

Abstract
Pancreatic agenesis is a human disorder caused by defects in pancreas development. To date, only a few genes have been linked to pancreatic agenesis in humans, with mutations in pancreatic and duodenal homeobox 1 (PDX1) and pancreas-specific transcription factor 1a (PTF1A) reported in only 5 families with described cases. Recently, mutations in GATA6 have been identified in a large percentage of human cases, and a GATA4 mutant allele has been implicated in a single case. In the mouse, Gata4 and Gata6 are expressed in several endoderm-derived tissues, including the pancreas. To analyze the functions of GATA4 and/or GATA6 during mouse pancreatic development, we generated pancreas-specific deletions of Gata4 and Gata6. Surprisingly, loss of either Gata4 or Gata6 in the pancreas resulted in only mild pancreatic defects, which resolved postnatally. However, simultaneous deletion of both Gata4 and Gata6 in the pancreas caused severe pancreatic agenesis due to disruption of pancreatic progenitor cell proliferation, defects in branching morphogenesis, and a subsequent failure to induce the differentiation of progenitor cells expressing carboxypeptidase A1 (CPA1) and neurogenin 3 (NEUROG3). These studies address the conserved and nonconserved mechanisms underlying GATA4 and GATA6 function during pancreas development and provide a new mouse model to characterize the underlying developmental defects associated with pancreatic agenesis.
Introduction
In vertebrates, the mature pancreas comprises primarily 3 morphologically and functionally distinct tissues that are derived from cells within the embryonic foregut endoderm. Nearly 90% of the pancreas is composed of acinar cells, which synthesize and secrete digestive enzymes for food processing. The endocrine tissue is organized into islets composed of α, β, δ, PP, and ε cells, which produce the hormones glucagon, insulin, somatostatin, pancreatic polypeptide, and ghrelin, respectively. The endocrine pancreas regulates broad aspects of metabolism, especially glucose homeostasis. The pancreatic ductal tissue is a branched tubular network that serves as the conduit for transporting enzymes synthesized in the acinar tissue to the duodenum (reviewed in ref. 1).
During mouse embryogenesis, morphological development of the pancreas begins with patterning of the foregut endoderm at E8.5 (2). A small group of endodermal cells commit to the pancreatic fate as a result of their unique positions within the foregut endoderm and their intrinsic responses to individual signaling pathways (3, 4). The subsequent evagination of the pancreatic endoderm leads to the formation of dorsal and ventral pancreatic buds between E8.75 and E9.5. Under the regulation of a series of transcription factors, the embryonic pancreas undergoes a period of exponential growth between E11.5 and E15.5, also called the secondary transition, to form the 3 primary structures of the pancreas (5, 6).
Numerous transcription factors have been implicated in the complex regulatory process of pancreas development. During the initial stages of pancreas bud formation, 2 of the primary pancreatic transcriptional regulators, pancreatic and duodenal homeobox 1 (Pdx1) and pancreas-specific transcription factor 1a (Ptf1A), are expressed with the onset of dorsal and ventral pancreatic bud formation (2). Consistent with the early expression pattern, null mutations in either Pdx1 or Ptf1a in mice result in severe disruption of pancreas formation; however, in each case, a rudimentary pancreas is able to form (7–10). This would suggest that additional, as of yet unidentified, transcription factors are required upstream of Pdx1 and Ptf1a to initiate pancreas induction from the foregut endoderm. In humans, mutations of PDX1 or PTF1A also result in pancreatic agenesis, suggesting a conserved function for these factors in pancreas outgrowth (11–13). Functioning downstream of Pdx1 and Ptf1a, the SRY/HMG transcription factor Sox9 regulates the maintenance of the early pancreatic progenitor cells and influences endocrine cell formation; in the mouse, loss of Sox9 in the pancreas leads to pancreatic hypoplasia (14, 15). At a slightly later stage, when pancreatic progenitor cells begin to differentiate into the 3 pancreatic cell lineages, carboxypeptidase A1 (CPA1) delineates a population of progenitor cells that reside in the tips of the branching pancreatic epithelium (16), and the basic helix-loop-helix (bHLH) transcription factor neurogenin 3 (NEUROG3) becomes activated in the endocrine precursor population, where it is necessary and sufficient to induce endocrine cell development (17–19). These events are followed by the secondary transition, when the endocrine and exocrine cell populations expand and differentiate to generate the mature hormone- and enzyme-producing cell types of the islet and acini, respectively (reviewed in refs. 1, 3).
The Gata factors are a 6-member family that share a common DNA-binding motif characterized by 2 tandem zinc-finger domains. All members recognize the consensus sequence A/T-GATA-A/G. Gata1, Gata2, and Gata3 represent a subgroup of the family that is preferentially expressed in hematopoietic cells and predominantly important for cellular proliferation and differentiation during hematopoiesis (20–22). Gata4, Gata5, and Gata6 represent the second subgroup, which is important for the differentiation of tissues derived from endoderm and mesoderm (23, 24). In the embryonic mouse, Gata4 is expressed in the primitive endoderm, heart, liver, small intestine, and pancreas (25, 26). Gata4 plays a key role in heart development in mice and humans (27–29); Gata4-null mice die between E7.5 and E9.5 due to defects in heart development and ventral morphogenesis (30–32). Gata5 is also expressed in the developing heart, the pulmonary mesenchyme, and tissue-restricted smooth muscle cells (33). Unlike Gata4- and Gata6-null mice, Gata5-deficient mice are viable and fertile (34). Gata6 is expressed in the primitive streak, allantois, muscle, heart, lung, and gut and plays an important role in lung development, specifically in branching morphogenesis and later stage differentiation of the lung epithelium (35–38). Gata6-null mice die between E5.5 and E7.5 due to defects in the primitive endoderm cell lineages during blastocyst formation (39, 40).
In the mouse, Gata4 and Gata6, but not Gata5, are expressed in overlapping domains within the foregut endoderm, including the regions that give rise to liver and pancreas, where they have been implicated as pioneer factors in initiating tissue-specific gene expression (26, 41–43). Both factors continue to be expressed throughout the developing dorsal and ventral pancreatic bud epithelia; however, as pancreatic development proceeds, Gata6 becomes restricted to the endocrine and ductal compartments of the pancreas, while Gata4 remains strongly expressed in the acinar tissue (26, 44). Although the early embryonic lethality associated with the Gata4–/– and Gata6–/– alleles has precluded standard loss-of-function analysis in the pancreas, in vivo mouse studies using tetraploid complementation and a transgenic Gata-engrailed fusion protein have indicated that Gata4 and/or Gata6 contribute to the regulation of pancreas development (26, 45). Furthermore, in vitro studies have implicated Gata4 and Gata6 in pancreatic gene regulation (46, 47).
Recent human genetic studies have indicated that the individual GATA4 and GATA6 proteins have essential roles in human pancreas development. Several inactivating mutant alleles of GATA6 and a mutant allele of GATA4 have been associated with human pancreatic agenesis (48, 49). To more precisely define the roles of Gata4 and Gata6 during mouse pancreas development and to create a potential model of human pancreatic agenesis, we generated mice in which Gata4 or Gata6 were deleted specifically in the pancreas (Pdx1Cre;Gata4fl/fl [Gata4-CKO, where CKO indicates conditional KO] and Pdx1Cre;Gata6fl/fl [Gata6-CKO] mice). During embryogenesis, the Gata4-CKO mice exhibited subtle defects in acinar morphology and the Gata6-CKO mice displayed minor ductal defects; however, both single CKO lines survived for a normal life span and did not display obvious physiological defects. In contrast, simultaneous deletion of both Gata4 and Gata6 (double-KO [DKO] mice) resulted in pancreatic agenesis at birth. The DKO mice initiated dorsal and ventral pancreatic outgrowth, but further pancreatic development and differentiation were arrested by E11.5. Reduced proliferation of the pancreatic progenitor cells and failed branching morphogenesis contributed to the pancreatic developmental defects in the DKO embryos. Furthermore, differentiation of the multipotent progenitor Cpa1+ tip cells and Neurog3+ endocrine precursor cells was not initiated in the DKO embryos. These studies demonstrate the important and redundant roles of Gata4 and Gata6 in pancreas development and differentiation and highlight the conserved and nonconserved functions of these factors in the mouse and human pancreata.
Results
Mild defects in pancreas-specific deletion of either Gata4 or Gata6.
To study the respective functions of Gata4 and Gata6 during pancreas development, we generated pancreas-specific deletions of Gata4 and Gata6 using Pdx1Cre;Gata4fl/fl and Pdx1Cre;Gata6fl/fl single-CKO mice (50–52). Cre recombinase activity is highly penetrant throughout the pancreatic epithelium as early as E9.5 in this Pdx1Cre line (Tg[Ipf1-cre]1Tuv) (ref. 53 and data not shown). Mice carrying pancreas-specific deletions of either Gata4 or Gata6 lived a normal life span, were euglycemic, and displayed normal glucose tolerance at 6–8 weeks of age. Morphological and histological analysis of adult pancreata from each single CKO line appeared grossly normal, displaying the appropriate formation and differentiation of all pancreatic cell types (data not shown and Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI63352DS1). On occasion, we observed the presence of enlarged or cystic ducts in Gata6-CKO pancreas (Supplemental Figure 1G); however, these defects did not appear to interfere with normal pancreatic function. Consistent with the lack of overt morphological defects, 6- to 8-week-old Gata4-CKO and Gata6-CKO mice displayed normal insulin levels in comparison with control littermates under fed, fasted, and glucose-challenged conditions (Supplemental Figure 2, A–C, and data not shown).
Although we were unable to detect significant phenotypes in the adult Gata4-CKO and Gata6-CKO mice, morphological analysis of their pancreas prior to birth revealed subtle developmental defects. In E18.5 Gata4-CKO embryos, acini appeared smaller, less eosinophilic, and less mature compared with those of littermate controls (Figure 1, A–D). The presence of Cpa1 expression in the exocrine compartment suggested that appropriate acinar differentiation was initiated; however, the subcellular localization of Cpa1 appeared to be unevenly distributed within the cytoplasm compared with control acini cells (Figure 1, E and F). Similar defects were observed in Ptf1aCre;Gata4-CKO embryos (data not shown). The persistence of low-level Sox9 expression in normal acinar cells was generally absent from the Gata4-CKO pancreas (Figure 1, E–H). Furthermore, small pancreatic ducts were more prominent and disorganized (Figure 1, G and H). The endocrine compartment of these mice was indistinguishable from that of their control littermates (data not shown).
Figure 1. Pancreas-specific deletion of either Gata4 or Gata6 causes mild embryonic pancreas defects.

(A–D) H&E staining of representative sections of E18.5 dorsal pancreas from control Gata4fl/fl (A and C) and Pdx1Cre;Gata4fl/fl embryos (B and D). (E–H) Immunofluorescence staining for Sox9 (red) and Cpa1 (green) of E18.5 dorsal pancreas from control Gata4fl/fl (E and G) and Pdx1Cre;Gata4fl/fl embryos (F and H). Cpa1 is expressed in acinar cells (E and F). Sox9 is expressed in ductal cells and centroacinar cells in both control and Pdx1Cre;Gata4fl/fl embryos (E and F). In acinar cells, Sox9 is expressed in control pancreas (G), but is absent in most Gata4-depleted acinar cells of Pdx1Cre;Gata4fl/fl pancreas (H). Original magnification, ×200 (A and B); ×640 (C–H). (I and J) H&E staining of representative sections of E18.5 control Gata6fl/+ (I) and Pdx1Cre;Gata6fl/fl (J) embryos. (K–P) Immunofluorescence staining of E18.5 control Gata6fl/+; R26R-LacZ (K, M, and O) and Pdx1Cre;Gata6fl/fl (L, N, and P) embryos. Cpa1 (green) expression is in the acinar cells, and DBA (red) marks the epithelial ductal regions. Increased ductal tissues are observed in Pdx1Cre;Gata6fl/fl pancreas (arrows in J, N, and P). Original magnification, ×200 (I–N); ×640 (O and P).
The E18.5 Gata6-CKO embryos displayed grossly normal exocrine and endocrine pancreatic morphology at E18.5 (Figure 1 and data not shown); however, we observed a small increase in the number and size of dolichos biflorus agglutinin (DBA) lectin–labeled epithelial ducts throughout the pancreas, which was retained in the adult pancreas (Figure 1, I–P, and Supplemental Figure 1). These results suggest that Gata4 and Gata6 each have modest and somewhat differing functions during pancreas development, which may be a reflection of their overlapping and unique expression patterns (26).
Mice retaining only 1 allele of Gata4 or 1 allele of Gata6 display significant, but dissimilar pancreatic defects.
To determine whether the lack of significant phenotypes in either single KO was due to redundant functional activity of the 2 family members in the pancreas, we generated compound mutant mice retaining 1 allele of Gata4 (Gata4fl/+;Gata6flfl;Pdx1Cre+) or 1 allele of Gata6 (Gata4flfl;Gata6fl/+;Pdx1Cre+). These mice survive postnatally and are euglycemic; however, they each display unique pancreatic developmental phenotypes. Mice lacking both alleles of Gata6 and retaining 1 allele of Gata4 do not display obvious exocrine defects, but have increased numbers of large ducts (Figure 2, E and H). Furthermore, we often observed hormone-positive cells residing within the ductal epithelium (Figure 2, H and J). Islet size, number, or composition was not significantly altered compared with control littermates. In contrast, mice lacking both alleles of Gata4, but retaining 1 Gata6 allele, had a noticeable reduction in pancreatic mass due to a decrease in exocrine tissue (Figure 2F). The remaining acini were slightly disorganized, but expressed several exocrine enzymes, such as elastase and amylase, suggesting that they have undergone appropriate differentiation (Figure 2, F and I, and data not shown). The number and size of islets and islet cell composition in these mice appeared unchanged compared with control littermates at birth and in the adult (Figure 2, F and I, and data not shown). These results suggest that although there is functional redundancy between Gata4 and Gata6, Gata4 function appears to be more critical for the formation of exocrine tissue, whereas Gata6 has a dominant role in ductal formation. These findings are consistent with the diverging pancreatic expression patterns of Gata4 and Gata6 during the secondary transition; Gata4 becomes localized to the exocrine (tip) compartment, whereas Gata6 becomes more restricted to the ductal (trunk) compartment (16, 26).
Figure 2. Mice retaining only 1 allele of Gata4 or 1 allele of Gata6 display dissimilar pancreatic defects.

Representative E18.5 pancreata from embryos retaining only 1 allele of Gata4 or Gata6 were analyzed in whole mounts (A–C), H&E-stained sections (D–F), and coimmunofluorescence staining with CK19 (green) and insulin (red) (G–I). In comparison with control pancreata (A, D, and G), the pancreata in Pdx1Cre;Gata4fl/+;Gata6fl/fl mice display more ductal structures (E, H, and J) and have insulin-expressing cells residing within the ductal epithelium (arrows in J). In Pdx1Cre;Gata4fl/fl;Gata6fl/+ embryos, pancreas volume is significantly reduced due to decreased acinar tissue (C, F, and I). Original magnification, ×14 (A); ×20 (B and C); ×200 (D–I); ×400 (J).
Pdx1Cre;Gata4fl/fl;Gata6fl/fl (DKO) mice display pancreatic agenesis at birth.
Although Gata4 and Gata6 have differential expression later in pancreas development, they have overlapping expression within the prepancreatic endoderm and throughout the early pancreatic epithelium (26). To determine whether complete lack of Gata activity would affect early pancreas formation and/or development, we generated Pdx1Cre;Gata4fl/fl;Gata6fl/fl DKO mice. Littermates with the genotype Gata4fl/+;Gata6fl/+;Pdx1Cre+ or Gata4fl/+;Gata6fl/+ (without Cre) were phenotypically normal, euglycemic, fertile, and indistinguishable from WT littermates, and were used as “control” embryos. The intercross of Pdx1Cre;Gata4fl/+;Gata6fl/+ and Gata4fl/+;Gata6fl/+ mice yielded DKO mice at the expected ratios at birth; however, the majority of DKO mice died during the early postnatal period. When we examined freshly dissected abdominal organs from newborn DKO mice, we could only detect the presence of a fibrous stromal structure in the region where the pancreas should have been located, as seen in control littermates (Figure 3, A and B). H&E sections through the entire abdominal region of E18.5 DKO embryos (n > 12) failed to reveal structures that were histologically similar to a pancreas (Figure 3, C and D). Furthermore, using an R26R:LacZ reporter to visualize the Pdx1-derived lineage, we could only identify a few labeled cells within the pancreatic region of the DKO embryos (Figure 3, F and G). The majority of the LacZ staining observed in the DKO gut region corresponded to endogenous β-gal activity that could also be observed in mice lacking the R26R:LacZ reporter (Figure 3E). These findings indicate that Gata4 and Gata6 have essential and redundant functions in the developing pancreas of the mouse and that the absence of both factors results in severe pancreatic agenesis.
Figure 3. Pancreas-specific deletion of Gata4 and Gata6 causes pancreatic agenesis.

(A and B) Dissected pancreas, stomach, spleen, and intestine of newborn control (A) and DKO (B) pups. The DKO pancreas is predominantly made up of mesenchymal-like stromal tissue. (C and D) H&E staining of representative sections from E18.5 embryos shows pancreas tissue occupying an area from the body wall to the midline in the control (C), whereas at the same plane of section, there is no apparent pancreas in the DKO (D). (E–G) LacZ staining of pancreatic progeny in whole E18.5 pancreas tissue shows fully developed pancreas in control embryos (E and F) and no apparent pancreatic tissue in the DKO embryos (G). Control pancreas from animals lacking the ROSA26:LacZ allele display endogenous LacZ staining in the gut tube (E). P, pancreas; Stm, stomach; Sp, spleen; Liv, liver; Kid, kidney. Pancreas or the region that should contain the pancreas is delineated by dashed lines in each panel. Original magnification, ×15 (A and B); ×25 (C and D); ×40 (E, F, and G).
Although the majority of DKO mice died shortly after birth, a small number (<5%) survived for approximately 1 month postnatally, presumably due to inadequate Cre activity, which would result in incomplete Gata4 and/or Gata6 deletion. The rare surviving mice were severely hyperglycemic (Supplemental Figure 3A), with small morphologically abnormal pancreata. In addition, the pancreatic tissue that was present contained reduced numbers of differentiated acinar cells and only a few small islets (Supplemental Figure 3, B–G). The numbers of insulin-, glucagon-, and amylase-expressing cells were also severely reduced (Supplemental Figure 3, B–G).
Initiation of pancreatic bud outgrowth occurs normally in DKO embryos.
Since both Gata4 and Gata6 are expressed in the prepancreatic domain of the foregut endoderm and have been implicated as pioneer factors that initiate tissue-specific gene expression (reviewed in ref. 54), we sought to determine whether the absence of pancreas tissue in the DKO mice at birth was due to a failure in pancreas specification in the foregut endoderm. We analyzed pancreatic bud formation at E9.5 in Pdx1Cre;Gata4fl/fl;Gata6fl/fl;Rosa26:LacZ (DKO;R26R-LacZ) embryos compared with their littermate controls (control;R26R-LacZ); inclusion of the Rosa26:LacZ reporter allowed the visualization of Pdx1-labeled progenitor cells. Surprisingly, the DKO embryos displayed morphologically normal dorsal and ventral pancreatic buds that were indistinguishable from those of littermate controls (Figure 4, A–D), suggesting Gata4 and Gata6 were dispensable for initiation of the pancreatic developmental program.
Figure 4. Pancreatic buds are specified in the E9.5 DKO embryos.
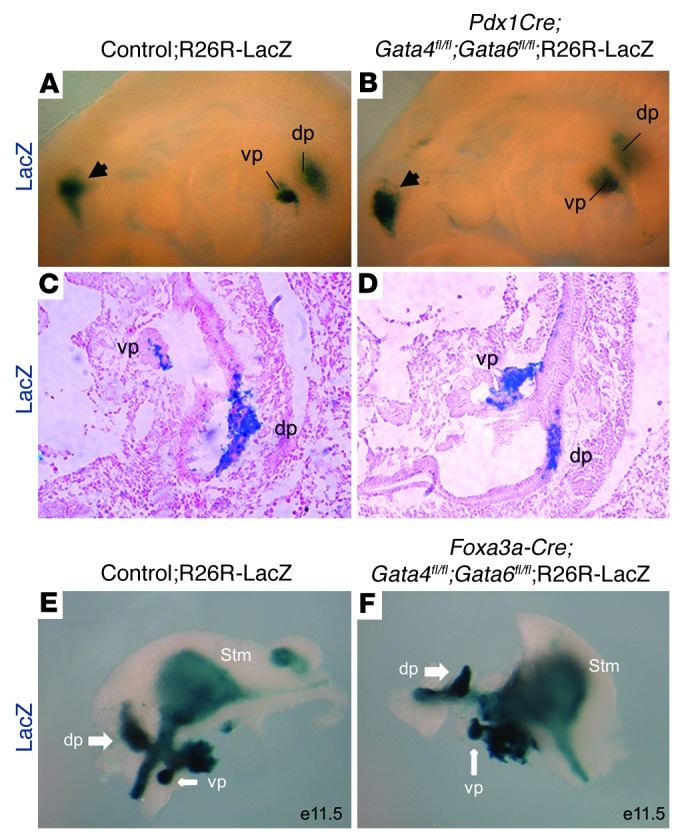
(A and B) Whole-mount LacZ labeling of Pdx1 progeny in E9.5 embryos shows distinctively formed pancreatic buds in both controls (A) and DKO embryos (B). A LacZ-positive region (arrowhead) was also detected in the hypothalamus region, as reported by Wicksteed et al. (53). (C and D) Representative sections of the pancreatic region from embryos shown in A and B confirm the presence of the dorsal and ventral buds in both control (C) and DKO embryos (D). (E and F) Whole-mount LacZ staining of Foxa3 lineage cells in control and embryos deleted for Gata4 and Gata6 in the Foxa3-Cre prepancreatic endodermal domain. Similar to the phenotype of embryos deleted for Gata4 and Gata6 in the later Pdx1 domain, dorsal and ventral pancreatic buds form in control E11.5 embryo (A) and in Foxa3-cre;Gata4fl/fl;Gata6fl/fl embryos (B). dp, dorsal pancreas; vp, ventral pancreas. Original magnification, ×40 (A and B); ×100 (C and D); ×80 (E and F).
A caveat of these studies is the use of Pdx1Cre to trigger the deletion of both Gata factors. By using the Pdx1Cre driver, deletion of Gata4 and Gata6 would not occur until after the pancreatic program had been initiated and Pdx1 expression activated. To resolve this issue, we deleted Gata4 and Gata6 from the foregut endoderm using the Foxa3-Cre mice (55). Foxa3-Cre becomes active at E8.5 in the anterior intestinal portal region and throughout the entire gut endoderm, which allows for efficient deletion of the Gata factors in the prepancreatic region of the foregut endoderm prior to bud initiation. Similar to the Pdx1:Cre DKO embryos, both dorsal and ventral pancreatic buds were able to form in the Foxa3-Cre;Gata4fl/fl;Gata6fl/fl;Rosa26:LacZ embryos (Figure 4, E and F), confirming that Gata4 and Gata6 function is not required for initial pancreas specification and bud outgrowth.
Pancreas differentiation becomes compromised in midgestation DKO embryos.
At E10.5, dorsal and ventral pancreatic bud outgrowth in the DKO;R26R-LacZ embryos remained indistinguishable from that in their littermate controls (Figure 5, A and B); however, there appeared to be a reduction in the number of Pdx1+ cells present in the pancreatic epithelial domain (Figure 5, D vs. C). Quantification of the total number of Pdx1+ cells relative to the number of cells expressing Foxa1 or Foxa2, transcription factors that are expressed throughout the foregut endoderm and pancreatic epithelium (reviewed in ref. 56), indicated that there was an approximately 40% reduction in the number of total pancreatic epithelial cells that maintain expression of Pdx1 in the DKO embryos (Figure 5E). To determine whether the reduction in Pdx1 expression was due to direct regulation by Gata activity, we used in silico analysis to identify 2 highly conserved Gata DNA binding consensus sequences, one within the Pdx1 distal enhancer (area IV; –6010 bp) and one within a proximal enhancer (area III; –1841 bp) (57–59). ChIP analysis using Gata4- and Gata6-specfic antibodies indicated that Gata4 and Gata6 occupy these sites both in 266-6 cell lines and E14.5 pancreatic tissue (Figure 5F and Supplemental Figure 5E). The accompanying study by Carrasco et al. confirms the binding of Gata4 and Gata6 to the Pdx1 proximal enhancer and further demonstrates direct binding and activation of Pdx1 by the Gata factors in vitro and in vivo (60). These analyses suggest that the Gata proteins positively regulate Pdx1 gene expression in the early pancreatic endoderm.
Figure 5. At E10.5, fewer endoderm cells are differentiated into Pdx1+ pancreatic progenitor cells, due partially to the loss of Gata4 regulation of Pdx1 gene expression.

(A–D) Representative sagittal sections from E10.5 LacZ-stained control (A) and DKO (B) embryos. The regions containing the pancreatic buds are highlighted by boxes. Pancreatic bud formation appears morphologically normal in the DKO embryo (boxed areas in A and B). (C and D) Immunofluorescence costaining using anti-Pdx1 and anti-Foxa1/2 antibodies shows that there is a smaller percentage of Pdx1+ pancreatic progenitor cells relative to the number of Foxa1/a2+ cells in the pancreatic endodermal region of DKO embryos (D vs. C). (E) Cell quantification of the relative number of Pdx1+ cells within the Foxa1/a2+ foregut endoderm (n = 4), p, pancreas; g, gut. ***P < 0.001. (F) Real-time qPCR analysis of DNA products immunoprecipitated by an anti-Gata4 antibody in 266-6 cells. Gata consensus sequences within Pdx1 enhancer area III (–1841 bp from the ATG) and Pdx1 enhancer area IV (–6010 bp from the ATG) are bound by the Gata4 protein. Gata4 does not bind the Arx promoter, which serves as a negative control. *P < 0.05. Original magnification, ×50 (A and B); ×200 (C and D).
By E11.5, shortly after the observed reduction in Pdx1-expressing cells and at the beginning of the secondary transition, there was a notable reduction in pancreatic bud size in the DKO;R26R-LacZ embryos compared with control embryos (Figure 6, A–F). The decrease in pancreatic area in the DKO embryos was not associated with increased apoptosis (Figure 6, C and D); however, there was a significant reduction in cell proliferation, as shown by decreased BrdU labeling throughout the pancreatic epithelium (Figure 6, E and F). Quantification of the BrdU+ cells indicated there was a 28% reduction of proliferating Pdx1 cells in the DKO pancreata (Figure 6G). Consistently, FACS analysis of E12.5 dorsal and ventral pancreatic bud tissue labeled with propidium iodide (PI) demonstrated a 38% reduction in the number of total pancreatic cells in S phase in the DKO pancreata compared with their littermate controls (Figure 6H). This indicates that reduced proliferation contributes to the defective pancreas development in the DKO embryos.
Figure 6. Morphological and molecular defects in DKO pancreas are apparent during secondary transition.

(A and B) Whole-mount LacZ lineage labeling of Pdx1 progeny in control (A) versus DKO (B) embryos indicates that by E11.5, the DKO embryos have smaller pancreatic buds. (C and D) Coimmunofluorescence staining of caspase 3 (red) and Pdx1 (green) in E11.5 sections of control (C) vs. DKO embryos (D) suggests that there is no obvious change in apoptosis in the DKO pancreas. Insets show positively caspase 3–labeled tissue from the same sections. (E and F) Coimmunofluorescence staining of BrdU (red) and Pdx1 (green) in E11.5 sections of control (E) vs. DKO embryos (F) indicates there are fewer proliferating Pdx1+ pancreatic cells. (G) Quantification of BrdU-labeled Pdx1+ cells indicates a significant (28%) reduction in the number of replicating Pdx1+ progenitor cells in DKO embryos. (H) FACS analysis of PI-stained cells to quantify overall cellular proliferation shows significant (38%) reduction of S phase cells in the DKO pancreatic buds. (I and J) Immunofluorescence staining of Sox9 (green) in the E12.5 ventral pancreatic bud. (K and L) Coimmunofluorescence staining of Cpa1 (green), Pdx1 (red), and Neurog3 (white) in the ventral pancreatic bud indicates that the DKO embryos are defective in pancreatic cell differentiation at E12.5, with the loss of multipotent progenitor marker Cpa1 and endocrine progenitor marker Neurog3 (n = 4; 20 sections evenly distributed through the entire pancreas were analyzed for each n). (M and N) Coimmunofluorescence staining of Ptf1a (red) and glucagon (green) in the E12.5 ventral pancreatic bud shows more centralized Pft1a distribution and much reduced glucagon-positive cells (N vs. M) *P < 0.05. Original magnification, ×60 (A and B); ×200 (C–F); ×400 (I–N, and insets).
Since we used Pdx1Cre or Foxa3-Cre to delete the Gata factors specifically from the pancreatic epithelium, we believe that the early defects observed in pancreas development in the DKO embryos are likely to be intrinsic to the epithelium. Consistently, we did not observe any obvious defects associated with the Islet1/2-expressing pancreatic mesenchyme between E9.5 and E12.5 (Supplemental Figure 4, A and B, and data not shown).
By E12.5, the pancreatic buds of the DKO embryos were further reduced in size compared with littermate controls, with the dorsal pancreas becoming either absent or significantly decreased in area (data not shown). While the control pancreatic epithelium continued to grow and branch at E12.5, residual DKO ventral pancreatic epithelium remained in a compact ball of cells (Figure 6, J, L, and N). To determine whether the apparent absence of branching morphogenesis in the DKO embryos correlated with differentiation defects, we assessed the expression of pancreatic progenitor and differentiation markers in the ventral pancreas. This analysis demonstrated that while Sox9+ progenitor cells were maintained in the DKO embryos, Cpa1- and Neurog3-expressing cells were mostly absent (n = 4), suggesting that the multipotent progenitor tip cells and endocrine precursor cells failed to differentiate (Figure 6, I–N, and Supplemental Figure 5). We were able to detect a very small number of glucagon-producing cells in the central region of the DKO ventral pancreas, which may either represent the Neurog3-independent glucagon+ cell population (61, 62) or be derived from cells that were not efficiently deleted for both Gata4 and Gata6 (Figure 6, M and N). We were unable to detect insulin+ β cells in the E12.5 DKO pancreata (data not shown).
The loss of Cpa1 and Neurog3 expression in the DKO embryos could be due to a block in differentiation of the respective progenitor populations and/or due to direct regulation of the Cpa1 and Neurog3 genes by the Gata factors. Sequence analysis of the Cpa1 and Neurog3 promoter identified 2 closely linked evolutionarily conserved Gata consensus sites within the Cpa1 promoter region and a single conserved site in the Neurog3 promoter region (Supplemental Figure 5E). ChIP analysis on chromatin isolated from E14.5 pancreatic tissue demonstrated Gata4 binding to the Cpa1 and Neurog3 promoters and weak Gata6 binding to the Neurog3 promoter (Supplemental Figure 5E). Since we have found the Gata6-specific antibody to be consistently less reliable in these assays, we are not able to make conclusions about Gata6 binding to these promoters; however, these findings suggest that Gata activity may be required to activate Cpa1 and Neurog3 to allow further differentiation of the pancreas during the secondary transition.
Attrition of the dorsal and ventral pancreas in DKO embryos becomes more pronounced throughout the secondary transition.
By E13.5, the only pancreatic tissue that could be detected by LacZ staining in the DKO;R26R-LacZ embryos was a small region of the ventral pancreas (Figure 7, A–D). The epithelium remained completely unbranched, and there was no longer evidence of pancreatic endocrine or exocrine markers (Figure 7D and data not shown). Interestingly, at this stage of development we observed an extension of LacZ+ Pdx1-derived cells into the proximal stomach (Figure 7, B and F). Since the continual reduction in ventral and dorsal pancreatic tissue in the DKO embryos did not appear to correlate with increased apoptosis (Figure 6D and data not shown), it is possible that, in the absence of appropriate differentiation, the Pdx1-derived cells migrate more anteriorly and become incorporated into the stomach epithelium. With the exception of a few scattered glucagon+ cells occasionally observed in the posterior stomach, the lineage-labeled cells in the stomach did not express mature endocrine or exocrine pancreatic markers. To determine the identity of the Pdx1-derived cells present in the stomach, we generated DKO;R26R-tomato mice (63) to facilitate coimmunofluorescence analysis (Supplemental Figure 6). This analysis demonstrated that the Pdx1-derived cells did not express pancreatic progenitor markers such as Pdx1, Neurog3, and Cpa1 (Supplemental Figure 6, C–F), suggesting that they have extinguished a pancreatic differentiation program. It is also possible that these cells are also mislocalized Pdx1+ duodenal cells; however, they do not express the intestinal marker Cdx2 (Supplemental Figure 6, I and J). On the other hand, it appears that Pdx1-derived cells have adopted local stomach cell fates, as they express the stomach progenitor marker Sox2 (Supplemental Figure 6, G and H). Consistently, by E17.5, the Pdx1-derived cells in the DKO;R26R-LacZ embryos became incorporated into the stomach epithelium, where they have acquired histological features of squamous epithelium (Supplemental Figure 7, A and C, and B and D).
Figure 7. Outgrowth of the pancreas during the secondary transition is defective in DKO embryos.
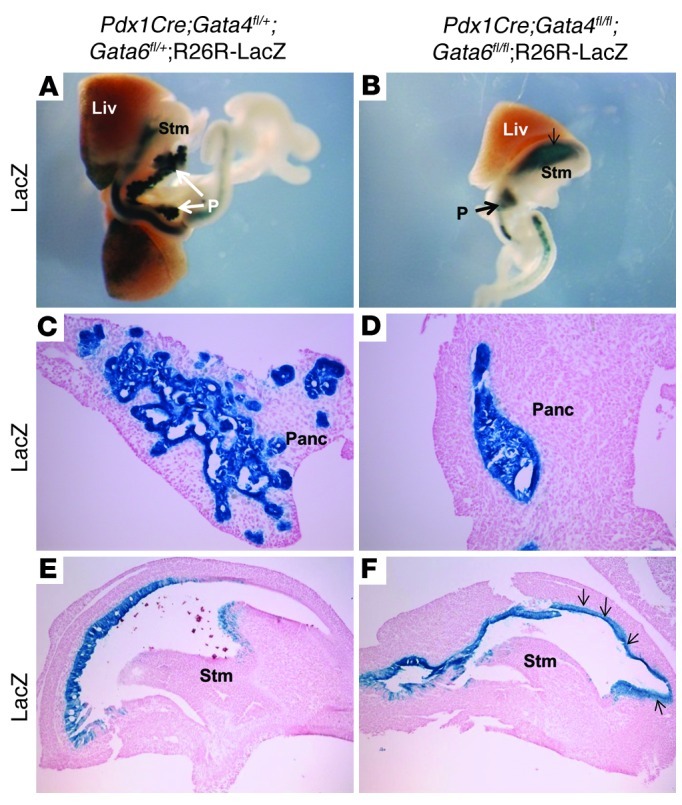
(A–D) LacZ staining of Pdx1 progeny at E13.5. LacZ whole-mount staining shows the normal branched structure of the pancreas in a representative control embryo (A), whereas only a small group of labeled cells are present in the ventral bud region of DKO pancreas (B). Sections of the ventral pancreatic regions show extensive branching morphogenesis in the control pancreas (C) and an unbranched structure of tightly clustered cells in the DKO pancreas (D). Note the expansion of LacZ-labeled Pdx1 progeny cells into the proximal stomach (B). Sections of the stomachs from control (E) and DKO (F) embryos demonstrate that Pdx1 descendent cells are found in the anterior part of the stomach (arrows in F). Panc, pancreas; Liv, liver; Stm, stomach. Original magnification, ×40 (A and B); ×200 (C and D); ×100 (E and F).
At late gestation, pancreas remnants in the DKO embryos remain unbranched and undifferentiated.
After E15.5, the dorsal pancreas of DKO;R26R-LacZ or R26R-tomato embryos was undetectable, and the remaining Pdx1-derived cells within the ventral pancreas resolved into a single-cell–layer epithelial duct-like epithelium (Figure 8, A and B, and data not shown). By the end of gestation, as the surrounding epithelium and mesenchyme continued to grow and differentiate, the Pdx1 lineage–labeled cells became further dispersed and, in some embryos, disappeared from the foregut region completely (Figure 8, A–F). The variable penetrance was likely due to the timing and degree of Gata4 and Gata6 deletion in the pancreas. Interestingly, there remains a large number of Pdx1-derived cells present in the proximal stomach region (Figure 8D), further suggesting that the undifferentiated Pdx1-dervied dorsal pancreas cells do not undergo apoptosis, but migrate to a new location.
Figure 8. At E17.5, pancreatic lineage cells remain unbranched and undifferentiated.
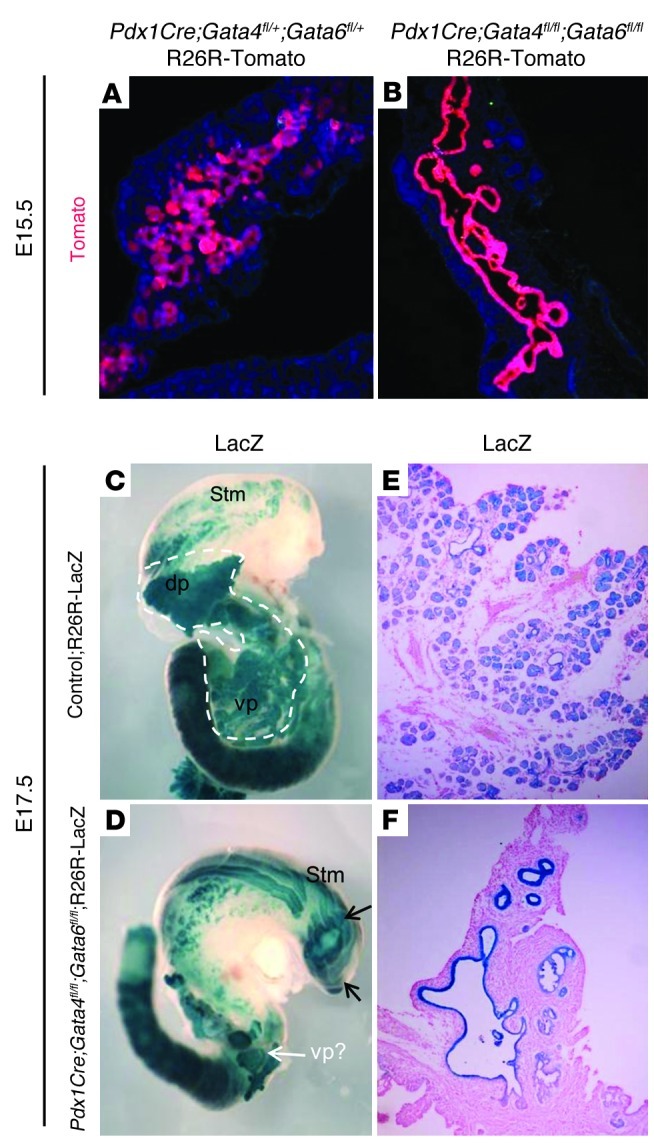
(A and B) The Rosa26:Tomato reporter (red) labels the Pdx1+ lineage in control (A) and DKO (B) pancreatic epithelium at E15.5. By this stage of development, the Pdx1+ progenies are restricted to a relatively unbranched single-cell–layered epithelium in the DKO pancreas. (C and D) Whole-mount and sections of LacZ-stained E17.5 control (C and E) and DKO (D and F) abdominal organs show well-extended glandular structure in the control pancreas (C) and only a small pancreatic remnant in the DKO (D). Sections through the remaining (ventral) pancreatic remnant in the DKO show LacZ-labeled Pdx1 progenies present in unbranched cystic ductal structures, and there is no morphological evidence of differentiated pancreatic tissue (F) as compared with the control sections (E). Note the extension of Pdx1-lineage–labeled cells in the proximal stomach in the DKO embryo (arrows in D). Original magnification, ×200 (A and B); ×20 (C and D); ×200 (E and F).
Discussion
In the current study, we investigated the functional roles of Gata4 and Gata6 during mouse pancreas development using a Cre-lox system to delete Gata4 and/or Gata6 specifically from the prepancreatic endoderm and the pancreas. Surprisingly, we found that deletion of either Gata4 or Gata6 results in only minor cellular defects that appear to resolve postnatally and do not result in apparent physiological defects. However, simultaneous deletion of both Gata4 and Gata6 leads to severe pancreatic agenesis by birth, suggesting that, unlike their human counterparts, mouse Gata4 and Gata6 are partially redundant in their regulation of pancreas development. Furthermore, despite the early overlapping expression of Gata4 and Gata6 in the prepancreatic endoderm and their putative roles as tissue-specific pioneer transcription factors, initial specification and evagination of the dorsal and ventral pancreatic buds were not affected in the DKO embryos. However, subsequent growth and branching morphogenesis of the pancreatic buds became severely compromised, and differentiation of the multipotent tip cells and endocrine progenitor populations did not occur. Therefore, Gata4 and Gata6 play essential and redundant roles in the morphogenesis, growth, and differentiation of the mouse pancreas, and loss of both factors ultimately leads to pancreatic agenesis at birth.
Despite the early expression of Gata4 and Gata6 throughout the prepancreatic endoderm, pancreas specification was not affected in the DKO embryos. It is possible that the absence of an earlier phenotype was due to inactivation of Gata4 and Gata6 by Pdx1Cre after the onset of pancreatic specification, Pdx1 induction, and Cre activation. However, use of Foxa3-Cre to inactivate Gata4 and Gata6 prior to Pdx1 expression (56) also did not prevent pancreas specification and resulted in impaired pancreas development at E11.5, similar to the Pdx1Cre DKO embryos. The absence of an early phenotype could also be due to compensation by the third sub-family member, Gata5. However, although Gata5 has also been implicated in pancreas specification and development in Xenopus and zebrafish (64, 65), Gata5 expression in the pancreatic foregut region of the mouse has not been reported (33). Therefore, the data suggest that Gata factor function is not required in the foregut endoderm to initiate the pancreatic developmental program.
Interestingly, the DKO phenotype is very similar to the null phenotypes associated with the Pdx1- and Ptf1a-null mice, in which pancreas development is arrested shortly after bud formation. However, it appears that the disruption of pancreatic development in the Gata DKO embryos occurs at a somewhat later stage than seen in the Pdx1- and Ptf1a-null mutants, which would be consistent with the observation that early Pdx1 and Ptf1a expression is maintained in the DKO pancreata until approximately E11.5. Molecular analysis of the DKO pancreas suggests that Gata function is first required after specification of the pancreatic multipotent progenitor population (Pdx1+Ptf1a+Sox9+), but before the subsequent formation of lineage-restricted endocrine (Neurog3+) and exocrine (Cpa1+) precursor populations. Identification of downstream targets of the Gata factors within the E11.5 pancreatic epithelium will begin to elucidate the important molecular events that are required to induce pancreatic subtype differentiation. Although pancreatic development appears to proceed relatively normally until approximately E10.5, shortly after this stage, Pdx1 and Ptf1 expression are downregulated and pancreatic cell proliferation is decreased. These molecular defects are consistent with the subsequent block in growth, branching morphogenesis, and cell differentiation, but do not explain the gradual loss of pancreatic tissue that has formed and the disappearance of Pdx1-lineage–labeled cells. The loss of these early Pdx1-lineage–labeled populations also cannot be attributed to cell death, since we were unable to detect significant apoptosis at any point in development of the DKO pancreas. However, we did observe Pdx1-lineage–labeled cells located in the proximal stomach, which suggests that in the absence of appropriate differentiation, these pancreas progenitor cells migrate to a new location. This is somewhat analogous to Ptf1a-null embryos; in the absence of Ptf1a protein, the Ptf1a-lineage–labeled pancreatic progenitor cells became reallocated to 2 distinct regions of the duodenal epithelium, where they suppressed their pancreas identity and productively contributed to all duodenal cell types (11). In the DKO embryos, the ectopically localized Pdx1-lineage–labeled cells were integrated within the stomach epithelium and did not appear to differentiate into pancreatic cell fates. We have yet to determine whether these cells differentiate into a subset of stomach cell fates, since the embryos die prior to significant cellular differentiation in the stomach.
In all DKO embryos examined, the pancreatic defects occurred earlier and appeared more severe in the dorsal pancreas than the ventral pancreas. This disparity is probably not due to a higher level of Pdx1Cre activity in the dorsal bud, since we observe a similar bias toward dorsal defects in the Foxa3-Cre DKO embryos. It is possible that the dorsal pancreatic defects are more obvious because the dorsal pancreas develops earlier and more rapidly than the ventral pancreas; however, this explanation is not fully consistent with the timing of the defects associated with the ventral pancreas, which continues to develop for approximately 24–36 hours after the dorsal pancreatic remnants have disappeared. This would suggest that the Gata factors have a more critical early function in the dorsal pancreas, which may reflect their known role as mediators of retinoic acid signaling (66, 67); retinaldehyde dehydrogenase-2 (RALDH2; Aldh1a2), the enzyme responsible for most retinoic acid production in the embryo, preferentially regulates dorsal pancreas development (68, 69). In the ventral pancreas, the Gata factors may be mediating pancreas differentiation in response to an alternative signaling pathway at a later time point. Interestingly, the more severe dorsal defect that we observe is inconsistent with the Gata4–/– and Gata6–/– tetraploid analysis, where there was a more severe impact on the formation of the ventral pancreatic bud (45). Since the septum transversum was absent in the tetraploid embryos, it is possible that the observed ventral pancreas defects were secondary to the loss of important signals from this tissue that are necessary for ventral pancreas induction (43). Alternatively, the different phenotypes could reflect a timing issue: the tetraploid embryos never express Gata4 and Gata6 in the endoderm, whereas in the conditional DKO mice, Gata factors are present in the endoderm prior to Cre-mediated deletion. For this reason, it remains possible that the initial presence of Gata factors in the endoderm, prior to Foxa3 activity, is sufficient to prime the pancreatic endoderm for ventral pancreas induction.
The phenotypes of the pancreas-specific Gata deletions are also not completely consistent with the phenotypes of the transient transgenic mice carrying Gata-engrailed fusions expressed from the Pdx1 promoter (27). Although a subset of these dominant negative transgenic embryos did result in pancreas agenesis, it appeared that pancreas formation was more sensitive to disruption of Gata6. This discrepancy was probably due to the caveats inherent to the transient transgenic dominant negative approach. In our initial transgenic studies, we were unable to establish viable transgenic lines carrying either Pdx1:Gata4-Engrailed or Pdx1:Gata6-Engrailed transgenes, suggesting that both factors could sufficiently disrupt pancreas development to be incompatible with survival, presumably by removing Gata activity in the Pdx1 expression domain. However, this outcome forced us to rely on the analysis of limited numbers of transient transgenic embryos, each with a different site of genomic integration and varied copy number, which precluded consistent assessment of gene function; within each transgenic group, the phenotypes ranged from minimum to substantial effects. It is possible that analysis of additional Pdx1:Gata4-Engrailed transgenic embryos with the appropriate temporal and spatial expression would have revealed phenotypes comparable to the Pdx1:Gata6-Engrailed transgenic embryos. Although flawed, these initial studies provided insightful information regarding the essential functions of Gata4 and Gata6 during pancreas development and led to this current analysis of Gata function in the pancreas using the better defined and controlled Cre-lox experimental system.
Many genes, mostly encoding transcription factors, have now been shown to affect pancreas development and growth in mice (reviewed in ref. 2). However, far fewer genes have been associated with human pancreatic agenesis. PDX1 and PTF1A mutations have been reported in only 5 families with described cases of pancreatic agenesis (11–13). Recently, Allen et al. (48) identified GATA6 mutations in 15 of 27 individuals displaying pancreatic agenesis, accounting for 56% of all cases and suggesting there is an essential and nonredundant role of GATA6 in human pancreas development. In addition, a GATA4 missense mutation was identified in a child with heart defects and pancreatic agenesis, although a direct link between the GATA4 missense mutation and pancreatic agenesis was not established (49). Interestingly, in human cases, pancreatic agenesis was caused by haploinsufficiency of individual GATA factors. In contrast, mice carrying homozygous pancreas-specific deletions of the individual Gata factors display only subtle cellular defects and appear physiologically normal throughout life. It is only when all Gata4 and Gata6 alleles are deleted that pancreatic agenesis can be observed. The discrepancy in phenotypes could be due to the nature of the Gata mutations. Humans carrying mutations in GATA6 are defective for GATA6 activity in all tissues, including nonpancreatic tissues that may provide instructive signals for induction of appropriate pancreas formation and development. In this case, the tetraploid complementation studies performed by Watt et al. (45) may more accurately replicate the human situation. However, the human data would still suggest that pancreas development in humans is more sensitive to GATA gene dosage than in mice, since neither the Gata4 nor Gata6 heterozygous mice develop significant pancreatic phenotypes. This is similar to the phenotypic differences caused by heterozygous mutations in the hepatocyte nuclear factors [Hnf] Hnf1alpha, Hnf3beta (Forkhead box a2; Foxa2), and Hnf4alpha (70). Heterozygous mutations in each of these factors in humans can lead to impaired glucose response and cause maturity-onset diabetes of the young (MODY) (13, 71), whereas heterozygous mutations in the orthologos genes in mice do not induce early onset diabetes (72). It is currently unclear whether the more severe phenotypes observed in humans are due to additional defects in modifier genes, less overlap in expression domains between closely related gene family members, or simply increased sensitivity to gene dosage. Despite these species differences, the development of this mouse model of pancreatic agenesis will provide us with a useful experimental platform for identifying the downstream Gata factor targets and molecular events associated with this critical human birth defect.
Methods
Mice.
Derivation of the mouse strains used in this study has been reported previously: Gata4fl/fl (Gata4tm1.1Sad) (50), Gata6fl/fl (Gata6tm2.1) (51), Pdx1:Cre (Tg[Ipf1-cre]1Tuv) (52), Foxa3:Cre (Tg(Foxa3-cre)1Khk) (55), Rosa26:LacZ (Gt[ROSA]26Sortm1Sor) (73), and Rosa26:Tomato (Gt[ROSA]26Sortm14[CAG-tdTomato]Hze/J) (63). The Gata4 floxed allele was genotyped using the following primer set: forward, 5′-ACAGAAGGCTGTGAGGACA-3′; reverse, 5′-ATCTTGTGATAGAGGCCACAGGCA-3′ (WT: 450 bp; Flox: 556 bp). The Gata6 floxed allele was genotyped using the following primer set: forward, 5′-CACCCCCTCCAAGTCTCCCTGTCA-3′, reverse, 5′-GGCAACCTGTCCCCGAAAACGAA-3′ (WT: 194 bp, flox: 252 bp). The remaining alleles were genotyped according to previously published protocols (50–52, 55, 63, 73). Timed matings were staged based on the appearance of vaginal plugs, which was considered to be E0.5. To measure blood glucose, a drop of tail vein blood was drawn from mice fed ad libitum and applied to Accu-CHEK Compact Plus Blood Glucose Meter (Roche).
Whole-mount LacZ staining.
Embryos younger than E13.5 were dissected and fixed directly for 1 hour at room temperature in a solution containing 0.2% glutaraldehyde, 2% formaldehyde, 5 mM EGTA, and 2 mM MgCl2. For embryos older than E13.5, the entire pancreas and surrounding tissues, including stomach, liver, spleen, and duodenum, were removed and fixed. Specimens were washed 3 times, 30 minutes each at room temperature in a rinse solution containing 2 mM MgCl2, 0.1% sodium deoxycholate. The samples were then stained overnight in the rinse solution containing 1 mg/ml X-gal, 5 mM potassium ferricyanide, and 5 mM potassium ferrocyanide. Whole-mount pictures were photographed using a digital Nikon dissecting scope. LacZ-stained tissues were fixed with 4% paraformaldehyde overnight and embedded in OCT for sectioning. The sections were counterstained by nuclear fast red.
Histological and immunohistochemistry analysis.
Embryos or pancreas tissues were dissected and fixed in 4% paraformaldehyde overnight at 4°C. Fixed specimens were either transferred to 70% ethanol and processed for paraffin embedding (for adult tissues) or transferred into 30% sucrose and cryopreserved in OCT (for embryos). Transverse or sagittal sections were cut at 7 εm. H&E staining was performed on every tenth section. Immunohistochemistry was performed using rabbit anti-Pdx1 (1:1000; Millipore), guinea pig anti-Pdx1 (1:1000; C. Wright, Vanderbilt University, Nashville, Tennessee, USA; BCBC), rabbit anti-Ptf1a (1:1000; BCBC), rabbit anti-Neurog3 (1:500; BCBC), rabbit anti-Nkx6.1 (1:1000; BCBC), rabbit anti-Sox9 (1:1000; Millipore), rabbit anti-amylase (1:1000; Sigma-Aldrich), mouse anti-Gata4 (1:500; Santa Cruz Biotechnology Inc.), goat anti-CPA (1:800; R&D), mouse anti-Cdx2 (1:80; BioGenex), mouse anti-Isl1/2 (1:100; Hybridoma Bank), guinea pig anti-insulin (1:1000; Linco), chicken anti–β-gal (1:250; Abcam), rat anti-BrdU (1:300; Abcam), and fluorescein-conjugated DBA (1:100; Vector Laboratory). Secondary antibodies Cy2, Cy3, and Cy5 that matched the species of primary antibodies were used at 1:200 dilution (Jackson ImmunoResearch). DAPI (1:1000; Invitrogen) was used at the final step for nuclear staining. The Vectastain ABC Kit (Vector Laboratories) was used for peroxidase staining. Images were acquired on a fluorescent microscope (Leica DM5500).
Analysis of pancreatic cell proliferation.
For FACS analysis of cells in S phase, pancreatic buds from E12.5 embryos were isolated and placed in ice-cold PBS. To dissociate the tissue into single-cell suspension, the buds were transferred to 500 μl of Accumax (A7089; Sigma-Aldrich) solution supplemented with 25 μg/μl DNase I, and incubated at 37°C for 30 minutes with agitation every 5–10 minutes to facilitate dispersion. After dissociation, 500 μl of RPMI 1640 with 2% FBS and 25 μg/μl DNaseI was added to stop the enzymatic activity. The pancreatic cells were collected, washed with 1× DPBS, and resuspended in 200 μl PI staining solution containing 0.1% NP-40, 15 μg/ml PI, and 140 μg/ml RNaseA in PBS. The cells were stained in PI solution for 30 minutes at 37°C, and analyzed in a BD FACSCalibur Cell Analyzer. The distribution of the cells in G1 or S phase was analyzed by FlowJo software. Statistical analysis was performed using Prism software (control, n = 12; DKO, n = 6). To analyze the proliferation state of pancreatic progenitor cells, BrdU was injected at 100 μg/g of body weight of pregnant females and chased for 2 hours. E12.5 embryos were dissected and immediately fixed in 4% paraformaldehyde overnight. Embryos were then cryopreserved, sectioned, and stained as described above.
Glucose tolerance test and measurement of plasma insulin levels.
Glucose levels were measured at 4:00 pm in fed mice using a glucometer (Accu-chek; Roche). For the glucose tolerance test, mice were fasted for 16 hours, followed by injection of glucose solution at 2 mg/g body weight. Glucose levels were measured at time 0, 15, 30, 60, 90, 120, and 180 minutes. Insulin levels were measured in fed mice and in fasted mice at 0 and 15 minutes after glucose injection using the Mouse Insulin ELISA kit (10-1247-01; Mercodia).
ChIP analysis.
For ChIP analysis in the 266-6 cell line, cells were grown to confluency in a 15-cm plate and chromatin was prepared and immunoprecipitated using the ChIP-IT Express kit (Active Motif). Chromatin was immunoprecipitated with 2 ug of either mouse anti-Gata4 (Santa Cruz Biotechnology Inc.) or mouse anti-IgG (Millipore) according to the manufacturers’ guidelines. Enrichment of DNA fragments was calculated using qPCR with SYBR Green and the following primers: Pdx1 aivF, AGCACTTGCAAATGCTGGCTC; Pdx1 aivR, tctaactgtgaccaaacagcttc; Pdx1 aiiiFm, TCAACACCTTGCCGCTGATC; Pdx1 aiiiR, AGTCCATTGTTCAGGGTGGC; ArxF, TCCTCCACCATTTGAGGGTA; ArxR, GCAACTTGAGGGGGTACAG. For the ChIP analysis performed on E14.5 embryo tissue, chromatin was prepared and immunoprecipitated from single litters as described by Gao et al. (58). The chromatin sample was divided in half and immunoprecipitated with mouse anti-Gata4 antibody (Santa Cruz Biotechnology Inc.) or rabbit anti-Gata6 antibody (Santa Cruz Biotechnology Inc.). Endpoint PCR was used to assess binding with the Pdx1 aiii primers described above and the following primers: Cpa1F, TGGTCAAGGGTGAAAGCCTG; Cpa1R, AACACTGACGAGGGTCCCTG; Neurog3F, GGCAGAGCAGATAAAGCGTG; Neurog3R, CGCCTGGAGTAAATTGCGTC; Cpa1exF, CGGAGCTAGTAGCAACCCCT; Cpa1exR: CAGGAGCTGGTTCTGATGTG.
Statistics.
All values are expressed as mean ± SEM. Statistical analysis was performed using a 2-tailed Student’s unpaired t test. Results were considered significant at P < 0.05.
Study approval.
All mice were maintained on a Swiss Black (Taconic) background. All animal studies were approved by the Institutional Animal Care and Use Committee (Columbia University).
Supplementary Material
Acknowledgments
We would like to thank Jessica Friel, Delphine Baronnier, Irene Yu, Yabin Xiao, and the Columbia DERC Histology core for technical assistance. We thank Anabel Rojas for sharing her Gata data prior to publication. We also thank members of the Sussel lab for critical reading of the manuscript. We would like to acknowledge funding from the following sources: NIH DK087711, BCBC NIH DK089523, and Columbia DERC NIH P30 DK63608 (to L. Sussel); NIH DK55743, HL094857, and HG006398 (to S.A. Duncan); and NIH DK61220 (to R.J. Macdonald).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3516–3528. doi:10.1172/JCI63352.
See the related Commentary beginning on page 3469.
References
- 1.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240(3):530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 2.Jorgensen MC, Ahnfelt-Ronne J, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28(6):685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- 3.Bort R, Martinez-Barbera JP, Beddington RS, Zaret KS. Hex homeobox gene-dependent tissue positioning is required for organogenesis of the ventral pancreas. Development. 2004;131(4):797–806. doi: 10.1242/dev.00965. [DOI] [PubMed] [Google Scholar]
- 4.Wandzioch E, Zaret KS. Dynamic signaling network for the specification of embryonic pancreas and liver progenitors. Science. 2009;324(5935):1707–1710. doi: 10.1126/science.1174497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pictet R, Rutter WJ. Handbook of Physiology, Section 7: Endocrinology. 1972. Development of the embryonic endocrine pancreas. In: Steiner DF, Freinkel N, eds. pp. 25–66. Vol. 1. Washington, DC, USA: American Physiological Society; [Google Scholar]
- 6.Slack JM. Developmental biology of the pancreas. Development. 1995;121(6):1569–1580. doi: 10.1242/dev.121.6.1569. [DOI] [PubMed] [Google Scholar]
- 7.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 8.Krapp A, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752–3763. doi: 10.1101/gad.12.23.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Offield MF, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- 11.Mercer TR, Dinger ME, Mariani J, Kosik KS, Mehler MF, Mattick JS. Noncoding RNAs in long-term memory formation. Neuroscientist. 2008;14(5):434–445. doi: 10.1177/1073858408319187. [DOI] [PubMed] [Google Scholar]
- 12.Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106–110. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- 13.Shih DQ, Stoffel M. Molecular etiologies of MODY and other early-onset forms of diabetes. Curr Diab Rep. 2002;2(2):125–134. doi: 10.1007/s11892-002-0071-9. [DOI] [PubMed] [Google Scholar]
- 14.Seymour PA, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104(6):1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seymour PA, Freude KK, Dubois CL, Shih HP, Patel NA, Sander M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol. 2008;323(1):19–30. doi: 10.1016/j.ydbio.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13(1):103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 17.Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97(4):1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- 19.Schwitzgebel VM, et al. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127(16):3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- 20.Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem. 2010;285(41):31087–31093. doi: 10.1074/jbc.R110.159079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patient RK, McGhee JD. The GATA family (vertebrates and invertebrates). Curr Opin Genet Dev. 2002;12(4):416–422. doi: 10.1016/S0959-437X(02)00319-2. [DOI] [PubMed] [Google Scholar]
- 22.Orkin SH. GATA-binding transcription factors in hematopoietic cells. Blood. 1992;80(3):575–581. [PubMed] [Google Scholar]
- 23.Molkentin JD. The zinc finger-containing transcription factors GATA-4, -5, and -6. Ubiquitously expressed regulators of tissue-specific gene expression. J Biol Chem. 2000;275(50):38949–38952. doi: 10.1074/jbc.R000029200. [DOI] [PubMed] [Google Scholar]
- 24.Brewer A, Pizzey J. GATA factors in vertebrate heart development and disease. Expert Rev Mol Med. 2006;8(22):1–20. doi: 10.1017/S1462399406000093. [DOI] [PubMed] [Google Scholar]
- 25.Arceci RJ, King AA, Simon MC, Orkin SH, Wilson DB. Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol Cell Biol. 1993;13(4):2235–2246. doi: 10.1128/mcb.13.4.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decker K, Goldman DC, Grasch CL, Sussel L. Gata6 is an important regulator of mouse pancreas development. Dev Biol. 2006;298(2):415–429. doi: 10.1016/j.ydbio.2006.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garg V, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424(6947):443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 28.Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Dev Biol. 2004;275(1):235–244. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 29.Rajagopal SK, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43(6):677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo CT, et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11(8):1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- 31.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11(8):1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 32.Narita N, Bielinska M, Wilson DB. Wild-type endoderm abrogates the ventral developmental defects associated with GATA-4 deficiency in the mouse. Dev Biol. 1997;189(2):270–274. doi: 10.1006/dbio.1997.8684. [DOI] [PubMed] [Google Scholar]
- 33.Zhao J, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40(6):939–953. doi: 10.1016/j.molcel.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Molkentin JD, Tymitz KM, Richardson JA, Olson EN. Abnormalities of the genitourinary tract in female mice lacking GATA5. Mol Cell Biol. 2000;20(14):5256–5260. doi: 10.1128/MCB.20.14.5256-5260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keijzer R, et al. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development. 2001;128(4):503–511. doi: 10.1242/dev.128.4.503. [DOI] [PubMed] [Google Scholar]
- 36.Koutsourakis M, Keijzer R, Visser P, Post M, Tibboel D, Grosveld F. Branching and differentiation defects in pulmonary epithelium with elevated Gata6 expression. Mech Dev. 2001;105(1–2):105–114. doi: 10.1016/S0925-4773(01)00386-0. [DOI] [PubMed] [Google Scholar]
- 37.Liu C, Morrisey EE, Whitsett JA. GATA-6 is required for maturation of the lung in late gestation. Am J Physiol Lung Cell Mol Physiol. 2002;283(2):L468–L475. doi: 10.1152/ajplung.00044.2002. [DOI] [PubMed] [Google Scholar]
- 38.Yang H, Lu MM, Zhang L, Whitsett JA, Morrisey EE. GATA6 regulates differentiation of distal lung epithelium. Development. 2002;129(9):2233–2246. doi: 10.1242/dev.129.9.2233. [DOI] [PubMed] [Google Scholar]
- 39.Koutsourakis M, Langeveld A, Patient R, Beddington R, Grosveld F. The transcription factor GATA6 is essential for early extraembryonic development. Development. 1999;126(4):723–732. [PubMed] [Google Scholar]
- 40.Morrisey EE, et al. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12(22):3579–3590. doi: 10.1101/gad.12.22.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bossard P, Zaret KS. GATA transcription factors as potentiators of gut endoderm differentiation. Development. 1998;125(24):4909–4917. doi: 10.1242/dev.125.24.4909. [DOI] [PubMed] [Google Scholar]
- 42.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9(2):279–289. doi: 10.1016/S1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- 43.Zaret KS, Watts J, Xu J, Wandzioch E, Smale ST, Sekiya T. Pioneer factors, genetic competence, and inductive signaling: programming liver and pancreas progenitors from the endoderm. Cold Spring Harb Symp Quant Biol. 2008;73:119–126. doi: 10.1101/sqb.2008.73.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ketola I, et al. Transcription factor GATA-6 is expressed in the endocrine and GATA-4 in the exocrine pancreas. Mol Cell Endocrinol. 2004;226(1–2):51–57. doi: 10.1016/j.mce.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Watt AJ, Zhao R, Li J, Duncan SA. Development of the mammalian liver and ventral pancreas is dependent on GATA4. BMC Dev Biol. 2007;7:37. doi: 10.1186/1471-213X-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritz-Laser B, Mamin A, Brun T, Avril I, Schwitzgebel VM, Philippe J. The zinc finger-containing transcription factor Gata-4 is expressed in the developing endocrine pancreas and activates glucagon gene expression. Mol Endocrinol. 2005;19(3):759–770. doi: 10.1210/me.2004-0051. [DOI] [PubMed] [Google Scholar]
- 47.Swift GH, Rose SD, MacDonald RJ. An element of the elastase I enhancer is an overlapping bipartite binding site activated by a heteromeric factor. J Biol Chem. 1994;269(17):12809–12815. [PubMed] [Google Scholar]
- 48.Allen HL, et al. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44(1):20–22. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagano T, et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322(5908):1717–1720. doi: 10.1126/science.1163802. [DOI] [PubMed] [Google Scholar]
- 50.Watt AJ, Battle MA, Li J, Duncan SA. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. Proc Natl Acad Sci U S A. 2004;101(34):12573–12578. doi: 10.1073/pnas.0400752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sodhi CP, Li J, Duncan SA. Generation of mice harbouring a conditional loss-of-function allele of Gata6. BMC Dev Biol. 2006;6:19. doi: 10.1186/1471-213X-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 53.Wicksteed B, et al. Conditional gene targeting in mouse pancreatic ss-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes. 2010;59(12):3090–3098. doi: 10.2337/db10-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25(21):2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. 2005;278(2):484–495. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 56.Kaestner KH. The FoxA factors in organogenesis and differentiation. Curr Opin Genet Dev. 2010;20(5):527–532. doi: 10.1016/j.gde.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gannon M, Gamer LW, Wright CV. Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev Biol. 2001;238(1):185–201. doi: 10.1006/dbio.2001.0359. [DOI] [PubMed] [Google Scholar]
- 58.Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008;22(24):3435–3448. doi: 10.1101/gad.1752608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerrish K, Van Velkinburgh JC, Stein R. Conserved transcriptional regulatory domains of the pdx-1 gene. Mol Endocrinol. 2004;18(3):533–548. doi: 10.1210/me.2003-0371. [DOI] [PubMed] [Google Scholar]
- 60.Carrasco M, Delgado I, Soria B, Martin F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest. 2012;122(10):3504–3515. doi: 10.1172/JCI63240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang S, et al. Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol. 2008;317(2):531–540. doi: 10.1016/j.ydbio.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mastracci TL, et al. Nkx2.2 and Arx genetically interact to regulate pancreatic endocrine cell development and endocrine hormone expression. Dev Biol. 2011;359(1):1–11. doi: 10.1016/j.ydbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Madisen L, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13(1):133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pack M, et al. Mutations affecting development of zebrafish digestive organs. Development. 1996;123:321–328. doi: 10.1242/dev.123.1.321. [DOI] [PubMed] [Google Scholar]
- 65.Weber H, Symes CE, Walmsley ME, Rodaway AR, Patient RK. A role for GATA5 in Xenopus endoderm specification. Development. 2000;127(20):4345–4360. doi: 10.1242/dev.127.20.4345. [DOI] [PubMed] [Google Scholar]
- 66.Capo-Chichi CD, et al. Perception of differentiation cues by GATA factors in primitive endoderm lineage determination of mouse embryonic stem cells. Dev Biol. 2005;286(2):574–586. doi: 10.1016/j.ydbio.2005.07.037. [DOI] [PubMed] [Google Scholar]
- 67.Mauney JR, Ramachandran A, Yu RN, Daley GQ, Adam RM, Estrada CR. All-trans retinoic acid directs urothelial specification of murine embryonic stem cells via GATA4/6 signaling mechanisms. PLoS One. 2010;5(7):E11513. doi: 10.1371/journal.pone.0011513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martin M, et al. Dorsal pancreas agenesis in retinoic acid-deficient Raldh2 mutant mice. Dev Biol. 2005;284(2):399–411. doi: 10.1016/j.ydbio.2005.05.035. [DOI] [PubMed] [Google Scholar]
- 69.Molotkov A, Molotkova N, Duester G. Retinoic acid generated by Raldh2 in mesoderm is required for mouse dorsal endodermal pancreas development. Dev Dyn. 2005;232(4):950–957. doi: 10.1002/dvdy.20256. [DOI] [PubMed] [Google Scholar]
- 70.Shih DQ, Heimesaat M, Kuwajima S, Stein R, Wright CV, Stoffel M. Profound defects in pancreatic beta-cell function in mice with combined heterozygous mutations in Pdx-1, Hnf-1alpha, and Hnf-3beta. Proc Natl Acad Sci U S A. 2002;99(6):3818–3823. doi: 10.1073/pnas.062605899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Froguel P, Velho G. Molecular genetics of maturity-onset diabetes of the young. Trends Endocrinol Metab. 1999;10(4):142–146. doi: 10.1016/s1043-2760(98)00134-9. [DOI] [PubMed] [Google Scholar]
- 72.Shih DQ, Stoffel M. Dissecting the transcriptional network of pancreatic islets during development and differentiation. Proc Natl Acad Sci U S A. 2001;98(25):14189–14191. doi: 10.1073/pnas.251558998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.