

Abstract
The hallmark t(14;18)(q32;q21) in follicular lymphoma (FL) results in constitutive overexpression of the BCL2 protein, allowing B cells to abrogate the default germinal center apoptotic program. Most tumors are characterized by recurrent secondary genetic alterations including genomic gains, losses, and mutations, some providing a growth advantage, including alterations in MLL2, EPHA7, TNFRSF14, and EZH2. The sequence in which these events occur and how they contribute to progression and ultimately to transformation is unclear. Lastly, crosstalk between neoplastic B cells and non-neoplastic immune and stromal cells in the microenvironment plays an important role in sustaining tumor cell growth, cultivating immune privilege, and promoting transformation.
Introduction
Follicular lymphoma (FL) is the second most common nodal lymphoma and occurs at a median age of 60 years (1, 2). Despite the fact that most patients present with disseminated disease at diagnosis and FL is generally considered incurable, it is the prototype of indolent lymphomas and, as such, is characterized by slow progression and high response rates to therapy. As a result, median overall survival times reach 10 years or more with current therapies (3). Nonetheless, most patients eventually develop increasingly resistant disease over time, and in up to 45% of cases, with a risk of 3% per year, the original indolent lymphoma transforms into an aggressive subtype, an event that is associated with poor outcome (4–6).
Current treatment strategies include radiation for limited-stage patients and monoclonal anti-CD20 antibodies (e.g., rituximab), alone or in combination with chemotherapy, for patients that present with advanced-stage disease (7). Asymptomatic and/or low-tumor-burden patients may be observed under a “watch and wait” approach (7). The introduction of rituximab has markedly improved outcomes and was the first drug to show an improvement in overall survival in FL in many years (8–10). Rituximab maintenance or consolidation with radio-immunotherapy after first-line therapy has further shown clear benefit for progression-free survival and potentially for overall survival (11–14). More aggressive treatment approaches with high-dose chemotherapy and autologous or allogeneic stem cell transplantation are reserved for more resistant disease in patients with good performance status (7).
Here we focus on the pathogenesis of FL and highlight recent advances in our understanding of the origin of FL, the frequent highly recurrent genetic alterations as well as the perturbations of the FL-associated microenvironment.
Pathology
Morphologically, FL is defined as a proliferation of malignant germinal center B cells (GCBs) that are admixed with nonmalignant cells such as T cells, follicular dendritic cells (FDCs), and macrophages and whose normal counterparts, i.e., centrocytes and centroblasts, represent the predominant cell types of the germinal center reaction (15). The relative proportion of centrocytes to centroblasts underlies the current grading scheme with, at its extremes, grade 1 FL comprising low numbers of centroblasts (0–5 per high-power field) and grade 3B FL marked by solid sheets of these same cells. Unlike many other malignancies, however, grading of FL does not provide a clear prognostic impact, nor is there a consensus on the nature of grade 3B FL, as these cases often resemble de novo diffuse large B cell lymphoma (DLBCL) and harbor somewhat different molecular characteristics than grades 1–3A FL (e.g., grade 3B FL is often negative for t(14;18) and variably expresses CD10) (16). The classical follicular growth pattern of FL can be revealed by staining for FDC markers such as CD21 or CD23. The t(14;18)(q32;q21) translocation can be detected by fluorescence in situ hybridization in 85% of all cases, results in overexpression of anti-apoptotic BCL2, and is considered a molecular hallmark of the disease. Nonetheless, this translocation is neither necessary nor sufficient for diagnosis, as it is absent in 15% of FLs and present in about 20%–30% of GCB-type DLBCLs (15, 17).
Cell of origin
The observations that the neoplastic cells are organized in follicles, express germinal center surface markers such as BCL6 and CD10, and have a characteristic gene expression profile of centrocytes and/or centroblasts (15, 18) all suggest that FL B cells are able to achieve a stage of differentiation reminiscent of GCB cells (19). The first hit of the oncogenic cascade leading to FL is generally attributed to the t(14;18), based on the frequency of this genetic aberration and its supposed mechanism of action. The translocation occurs following a double-strand break at the IGH locus on chromosome 14 attributed to defective RAG-mediated VDJ recombination and a break at the BCL2 locus on chromosome 18 thought to be linked to an inherent fragility at CpG sites (20, 21). As VDJ recombination occurs at an early B cell developmental stage in the bone marrow, the t(14;18) as a first genetic hit is similarly believed to occur in the bone marrow but not to prevent further differentiation. Naive B cells carrying the t(14;18) exit the bone marrow and colonize secondary lymphoid tissue, where they undergo the germinal center reaction but have a survival advantage due to their constitutive expression of BCL2, which is not normally expressed in the germinal center (22). BCL2 may also rescue these cells from apoptosis due to weak B cell receptor (BCR) affinity (22). Nonetheless, the expression of a structurally and likely functionally preserved BCR seems crucial as, despite ongoing somatic hypermutation, a selective bias against deleterious mutations in BCR is observed (23, 24). BCR signaling may be further stimulated by the interaction of mannosylated immunoglobulin variable regions with lectins expressed on the surface of non-neoplastic immune cells (25). The early FL progenitors then acquire secondary genetic alterations under the influence of activation-induced cytidine deaminase (AID), the importance of which has been suggested in a mouse model in which genetic deletion of AID prevented BCL6-driven germinal center–derived lymphomas (26).
This current hypothesis of FL pathogenesis recognizes naive t(14;18)-positive circulating B cells as potentially tumor-initiating cells but is challenged by the observation that similar cells can be detected in the blood of 50%–70% of healthy individuals who are not prone to developing FL (27–29). Interestingly, both the incidence of FL and the frequency of t(14;18)-positive cells in healthy individuals increases with age (29). Importantly, recent studies have shown that most of these cells are not naive B cells, but instead are germinal center–educated IgD+CD27+ (or IgM+CD27+) memory cells (30, 31). Similar to true FL tumor cells, these so-called FL-like cells in healthy individuals frequently show evidence of class-switch recombination of the translocated IGH allele, whereas the functional allele encodes a surface IgD (or IgM). The role of these FL-like cells in the pathogenesis of FL remains elusive. Nonetheless, precursor lymphoma-initiating cells must exist, as illustrated by two reports of FL arising in both the donor and the recipient after allogeneic hematopoietic stem cell transplantation (32, 33). In the most recent of these studies, the t(14;18) was retrospectively detected in the donor lymphocyte infusion, together with a low abundance of mutated alleles matching 14 of the 15 gene mutations that were shared between the tumors from the donor and the recipient (33). This case report provides evidence that the obligate lymphoma progenitor has far more complex genetic alterations than previously assumed.
Our understanding of the origin of FL is further complicated by the recognition of a loosely defined entity called FL in situ, which has been proposed as a true precursor lesion on the basis of relatively uncommon progression to overt FL (34, 35). However, this hypothesis will require the collection of a larger series of patients to gain insight into the natural history of FL in situ and to clearly delineate it from very early stage, incidentally detected FL and lymph nodes with partial involvement.
Genetic landscape of FL
The observations that BCL2 has anti-proliferative functions in addition to its anti-apoptotic properties (36), and that the t(14;18) is found in healthy individuals (27–29), suggest that overexpression of BCL2 is insufficient to induce lymphomagenesis. In addition, transgenic mouse models of BCL2 overexpression do not faithfully recapitulate FL pathogenesis. In the original Eμ-BCL2 model, mice developed follicular hyperplasia and, after prolonged latencies, high-grade lymphomas that were clearly not reminiscent of FL (37). Subsequently, Egle et al. developed the VavP-BCL2 model, in which BCL2 was overexpressed under the control of the pan-hematopoietic VavP promoter (38). These mice developed lymphomas that shared morphological features with FL but, on the molecular level, the high expression of the BCL2 transgene in the T cell compartment represents an artificial situation that is not found in FL (38). Furthermore, the consideration that clonal cytogenetic aberrations, copy number alterations, and uniparental disomy are almost universally found in FL provides additional support to the notion that other genetic events contribute to FL pathogenesis.
Apart from the t(14;18), the most common chromosomal aberrations include non-random losses of 1p36 and 6q as well as gains of 7, 18, and X (39–42). Recently, careful analysis of candidate genes within these regions has yielded insight into potential key players. For example, within the 1p36 region that is frequently targeted by heterozygous deletions and copy-neutral loss of heterozygosity, the TNFRSF14 gene was shown to be recurrently mutated (43, 44). In two studies, mutations were found in 46 of 251 cases (18.3%) (43) and in 37 of 81 samples (46%) (44). In both reports, 57% of TNFRSF14 mutations were predicted to result in a truncated protein devoid of its transmembrane domain, resulting in decreased surface expression. TNFRSF14 mutations provide the first insight into the link between the genetics of FL and the associated microenvironment. However, a multiplicity of ligands bind TNFRSF14, and the receptor can transmit both inhibitory and stimulatory signals to T cells depending on ligand interaction, so further functional studies are clearly warranted to elucidate the role of these mutations in FL pathogenesis.
More recently, Oricchio et al. performed a systematic screen of genes within commonly deleted regions on the long arm of chromosome 6 (45), motivated by the high frequency of 6q deletions in FL as well as their association with adverse outcome (41). This screen revealed TNFAIP3/A20, a negative regulator of NF-κB signaling, as well as the receptor tyrosine kinase EPHA7 as potential tumor suppressors in FL (45). The authors demonstrated that silencing of EPHA7 decreased lymphomagenesis latency in VavP-BCL2 transgenic mice, and that restoration of its tumor suppressor function through administration of exogenous EPHA7 delayed tumor formation in xenograft models. The authors proposed a model in which soluble EPHA7 binds EPHA2 on lymphoma cell surfaces and prevents activation of signaling through ERK as well as other SRC kinases. They further showed that EPHA7 expression was absent by immunohistochemistry in 72% of FL cases on a tissue microarray and suggested that absence of protein expression occurred through genomic deletions as well as promoter methylation, corroborating earlier findings from Dawson et al. (46).
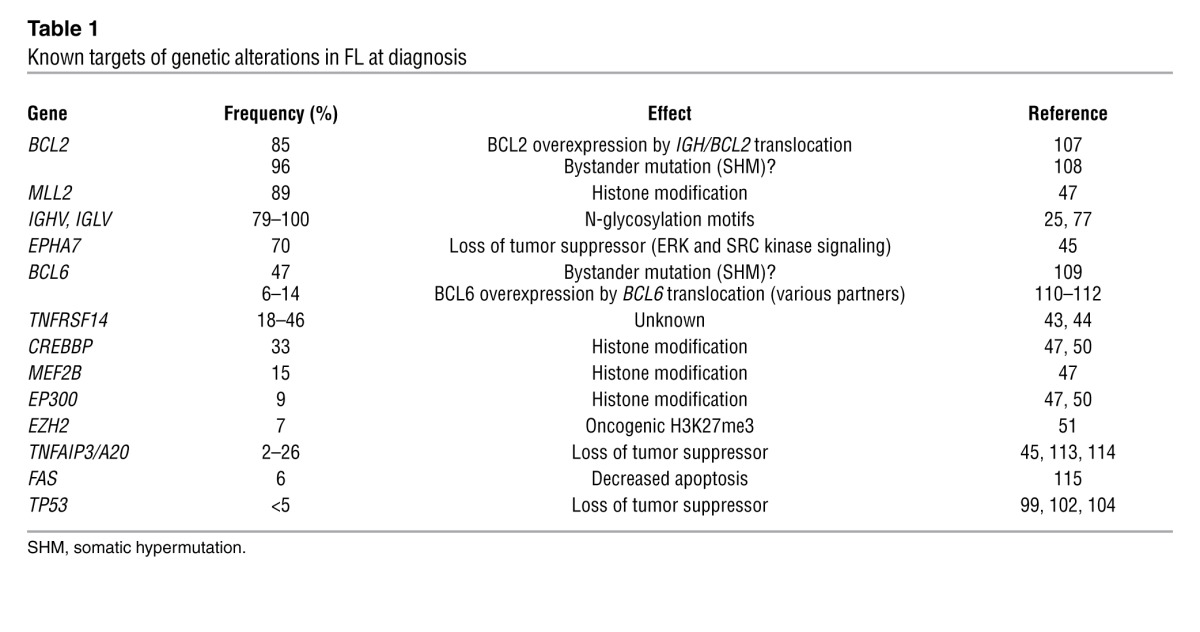
High-throughput sequencing technology has increased the pace of discoveries in cancer to an unprecedented level. Among the nodal lymphomas, the mutational landscape has thus far been most accurately defined in DLBCL and, to a lesser extent, in FL (47–50). Unexpectedly, it was shown that germinal center–derived lymphomas are characterized by frequent mutations of histone-modifying genes. In FL in particular, recurrent mutations have been reported in the histone methyltransferases MLL2 (89%) and EZH2 (7.2%), the histone acetylases CREBBP (32.6%), EP300 (8.7%), and MEF2B (15.3%) (refs. 47, 48, 50, 51, and Table 1). The high recurrence of these mutations illustrates that FL is likely a disease of the epigenome as well as the genome (52). EZH2, the best-characterized histone-modifying gene in lymphoma, is the catalytic unit of the polycomb repressive complex 2. Mutations of Tyr641 within the SET domain of EZH2 have been identified in 7% of FLs and 22% of GCB-like DLBCLs, leading to increased histone H3 Lys27 trimethylation (H3K27me3) (47, 53, 54). The finding that EZH2 is upregulated in B cells during the germinal center reaction (55) suggests that activating EZH2 mutations lock in a transcriptional profile that favors proliferation. EZH2-mediated silencing of anti-proliferative genes was subsequently documented in normal centroblasts (56), lending further support to the notion that FL subverts the physiological gene expression program of GCB cells. Although our knowledge of genetic aberrations in FL has dramatically increased over the last few years, further studies are necessary in order to provide a comprehensive mutational landscape of all genetic changes that contribute to tumorigenesis.
Table 1.
Known targets of genetic alterations in FL at diagnosis
Microenvironment
FL histology is marked by a varying proportion of nonmalignant immune-related cells. The importance of the microenvironment in FL is further highlighted by the fact that researchers have been unable to propagate FL cell lines, and even short-term growth in vitro requires survival signals derived from either feeder cells or cytokines. Several gene expression profiling studies highlighted that gene expression signatures derived from non-neoplastic immune cells were related to disease behavior (57–61). In the landmark study from the Lymphoma/Leukemia Molecular Profiling Project (LLMPP), the expression of T cell and macrophage-related genes was associated with favorable survival, whereas other macrophage or dendritic cell–derived transcripts were predictive of poor outcome (57). Distinct cellular subsets, however, cannot be easily attributed to one of these immune-response signatures, as illustrated by numerous immunohistochemistry studies that have tried to parse out the intricate relationship between non-neoplastic immune cells in FL (58, 62–74). This approach has led to highly contradictory results, in part explained by treatment heterogeneity within small studies as well as by spurious associations of biomarkers with outcome, resulting from a failure to control for multiple hypothesis testing.
We conceptualize the role of the microenvironment in FL as 2-fold, supporting tumor growth and survival, as well as suppressing the antitumoral immune response (Figure 1). FL cells express high levels of CXCR4 and CXCR5 and are attracted into follicles by cytokines such as CXCL13 that are released by follicular T helper (FTH) cells or FDCs (75, 76). As a result of their localization and the expression of Fc and complement receptors, FDCs are ideal antigen presenting cells, thus contributing to BCR signaling, which may also occur through the interaction of N-glycosylation motifs and lectins from the innate immune system (25, 77). FTH cells engage in cellular interaction with FL through their T cell receptor via MHC class II as well as CD40L/CD40. FTH cells are enriched in FL biopsies and secrete cytokines such as IL-4, which binds to the IL-4 receptor on lymphoma cells and triggers signaling by phosphorylation of ERK and STAT6 (78). The role of stromal cells in FL pathogenesis has emerged, as in vitro studies have shown that they increase neoplastic B cell survival, contribute to monocyte recruitment via the secretion of CCL2, and contribute to macrophage polarization (79, 80).
Figure 1. A model of the microenvironment in FL.

Tumor cells are nurtured by a variety of cells (orange) such as FTH cells, FDCs, and follicular reticular cells (FRCs). Beneficial signals for growth and survival include cytokines such as IL-4 and IL-21, which bind to interleukin receptors on lymphoma cells (IL-4R/IL-21R) or CXCL12 and CXCL13 secreted by stromal cell subsets. BCR signaling occurs through stimulation of the BCR by the innate immune system through N-glycans or by specific antigen presentation through professional APCs such as FDCs. Tumor cells subvert the antitumor immune response from T helper cells, CTLs, and macrophages (purple). For example, FL cells prevent lysis through CTLs by inducing a T cell immunologic synapse dysfunction, or by secretion of IL-12 they induce T cell exhaustion. Immune cell subsets that suppress an efficient immunological response against the tumor include Tregs and M2 polarized macrophages (TAMs) (blue). The former are enriched in the FL microenvironment and dampen the T cell response. FL contributes to the preferential conversion of T helper cells into Tregs by proteins such as TGF-β or CCL22. Whereas classically activated TAMs (M1 polarized) control malignant growth through induction of a Th1 response, M2-polarized TAMs exert a tumor-promoting function through angiogenesis and induction of an immunosuppressive Th2 response.
In most studies using specimens pre-dating the rituximab era, elevated numbers of CD68+ tumor-associated macrophages (TAMs) were associated with poor prognosis (57, 62, 67, 72). These findings are usually interpreted in the light of alternative or M2 polarization of macrophages, a phenotype that is associated with tumor dissemination, immunosuppression, and angiogenesis (81, 82). Two studies showed that increased microvessel density and angiogenic sprouting correlated both with increased numbers of macrophages and with poor outcome in FL (83, 84). FL B-cells divert the classical activation of the innate immune system and subvert the function of the adaptive immune response. For example, immune synapses between malignant B cells and T cells are defective, although CD8+ CTLs have been shown to localize at the follicle border and enter into contact with tumor cells (85, 86). It was also recently shown that IL-12 secretion by malignant B cells induces T cell exhaustion via expression of TIM3 (87). Moreover, FL cells induce the conversion of effector T cells into FOXP3-expressing Tregs, which suppress the proliferation and activity of both CD4+CD25– and CD8+ T cells (88–90). Taken together, these observations illustrate how the malignant B cells dampen the function of various T cell subsets in the FL microenvironment in order to escape immunosurveillance.
Complex pathways of progression and transformation
The observation that two-thirds of FL patients present with stage III or IV disease at diagnosis may reflect the ability of malignant lymphocytes to home to nodal and extranodal sites at an early stage during disease initiation (15). In FL, ongoing somatic hypermutation enables the tracking of multiple coexisting clones that differ by their mutational load on the variable regions of the immunoglobulin locus. The findings from studies using this approach suggest extensive trafficking of clones between adjacent follicles (91, 92) and also that progression occurs either by direct clonal evolution or by divergent evolution from a supposed common progenitor cell (refs. 32, 93, and Figure 2). Disease evolution in FL is best understood from a Darwinian perspective, wherein the dominant clone is in constant flux and under the selective pressure of survival signals from the lymphoma cells themselves as well as their associated microenvironment. This theory helps explain the observed pattern of clinical progression and the spontaneous waxing and waning of disease in patients in the absence of treatment.
Figure 2. A model of the complex pathways leading to progression and transformation in FL.
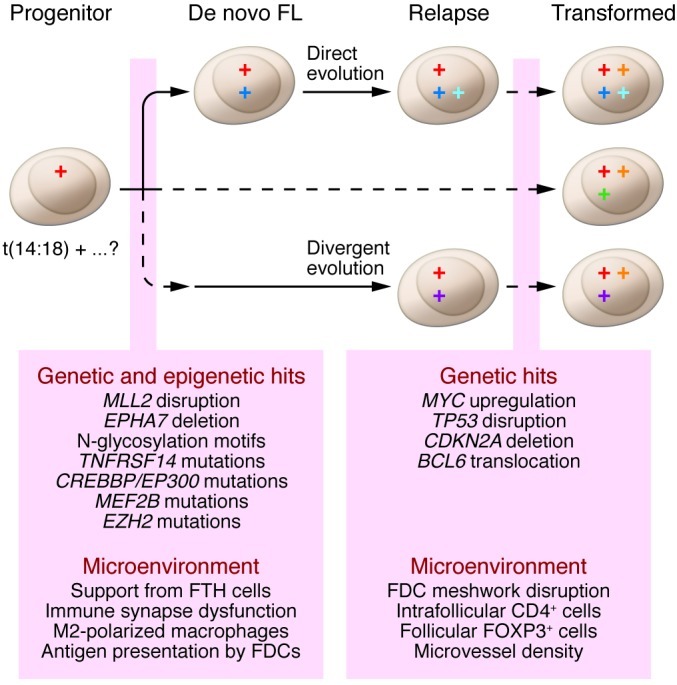
Evolution occurs through emergence of traits that provide a growth advantage under selective pressures such as therapy and tumor control by the microenvironment. The circles denote hits that drive somatic cells to malignancy. The progenitor cell may precede overt FL by many years and in roughly 85% of cases harbors a BCL2/IGH rearrangement in addition to putative secondary hits. The progenitor disseminates early during the course of the disease. Clinical FL develops with the acquisition of further genetic alterations and establishment of immune privilege. Relapse occurs either by direct clonal evolution from de novo FL or by indirect evolution from a common ancestral cell. Transformation is thought to develop along similar pathways, but may be dominated by one of a number of key genetic alterations.
An elegant study using single-cell phospho-flow cytometry recently showed that FL samples contain a variable proportion of cells with impaired BCR signaling (94). Increased numbers not only had a negative prognostic impact but were also associated with disease progression. Suppression of BCR signaling was not due to loss of BCR expression but rather explained by specific suppression of downstream activation. These data support the hypothesis that late stages of FL are autonomous and may not rely on external signals, including antigen-induced BCR activation.
Over the course of their illness, as many as 45% of FL patients develop aggressive disease (4–6). The incidence of transformation, however, may be underestimated, as up to 63% of patients had higher-grade transformation in autopsy series (95). Clinically, transformation is usually accompanied by a sudden onset of B symptoms (fever, night sweats, and weight loss) and the development of extranodal disease and/or rapid tumor growth. In population-based studies, the median survival post-transformation has been reported to range from 1.2 to 1.7 years (4, 5). The patients in these series were not treated initially with rituximab, however, and it is currently unclear whether rituximab may decrease the incidence of transformation and/or change its prognosis. The pathology of transformed FL (TFL) most often resembles DLBCL, but other histologies may be observed that overlap Burkitt lymphoma and DLBCL (so-called gray-zone lymphomas) (96). TFL shares a distinct gene expression profile and immunophenotype with the original FL (97). Similar to progression of FL, the process of transformation may occur by either direct clonal evolution or emergence from an ancestral clone that arises from a supposed common progenitor (32).
It is presently unresolved whether TFL occurs through an outgrowth of an existing subclone in the antecedent FL or results from random genetic, epigenetic, or microenvironmental hits during the clinical course of FL. Multiple studies have examined FL and TFL biopsies and provide associations of genetic aberrations with the process of transformation. One of the most commonly described findings is upregulation of MYC or its target genes, which was reported in 6 of 12 cases in a first series (97) and in 14 of 20 cases in a subsequent series (98). Increased activity of the MYC proto-oncogene by translocations or mutations is otherwise rare in FL but can define so-called double-hit lymphomas, which harbor both MYC and BCL2 rearrangements and are clearly associated with very poor outcome (99). Deregulation of MYC is also a key determinant of the pluripotency signature that was predictive of transformation when applied to de novo FL (100). Other genetic aberrations that have been associated with transformation include TP53 mutations (98, 101, 102), deletions or silencing of the CDKN2A locus (103), and BCL6 translocations (104). On the other hand, transformation has also been attributed to changes in the tumor microenvironment that include the disruption of FDC meshworks (58), increased intrafollicular CD4+ T cells (58), a follicular rather than a diffuse localization of FOXP3+ Tregs (63), and an increased microvessel density (83). However, all the preceding findings are correlative, and the key genetic alteration(s) that drive the process of transformation remain elusive.
Conclusions and future directions
FL is a disease of both the tumor genome/epigenome and tumor-infiltrating cells. While our understanding of the crosstalk between tumor and non-tumor cells steadily increases, the most significant recent advances undoubtedly stem from the application of genomic screens and next-generation sequencing technology that have revealed that FL is characterized, in addition to the t(14;18), by at least two highly recurrent genetic alterations present in 70% or more of FL cases (mutational disruption of MLL2 and EPHA7 deletion or silencing). The sequence of acquisition of these aberrations is currently unknown and will likely be elucidated in the near future by sequencing serial specimens. This will also improve our understanding of the events that lead to transformation.
Our increasing understanding of the biology of FL is already leading to novel treatments based on clearly identified molecular targets. Examples include small-molecule inhibitors that prevent signaling through the BCR by blocking downstream kinases (such as spleen tyrosine kinase or Bruton’s tyrosine kinase) or compounds that induce apoptosis, thus counteracting the overexpression of BCL2 (e.g., ABT-263) (105). It will be of great interest to follow the development of EPHA7 as a novel targeted approach in clinical trials. A further potential “druggable” target is EZH2, although mutations in its gene occur at a comparatively lower frequency than EPHA7 deletions or silencing. The introduction of agents that target the tumor microenvironment will also likely contribute to our therapeutic arsenal in the future. Our increasing understanding of the intricate relationship between tumor and immune cell subsets is already leading to novel treatment approaches such as in situ vaccination or blockade of tumor-promoting signaling such as CTLA4-mediated inhibition of T cells by Tregs (106).
Although FL is considered an indolent disease, the ultimate goal will be to achieve durable remissions and even cures with minimal toxicity, and to reduce the incidence of TFL, a clinically dominant event associated with markedly inferior survival.
Acknowledgments
R.D. Gascoyne is supported by National Cancer Institute of Canada (NCIC) Terry Fox Foundation Program Project 019001. R. Kridel is funded by the Ligue Genevoise Contre le Cancer et Fondation Dr Henri Dubois-Ferrière Dinu Lipatti.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3424–3431. doi:10.1172/JCI63186.
References
- 1. Table 19.23: All Lymphoid neoplasms with detailed non-Hodgkin lymphoma subtypes. In: National Cancer Institute.SEER Cancer Statistics Review 1975–2008 . Bethesda, Maryland, USA: NIH; 2008:23. [Google Scholar]
- 2.Anderson JR, Armitage JO, Weisenburger DD. Epidemiology of the non-Hodgkin’s lymphomas: distributions of the major subtypes differ by geographic locations. Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol. 1998;9(7):717–720. doi: 10.1023/A:1008265532487. [DOI] [PubMed] [Google Scholar]
- 3.Fisher RI, LeBlanc M, Press OW, Maloney DG, Unger JM, Miller TP. New treatment options have changed the survival of patients with follicular lymphoma. J Clin Oncol. 2005;23(33):8447–8452. doi: 10.1200/JCO.2005.03.1674. [DOI] [PubMed] [Google Scholar]
- 4.Al-Tourah AJ, et al. Population-based analysis of incidence and outcome of transformed non-Hodgkin’s lymphoma. J Clin Oncol. 2008;26(32):5165–5169. doi: 10.1200/JCO.2008.16.0283. [DOI] [PubMed] [Google Scholar]
- 5.Bastion Y, et al. Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol. 1997;15(4):1587–1594. doi: 10.1200/JCO.1997.15.4.1587. [DOI] [PubMed] [Google Scholar]
- 6.Montoto S, et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B-cell lymphoma. J Clin Oncol. 2007;25(17):2426–2433. doi: 10.1200/JCO.2006.09.3260. [DOI] [PubMed] [Google Scholar]
- 7. Dreyling M, Ghielmini M, Marcus R, Salles G, Vitolo U. Newly diagnosed and relapsed follicular lymphoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2011. 22 suppl 6: vi59 vi63 10.1093/annonc/mdr388 [DOI] [PubMed] [Google Scholar]
- 8.Marcus R, et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol. 2008;26(28):4579–4586. doi: 10.1200/JCO.2007.13.5376. [DOI] [PubMed] [Google Scholar]
- 9.Hiddemann W, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: Results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood. 2005;106(12):3725–3732. doi: 10.1182/blood-2005-01-0016. [DOI] [PubMed] [Google Scholar]
- 10.Herold M, et al. Rituximab added to first-line mitoxantrone, chlorambucil, and prednisolone chemotherapy followed by interferon maintenance prolongs survival in patients with advanced follicular lymphoma: an East German Study Group Hematology and Oncology Study. J Clin Oncol. 2007;25(15):1986–1992. doi: 10.1200/JCO.2006.06.4618. [DOI] [PubMed] [Google Scholar]
- 11.Salles G, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011;377(9759):42–51. doi: 10.1016/S0140-6736(10)62175-7. [DOI] [PubMed] [Google Scholar]
- 12.Morschhauser F, et al. Phase III trial of consolidation therapy with yttrium-90-ibritumomab tiuxetan compared with no additional therapy after first remission in advanced follicular lymphoma. J Clin Oncol. 2008;26(32):5156–5164. doi: 10.1200/JCO.2008.17.2015. [DOI] [PubMed] [Google Scholar]
- 13.Vidal L, et al. Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst. 2011;103(23):1799–1806. doi: 10.1093/jnci/djr418. [DOI] [PubMed] [Google Scholar]
- 14.van Oers MH, et al. Rituximab maintenance treatment of relapsed/resistant follicular non-Hodgkin’s lymphoma: long-term outcome of the EORTC 20981 phase III randomized intergroup study. J Clin Oncol. 2010;28(17):2853–2858. doi: 10.1200/JCO.2009.26.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swerdlow SH, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Geneva, Switzerland: World Health Organization; 2008. [Google Scholar]
- 16.Salaverria I, Siebert R. Follicular lymphoma grade 3B. Best Pract Res Clin Haematol. 2011;24(2):111–119. doi: 10.1016/j.beha.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med. 1987;317(19):1185–1189. doi: 10.1056/NEJM198711053171904. [DOI] [PubMed] [Google Scholar]
- 18.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 19.Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nat Rev Cancer. 2005;5(4):251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- 20.Tsujimoto Y, Gorham J, Cossman J, Jaffe E, Croce CM. The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science. 1985;229(4720):1390–1393. doi: 10.1126/science.3929382. [DOI] [PubMed] [Google Scholar]
- 21.Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135(6):1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roulland S, Faroudi M, Mamessier E, Sungalee S, Salles G, Nadel B. Early steps of follicular lymphoma pathogenesis. Adv Immunol. 2011;111:1–46. doi: 10.1016/B978-0-12-385991-4.00001-5. [DOI] [PubMed] [Google Scholar]
- 23.Zuckerman NS, et al. Ig gene diversification and selection in follicular lymphoma, diffuse large B cell lymphoma and primary central nervous system lymphoma revealed by lineage tree and mutation analyses. Int Immunol. 2010;22(11):875–887. doi: 10.1093/intimm/dxq441. [DOI] [PubMed] [Google Scholar]
- 24.Bahler DW, Levy R. Clonal evolution of a follicular lymphoma: evidence for antigen selection. Proc Natl Acad Sci U S A. 1992;89(15):6770–6774. doi: 10.1073/pnas.89.15.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coelho V, et al. Glycosylation of surface Ig creates a functional bridge between human follicular lymphoma and microenvironmental lectins. Proc Natl Acad Sci U S A. 2010;107(43):18587–18592. doi: 10.1073/pnas.1009388107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasqualucci L, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40(1):108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- 27.Limpens J, et al. Lymphoma-associated translocation t(14;18) in blood B cells of normal individuals. Blood. 1995;85(9):2528–2536. [PubMed] [Google Scholar]
- 28.Dolken G, Illerhaus G, Hirt C, Mertelsmann R. BCL-2/JH rearrangements in circulating B cells of healthy blood donors and patients with nonmalignant diseases. J Clin Oncol. 1996;14(4):1333–1344. doi: 10.1200/JCO.1996.14.4.1333. [DOI] [PubMed] [Google Scholar]
- 29.Schüler F, et al. Prevalence and frequency of circulating t(14;18)-MBR translocation carrying cells in healthy individuals. Int J Cancer. 2009;124(4):958–963. doi: 10.1002/ijc.23958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirt C, Dolken G, Janz S, Rabkin CS. Distribution of t(14;18)-positive, putative lymphoma precursor cells among B-cell subsets in healthy individuals. Br J Haematol. 2007;138(3):349–353. doi: 10.1111/j.1365-2141.2007.06671.x. [DOI] [PubMed] [Google Scholar]
- 31.Roulland S, et al. Follicular lymphoma-like B cells in healthy individuals: a novel intermediate step in early lymphomagenesis. J Exp Med. 2006;203(11):2425–2431. doi: 10.1084/jem.20061292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carlotti E, et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma may occur by divergent evolution from a common progenitor cell or by direct evolution from the follicular lymphoma clone. Blood. 2009;113(15):3553–3557. doi: 10.1182/blood-2008-08-174839. [DOI] [PubMed] [Google Scholar]
- 33.Weigert O, et al. Molecular ontogeny of donor-derived follicular lymphomas occurring after hematopoietic cell transplantation. Cancer Discovery. 2012;2:47–55. doi: 10.1158/2159-8290.CD-11-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jegalian AG, et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood. 2011;118(11):2976–2984. doi: 10.1182/blood-2011-05-355255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cong P, Raffeld M, Teruya-Feldstein J, Sorbara L, Pittaluga S, Jaffe ES. In situ localization of follicular lymphoma: description and analysis by laser capture microdissection. Blood. 2002;99(9):3376–3382. doi: 10.1182/blood.V99.9.3376. [DOI] [PubMed] [Google Scholar]
- 36.Zinkel S, Gross A, Yang E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ. 2006;13(8):1351–1359. doi: 10.1038/sj.cdd.4401987. [DOI] [PubMed] [Google Scholar]
- 37.McDonnell TJ, Korsmeyer SJ. Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14;18). Nature. 1991;349(6306):254–256. doi: 10.1038/349254a0. [DOI] [PubMed] [Google Scholar]
- 38.Egle A, Harris AW, Bath ML, O’Reilly L, Cory S. VavP-Bcl2 transgenic mice develop follicular lymphoma preceded by germinal center hyperplasia. Blood. 2004;103(6):2276–2283. doi: 10.1182/blood-2003-07-2469. [DOI] [PubMed] [Google Scholar]
- 39.Schwaenen C, et al. Microarray-based genomic profiling reveals novel genomic aberrations in follicular lymphoma which associate with patient survival and gene expression status. Genes Chromosomes Cancer. 2009;48(1):39–54. doi: 10.1002/gcc.20617. [DOI] [PubMed] [Google Scholar]
- 40.Johnson NA, Al-Tourah A, Brown CJ, Connors JM, Gascoyne RD, Horsman DE. Prognostic significance of secondary cytogenetic alterations in follicular lymphomas. Genes Chromosomes Cancer. 2008;47(12):1038–1048. doi: 10.1002/gcc.20606. [DOI] [PubMed] [Google Scholar]
- 41.Cheung KJ, et al. Genome-wide profiling of follicular lymphoma by array comparative genomic hybridization reveals prognostically significant DNA copy number imbalances. Blood. 2009;113(1):137–148. doi: 10.1182/blood-2008-02-140616. [DOI] [PubMed] [Google Scholar]
- 42.Horsman DE, Connors JM, Pantzar T, Gascoyne RD. Analysis of secondary chromosomal alterations in 165 cases of follicular lymphoma with t(14;18). Genes Chromosomes Cancer. 2001;30(4):375–382. doi: 10.1002/gcc.1103. [DOI] [PubMed] [Google Scholar]
- 43.Cheung KJ, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70(22):9166–9174. doi: 10.1158/0008-5472.CAN-10-2460. [DOI] [PubMed] [Google Scholar]
- 44.Launay E, et al. High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia. 2012;26(3):559–562. doi: 10.1038/leu.2011.266. [DOI] [PubMed] [Google Scholar]
- 45.Oricchio E, et al. The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell. 2011;147(3):554–564. doi: 10.1016/j.cell.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawson DW, et al. Global DNA methylation profiling reveals silencing of a secreted form of Epha7 in mouse and human germinal center B-cell lymphomas. Oncogene. 2007;26(29):4243–4252. doi: 10.1038/sj.onc.1210211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pasqualucci L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lohr JG, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sneeringer CJ, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107(49):20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yap DB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117(8):2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Galen JC, et al. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol. 2004;34(7):1870–1881. doi: 10.1002/eji.200424985. [DOI] [PubMed] [Google Scholar]
- 56.Velichutina I, et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116(24):5247–5255. doi: 10.1182/blood-2010-04-280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dave SS, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 58.Glas AM, et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. J Clin Oncol. 2007;25(4):390–398. doi: 10.1200/JCO.2006.06.1648. [DOI] [PubMed] [Google Scholar]
- 59.Husson H, et al. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood. 2002;99(1):282–289. doi: 10.1182/blood.V99.1.282. [DOI] [PubMed] [Google Scholar]
- 60.Glas AM, et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood. 2005;105(1):301–307. doi: 10.1182/blood-2004-06-2298. [DOI] [PubMed] [Google Scholar]
- 61.Harjunpaa A, et al. Differential gene expression in non-malignant tumour microenvironment is associated with outcome in follicular lymphoma patients treated with rituximab and CHOP. Br J Haematol. 2006;135(1):33–42. doi: 10.1111/j.1365-2141.2006.06255.x. [DOI] [PubMed] [Google Scholar]
- 62.Farinha P, et al. Analysis of multiple biomarkers shows that lymphoma-associated macrophage (LAM) content is an independent predictor of survival in follicular lymphoma (FL). Blood. 2005;106(6):2169–2174. doi: 10.1182/blood-2005-04-1565. [DOI] [PubMed] [Google Scholar]
- 63.Farinha P, Al-Tourah A, Gill K, Klasa R, Connors JM, Gascoyne RD. The architectural pattern of FOXP3-positive T cells in follicular lymphoma is an independent predictor of survival and histologic transformation. Blood. 2010;115(2):289–295. doi: 10.1182/blood-2009-07-235598. [DOI] [PubMed] [Google Scholar]
- 64.Wahlin BE, et al. A unifying microenvironment model in follicular lymphoma: outcome is predicted by programmed death-1--positive, regulatory, cytotoxic, and helper T cells and macrophages. Clin Cancer Res. 2010;16(2):637–650. doi: 10.1158/1078-0432.CCR-09-2487. [DOI] [PubMed] [Google Scholar]
- 65.Carreras J, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood. 2006;108(9):2957–2964. doi: 10.1182/blood-2006-04-018218. [DOI] [PubMed] [Google Scholar]
- 66.Carreras J, et al. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J Clin Oncol. 2009;27(9):1470–1476. doi: 10.1200/JCO.2008.18.0513. [DOI] [PubMed] [Google Scholar]
- 67.Alvaro T, et al. The presence of STAT1-positive tumor-associated macrophages and their relation to outcome in patients with follicular lymphoma. Haematologica. 2006;91(12):1605–1612. [PubMed] [Google Scholar]
- 68.Alvaro T, et al. Immunohistochemical patterns of reactive microenvironment are associated with clinicobiologic behavior in follicular lymphoma patients. J Clin Oncol. 2006;24(34):5350–5357. doi: 10.1200/JCO.2006.06.4766. [DOI] [PubMed] [Google Scholar]
- 69.Sweetenham JW, et al. Prognostic value of regulatory T cells, lymphoma-associated macrophages, and MUM-1 expression in follicular lymphoma treated before and after the Introduction of monoclonal antibody therapy: a Southwest Oncology Group Study. Ann Oncol. 2010;21(6):1196–1202. doi: 10.1093/annonc/mdp460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee AM, et al. Number of CD4+ cells and location of forkhead box protein P3-positive cells in diagnostic follicular lymphoma tissue microarrays correlates with outcome. J Clin Oncol. 2006;24(31):5052–5059. doi: 10.1200/JCO.2006.06.4642. [DOI] [PubMed] [Google Scholar]
- 71.Taskinen M, Karjalainen-Lindsberg ML, Leppa S. Prognostic influence of tumor-infiltrating mast cells in patients with follicular lymphoma treated with rituximab and CHOP. Blood. 2008;111(9):4664–4667. doi: 10.1182/blood-2007-11-125823. [DOI] [PubMed] [Google Scholar]
- 72.Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppa S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res. 2007;13(19):5784–5789. doi: 10.1158/1078-0432.CCR-07-0778. [DOI] [PubMed] [Google Scholar]
- 73.de Jong D, et al. Impact of the tumor microenvironment on prognosis in follicular lymphoma is dependent on specific treatment protocols. Haematologica. 2009;94(1):70–77. doi: 10.3324/haematol.13574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Canioni D, et al. High numbers of tumor-associated macrophages have an adverse prognostic value that can be circumvented by rituximab in patients with follicular lymphoma enrolled onto the GELA-GOELAMS FL-2000 trial. J Clin Oncol. 2008;26(3):440–446. doi: 10.1200/JCO.2007.12.8298. [DOI] [PubMed] [Google Scholar]
- 75.Trentin L, et al. Homeostatic chemokines drive migration of malignant B cells in patients with non-Hodgkin lymphomas. Blood. 2004;104(2):502–508. doi: 10.1182/blood-2003-09-3103. [DOI] [PubMed] [Google Scholar]
- 76.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114(16):3367–3375. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu D, McCarthy H, Ottensmeier CH, Johnson P, Hamblin TJ, Stevenson FK. Acquisition of potential N-glycosylation sites in the immunoglobulin variable region by somatic mutation is a distinctive feature of follicular lymphoma. Blood. 2002;99(7):2562–2568. doi: 10.1182/blood.V99.7.2562. [DOI] [PubMed] [Google Scholar]
- 78.Pangault C, et al. Follicular lymphoma cell niche: identification of a preeminent IL-4-dependent T(FH)-B cell axis. Leukemia. 2010;24(12):2080–2089. doi: 10.1038/leu.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guilloton F, et al. Mesenchymal stromal cells orchestrate follicular lymphoma cell niche through the CCL2–dependent recruitment and polarization of monocytes. Blood. 2012;119(11):2556–2567. doi: 10.1182/blood-2011-08-370908. [DOI] [PubMed] [Google Scholar]
- 80.Ame-Thomas P, et al. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood. 2007;109(2):693–702. doi: 10.1182/blood-2006-05-020800. [DOI] [PubMed] [Google Scholar]
- 81.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi: 10.1016/S1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 82.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farinha P, Kyle AH, Minchinton AI, Connors JM, Karsan A, Gascoyne RD. Vascularization predicts overall survival and risk of transformation in follicular lymphoma. Haematologica. 2010;95(12):2157–2160. doi: 10.3324/haematol.2009.021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clear AJ, et al. Increased angiogenic sprouting in poor prognosis FL is associated with elevated numbers of CD163+ macrophages within the immediate sprouting microenvironment. Blood. 2010;115(24):5053–5056. doi: 10.1182/blood-2009-11-253260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Laurent C, et al. Distribution, function, and prognostic value of cytotoxic T lymphocytes in follicular lymphoma: a 3-D tissue-imaging study. Blood. 2011;118(20):5371–5379. doi: 10.1182/blood-2011-04-345777. [DOI] [PubMed] [Google Scholar]
- 86.Ramsay AG, et al. Follicular lymphoma cells induce T-cell immunologic synapse dysfunction that can be repaired with lenalidomide: implications for the tumor microenvironment and immunotherapy. Blood. 2009;114(21):4713–4720. doi: 10.1182/blood-2009-04-217687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang ZZ, et al. IL–12 upregulates TIM–3 expression and induces T cell exhaustion in patients with follicular B cell non–Hodgkin lymphoma. J Clin Invest. 2012;122(4):1271–1282. doi: 10.1172/JCI59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang ZZ, Novak AJ, Stenson MJ, Witzig TE, Ansell SM. Intratumoral CD4+CD25+ regulatory T-cell-mediated suppression of infiltrating CD4+ T cells in B-cell non-Hodgkin lymphoma. Blood. 2006;107(9):3639–3646. doi: 10.1182/blood-2005-08-3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. Attenuation of CD8(+) T-cell function by CD4(+)CD25(+) regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2006;66(20):10145–10152. doi: 10.1158/0008-5472.CAN-06-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ai WZ, Hou JZ, Zeiser R, Czerwinski D, Negrin RS, Levy R. Follicular lymphoma B cells induce the conversion of conventional CD4+ T cells to T-regulatory cells. Int J Cancer. 2009;124(1):239–244. doi: 10.1002/ijc.23881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Oeschger S, Brauninger A, Kuppers R, Hansmann ML. Tumor cell dissemination in follicular lymphoma. Blood. 2002;99(6):2192–2198. doi: 10.1182/blood.V99.6.2192. [DOI] [PubMed] [Google Scholar]
- 92.Aarts WM, et al. In situ analysis of the variable heavy chain gene of an IgM/IgG-expressing follicular lymphoma: evidence for interfollicular trafficking of tumor cells. Am J Pathol. 2002;160(3):883–891. doi: 10.1016/S0002-9440(10)64911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruminy P, et al. S(mu) mutation patterns suggest different progression pathways in follicular lymphoma: early direct or late from FL progenitor cells. Blood. 2008;112(5):1951–1959. doi: 10.1182/blood-2007-11-124560. [DOI] [PubMed] [Google Scholar]
- 94.Irish JM, et al. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc Natl Acad Sci U S A. 2010;107(29):12747–12754. doi: 10.1073/pnas.1002057107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garvin AJ, Simon RM, Osborne CK, Merrill J, Young RC, Berard CW. An autopsy study of histologic progression in non-Hodgkin’s lymphomas. 192 cases from the National Cancer Institute. Cancer. 1983;52(3):393–398. doi: 10.1002/1097-0142(19830801)52:3<393::AID-CNCR2820520302>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 96.Lossos IS, Gascoyne RD. Transformation of follicular lymphoma. Best Pract Res Clin Haematol. 2011;24(2):147–163. doi: 10.1016/j.beha.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lossos IS, et al. Transformation of follicular lymphoma to diffuse large-cell lymphoma: alternative patterns with increased or decreased expression of c-myc and its regulated genes. Proc Natl Acad Sci U S A. 2002;99(13):8886–8891. doi: 10.1073/pnas.132253599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davies AJ, et al. Transformation of follicular lymphoma to diffuse large B-cell lymphoma proceeds by distinct oncogenic mechanisms. Br J Haematol. 2007;136(2):286–293. doi: 10.1111/j.1365-2141.2006.06439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johnson NA, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114(11):2273–2279. doi: 10.1182/blood-2009-03-212191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gentles AJ, et al. A pluripotency signature predicts histologic transformation and influences survival in follicular lymphoma patients. Blood. 2009;114(15):3158–3166. doi: 10.1182/blood-2009-02-202465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davies AJ, et al. A limited role for TP53 mutation in the transformation of follicular lymphoma to diffuse large B-cell lymphoma. Leukemia. 2005;19(8):1459–1465. doi: 10.1038/sj.leu.2403802. [DOI] [PubMed] [Google Scholar]
- 102.Lo Coco F, Gaidano G, Louie DC, Offit K, Chaganti RS, Dalla-Favera R. p53 mutations are associated with histologic transformation of follicular lymphoma. Blood. 1993;82(8):2289–2295. [PubMed] [Google Scholar]
- 103.Fitzgibbon J, et al. Genome-wide detection of recurring sites of uniparental disomy in follicular and transformed follicular lymphoma. Leukemia. 2007;21(7):1514–1520. doi: 10.1038/sj.leu.2404696. [DOI] [PubMed] [Google Scholar]
- 104.Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood. 2003;102(4):1443–1448. doi: 10.1182/blood-2002-08-2482. [DOI] [PubMed] [Google Scholar]
- 105.Cheson BD. New agents in follicular lymphoma. Best Pract Res Clin Haematol. 2011;24(2):305–312. doi: 10.1016/j.beha.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 106.Brody J, Kohrt H, Marabelle A, Levy R. Active and passive immunotherapy for lymphoma: proving principles and improving results. J Clin Oncol. 2011;29(14):1864–1875. doi: 10.1200/JCO.2010.33.4623. [DOI] [PubMed] [Google Scholar]
- 107.Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 108.Schuetz JM, et al. Leukemia. BCL2 mutations in diffuse large B-cell lymphoma [published online ahead of print December 22, 2011]. doi: 10.1038/leu.2011.378 . [DOI] [PubMed] [Google Scholar]
- 109.Migliazza A, et al. Frequent somatic hypermutation of the 5′ noncoding region of the BCL6 gene in B-cell lymphoma. Proc Natl Acad Sci U S A. 1995;92(26):12520–12524. doi: 10.1073/pnas.92.26.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lo Coco F, et al. Rearrangements of the BCL6 gene in diffuse large cell non-Hodgkin’s lymphoma. Blood. 1994;83(7):1757–1759. [PubMed] [Google Scholar]
- 111.Bastard C, et al. LAZ3 rearrangements in non-Hodgkin’s lymphoma: correlation with histology, immunophenotype, karyotype, and clinical outcome in 217 patients. Blood. 1994;83(9):2423–2427. [PubMed] [Google Scholar]
- 112.Otsuki T, et al. Analysis of LAZ3 (BCL-6) status in B-cell non-Hodgkin’s lymphomas: results of rearrangement and gene expression studies and a mutational analysis of coding region sequences. Blood. 1995;85(10):2877–2884. [PubMed] [Google Scholar]
- 113.Kato M, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459(7247):712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 114.Honma K, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114(12):2467–2475. doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 115.Gronbaek K, et al. Somatic Fas mutations in non-Hodgkin’s lymphoma: association with extranodal disease and autoimmunity. Blood. 1998;92(9):3018–3024. [PubMed] [Google Scholar]