DOI] [PubMed] [Google Scholar]
- 97.Lemonnier F, et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood. 2012;120(7):1466–1469. doi: 10.1182/blood-2012-02-408542. [DOI] [PubMed] [Google Scholar]
- 98.Agnelli L, et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood. 2012;120(6):1274–1281. doi: 10.1182/blood-2012-01-405555. [DOI] [PubMed] [Google Scholar]
- 99.Rossi M, et al. BCL10 down-regulation in peripheral T-cell lymphomas [published online ahead of print July 18, 2012]. Hum Pathol. doi: 10.1016/j.humpath.2012.03.024. doi: 10.1016/j.humpath.2012.03.024 . [DOI] [PubMed] [Google Scholar]