

Abstract
The osteoporosis associated with human hyperthyroidism has traditionally been attributed to elevated thyroid hormone levels. There is evidence, however, that thyroid-stimulating hormone (TSH), which is low in most hyperthyroid states, directly affects the skeleton. Importantly, Tshr-knockout mice are osteopenic. In order to determine whether low TSH levels contribute to bone loss in hyperthyroidism, we compared the skeletal phenotypes of wild-type and Tshr-knockout mice that were rendered hyperthyroid. We found that hyperthyroid mice lacking TSHR had greater bone loss and resorption than hyperthyroid wild-type mice, thereby demonstrating that the absence of TSH signaling contributes to bone loss. Further, we identified a TSH-like factor that may confer osteoprotection. These studies suggest that therapeutic suppression of TSH to very low levels may contribute to bone loss in people.
Introduction
Hyperthyroidism, a common health risk affecting approximately 1 in 100 individuals, is often accompanied by worsening osteoporosis, especially in postmenopausal women (1). There is compelling evidence from early in vitro and more recent mouse genetic studies that both thyroxine (T4) and triiodothyronine (T3) stimulate the resorption of bone by osteoclasts (2). This leaves no doubt that thyroid hormone excess is a major contributor to the profound bone loss and high risk of fracture in hyperthyroidism. Physiology tells us that high serum thyroid hormone suppresses the production of thyroid-stimulating hormone (TSH) from the anterior pituitary, although in subclinical hyperthyroidism, thyroid hormone levels can be normal, while TSH is low or undetectable. The osteoporosis associated with subclinical hyperthyroidism is therefore unlikely to arise from thyroid hormone excess alone (3).
We and others have shown that, in addition to its known function in stimulating thyroid follicular cells, TSH can act directly on the skeleton (4–8). Activation of the TSH receptor (TSHR) on the osteoclast prevents the resorption of bone (4). When administered intermittently, TSH stimulates osteoblastic bone formation and, in rodent models, rescues ovariectomy-induced bone loss (5–8). In contrast, absence of the TSHR in the global Tshr–/– mouse causes high-turnover osteoporosis (4). That this osteoporosis is not reversed upon thyroid hormone replacement suggests that absent TSH signaling may have a permissive role in causing this bone loss. Epidemiologic evidence favors strong correlations between low TSH and high bone turnover; low TSH and low bone mineral density (BMD); and low TSH and high fracture risk in hyperthyroid patients (9–23). This raises the question of whether low TSH levels might contribute to the bone loss in hyperthyroidism that has been attributed solely to high thyroid hormone levels.
We therefore compared the skeletal phenotypes of wild-type and Tshr–/– mice that were rendered hyperthyroid by the implantation of slow-release 5-mg T4 pellets. We show that hyperthyroid Tshr–/– mice completely devoid of TSH signaling display higher levels of bone resorption and bone loss compared with hyperthyroid wild-type mice, even though TSH levels in the latter mice become undetectable. This finding not only establishes a role for TSH signaling in thyrotoxic osteoporosis but, equally importantly, suggests that a local TSH-like factor, which we identify as a novel Tshβ splice variant, might offer osteoprotection, even in the context of undetectable serum TSH.
Results and Discussion
To establish a role for attenuated TSHR signaling in hyperthyroid bone loss, we compared hyperthyroid wild-type and Tshr–/– mice for differences in bone mass and bone remodeling. The expectation was that hyperthyroid Tshr–/– mice would show greater bone loss than hyperthyroid wild-type mice, directly implicating low TSH signaling in the pathogenesis of hyperthyroid bone loss. For this, we rendered wild-type and Tshr–/– mice hypothyroid by adding 0.5% 2-mercapto-1-methyl-imidazole (methimazole) to drinking water for 14 days. Both T4 and T3 expectedly declined to low levels, whereas serum TSH rose sharply in both groups (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI63948DS1). Thereafter, both groups were implanted with either 0-mg or 5-mg sustained-release thyroid hormone (T4) pellets for 21 days, during which time the mice were fed normal chow. The 0-mg pellet produced no change in serum T4 levels, while, as expected from the induction of a hypothyroid state, TSH levels rose in wild-type and Tshr–/– mice. In contrast, serum T4 levels rose and serum TSH declined in mice receiving 5-mg pellets, essentially modeling human hyperthyroidism.
At 21 days, mice in all groups were sacrificed for histomorphometry and bone marker measurements. Figure 1A shows that compared with mice receiving the 0-mg pellet, those receiving a 5-mg pellet displayed a marked reduction in areal bone mineral density (aBMD). This decline was significantly greater in hyperthyroid Tshr–/– mice compared with hyperthyroid wild-type mice. That this difference was seen in the context of similar T4 levels (Supplemental Table 1) established that absent TSH signaling was the cause of the greater bone loss in Tshr–/– mice rendered hyperthyroid. Micro-CT–based volumetric measures, including volumetric BMD (vBMD), bone volume/trabecular volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and connectivity density (Conn.D), all declined significantly in mice receiving the 5-mg pellet versus those implanted with the 0-mg pellet (Figure 1, B and C). Importantly, decrements in vBMD, BV/TV, Tb.Th, and Conn.D were statistically significantly (P < 0.05) greater in Tshr–/– hyperthyroid mice than in wild-type hyperthyroid mice, while differences in Tb.N and trabecular spacing (Tb.Sp) approached significance (0.1 > P > 0.05). This suggests that the major effect of this relatively short treatment is on Tb.Th rather than on Tb.N. No differences were noted in wild-type versus Tshr–/– mice receiving the 0-mg T4 pellet (P > 0.05). Together the data reaffirm that absence of TSHR in experimentally induced hyperthyroidism can exaggerate the attendant bone loss.
Figure 1. Hyperthyroid TSHR-deficient mice have greater bone loss than wild-type hyperthyroid mice with intact TSH signaling.

aBMD (A), micro-CT images (B), and individual parameters from vertebral bodies (C) of wild-type (Tshr+/+) and Tshr–/– mice implanted with 0-mg (placebo) or 5-mg T4 pellets (TH) for 21 days. Mean percent changes (±SEM) in vBMD, BV/TV, Tb.N, Tb.Th, Tb.Sp, and Conn.D are shown. **P < 0.01; n = 6–8 mice/group.
As human hyperthyroidism is associated with high bone turnover (24), dynamic histomorphometry was performed by injecting mice with calcein (15 mg/kg, i.p.) 14 and 4 days prior to sacrifice. Figure 2, A and B, shows increased bone resorption and formation, as evidenced by tartrate-resistant acid phosphatase (TRAP) and calcein labeling, respectively, in Tshr–/– mice rendered hyperthyroid. Individual parameters, namely osteoclast surface (Oc.S/BPm), mineralizing surface (MS), bone formation rate (BFR), and double label fraction (dLS/BPm) increased significantly (P < 0.05) in Tshr–/– mice implanted with 5-mg pellets compared with those receiving 0 mg T4 (Table 1). The increases in Oc.S/BPm and BFR were highly significantly (P = 0.004) and marginally (P = 0.081) greater, respectively, in Tshr–/– hyperthyroid mice compared with wild-type hyperthyroid mice (Table 1), confirming that TSHR deficiency causes the increased bone resorption. The apparent discordance between bone formation and bone resorption was expected, as formation lags behind resorption, which is the primary stimulus causing hyperthyroid bone loss.
Figure 2. TSHR deficiency accelerates bone remodeling in hyperthyroidism.

Representative images of TRAP-labeled (A) or calcein-labeled bones (B), as well as serum C-telopeptide (CTX) (C) and serum osteocalcin (D) levels in wild-type and Tshr–/– mice implanted with 0-mg (placebo) or 5-mg sustained-release T4 pellets for 21 days. Scale bars in A and B: 100 μm; B inset, 10 μm. **P < 0.01, †P < 0.01; n = 4–9 mice/group.
Table 1.
Histomorphometry parameters
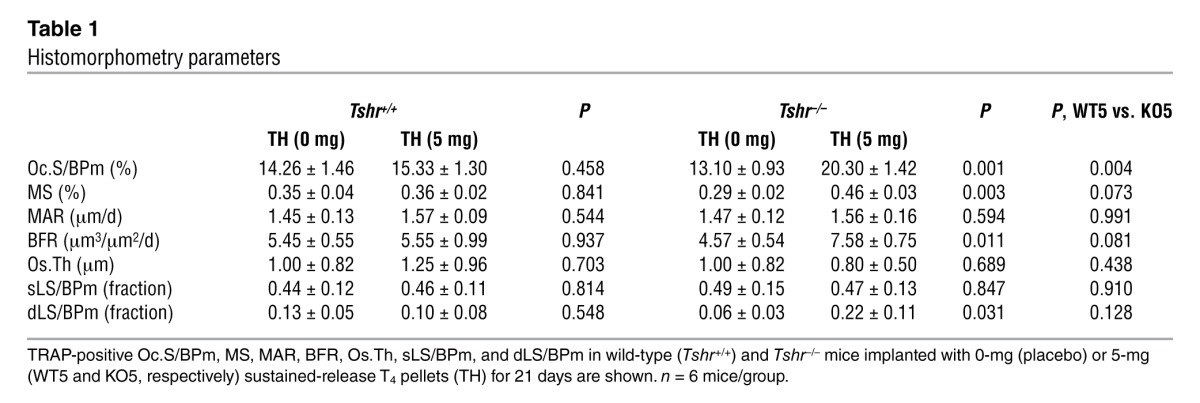
We further studied remodeling dynamically by measuring the serum markers C-telopeptide and osteocalcin. Consistent with the increased Oc.S/BPm (Table 1), serum C-telopeptide levels were markedly elevated in hyperthyroid Tshr–/– mice compared with wild-type hyperthyroid mice (Figure 2C). Concordant with the changes in BFR (Table 1), serum osteocalcin levels were also significantly higher in hyperthyroid Tshr–/– mice (Figure 2D). Interestingly, we did not note increments in remodeling parameters in wild-type mice implanted with 5-mg T4 pellets compared with those receiving 0-mg T4 pellets. This was not unexpected, as after ovariectomy in mice, for example, elevated remodeling occurs very rapidly, causing profound bone loss within 2–3 weeks, which is followed by a low remodeling state with slower bone loss. This means that we likely missed the hyper-resorption phase in our 21-day experiments. Overall, however, we show that net bone loss is more pronounced in mice deficient in Tshr, suggesting an osteoprotective function for TSH signaling, which is lost in hyperthyroidism.
This observation is relevant both clinically and biologically. There is growing evidence for an association between low TSH, low BMD, and increased bone turnover in hyperthyroidism. The risk of vertebral and non-vertebral fractures increases 4.5- and 3.2-fold, respectively, with serum TSH levels of 0.1 IU/l or less (9). Likewise, euthyroid women with serum TSH levels in the lowest tertile of the normal range have a higher incidence of vertebral fractures, independent of thyroid hormone levels (19). Analysis of the National Health and Nutrition Examination Survey (NHANES) data show that the odds ratio for correlations between TSH and bone mass range between 2 and 3.4 (18). Moreover, in the Tromso study, participants with serum TSH levels below 2 SD had significantly lower BMDs (15). In patients taking suppressive doses of T4 for thyroid cancer, serum cathepsin K levels were grossly elevated (16). Importantly, and of clinical relevance, greater bone loss occurs in T4-treated patients with suppressed TSH levels than in those without suppression (12, 13). Our mouse study attempts to reinforce the idea that these clinical observations may not simply be correlative; instead, low TSH may facilitate the excessive bone loss in human hyperthyroidism. There is therefore a need for caution when T4 supplementation is initiated so as not to unnecessarily lower serum TSH to undetectable levels.
The reverse, which is that a constantly elevated TSH level might be osteoprotective, may or may not be true. We did not find, at least in these short-term studies, significant BMD or BV/TV differences between wild-type and Tshr–/– mice rendered hypothyroid with 0-mg T4 pellets. Both groups of mice had elevated TSH levels, but with abrogated TSHR signaling in Tshr–/– mice. Nonetheless, in humans, the HUNT 2 study found a positive correlation between TSH and BMD at the distal forearm (17). It also appears from our data using osteoclasts from embryonic stem cells that the agonist TSHR antibody may offer antiresorptive osteoprotection against the bone loss in Graves disease, but this needs further validation in humans (25). Importantly, however, patients harboring the activating TSHR polymorphism D727E have high bone mass (20–22). However, hypothyroid patients can be at a high fracture risk, suggesting that persistently elevated serum TSH levels may, in fact, suppress bone resorption to the ultimate detriment of optimal skeletal remodeling and bone strength (26). Nonetheless, we and others have shown that TSH administered intermittently acts upon the osteoblast to promote bone formation in rodents and humans (5–7).
What is fascinating biologically, however, is that while the two hyperthyroid genotypes have undetectable serum TSH levels and similar T4 levels, Tshr–/– mice lose more bone than wild-type mice. This establishes the TSHR as the only determinant that could account for the bone loss differences between the two genotypes when rendered hyperthyroid. Underscoring this difference, we report the identification of a Tshβ splice variant, produced in bone marrow (27), which is regulated positively, rather than negatively, by T4 (Supplemental Figure 1). This molecule, we believe, is capable of exerting a bone-protective effect, which would be lost when the TSHR is deleted. The production of a pituitary hormone–like hormone in bone marrow is not surprising. We and others have shown that anterior pituitary hormones, such as adrenocorticotropic hormone (ACTH) (28), and posterior pituitary hormones, such as oxytocin (29), are produced by bone cells. While their physiologic importance remains unresolved, the possibility of simpler, shorter feedback or feed-forward loops within bone, where GPCRs for pituitary hormones have been identified (30, 31), might be biologically meaningful in the intricate control of skeletal integrity.
Methods
Ten week-old Tshr–/– mice, originally generated on a mixed C57BL/6 × 129Sv background, were backcrossed for more than 10 generations. The mice were maintained on a 12-hour light/12-hour dark photoperiod. We supplemented Tshr–/– mice with thyroid extract (100 ppm) at weaning (4), but supplementation was suspended at the start of the experiments. Mice were first rendered hypothyroid by addition of 0.5% 2-mercapto-1-methyl-imidazole (methimazole) to drinking water for 14 days. Thereafter, both groups were implanted with either 0-mg or 5-mg sustained-release thyroid hormone (T4) pellets (Innovative Research of America) for 21 days and were fed normal chow. At 21 days, mice in all groups were sacrificed for histomorphometry and bone marker measurements. Serum T4 and TSH measurements were made using Multiplex assays (Millipore). aBMD was measured using a small animal densitometer (PIXImus, GE-Lunar). Micro-CT (Desktop CT 40, SCANCO) allowed quantitation of vBMD, BV/TV, Tb.Th, Tb.N, Tb.Sp, and Conn.D (32). Histomorphometry using calcein (Sigma-Aldrich) and Bioquant 11 (RM Biometrics) provided estimates of MS, mineral apposition rate (MAR), BFR, osteoid thickness (Os.Th), single label fraction (sLS/BPm), and dLS/BPm, while TRAP staining provided Oc.S/BPm (32). Osteocalcin ELISA (Biomedical Technologies) and RatLaps ELISA (Nordic Bioscience Diagnostics) were used.
Statistics.
Data are expressed as mean ± SEM. Statistical comparisons were carried out using 1-way ANOVA with Bonferroni’s correction and 2-tailed Student’s t test. P values less than 0.05 were considered significant.
Study approval.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of Mount Sinai School of Medicine.
Supplementary Material
Acknowledgments
This work was supported in part by DK080459 (to M. Zaidi, L. Sun, and T.F. Davies), DK069713 and DK052464 (to T.F. Davies), and AG23186 and DK70526 (to M. Zaidi) from the NIH and by the VA Merit Review Program (to T.F. Davies and H.C. Blair). J. Cao is supported by the USDA ARS CRIS Program (5450-51000-046-00D).
Footnotes
Conflict of interest: Mone Zaidi is a named inventor of a pending patent application filed by Mount Sinai School of Medicine. In the event the pending patent is licensed, he would be entitled to a share of any proceeds. Terry F. Davies is a member of the Board of Kronus Inc. (Star, Idaho, USA).
Citation for this article: J Clin Invest. 2012;122(10):3737–3741. doi:10.1172/JCI63948.
References
- 1.Sawin CT, Geller A, Hershman JM, Castelli W, Bacharach P. The aging thyroid. The use of thyroid hormone in older persons. JAMA. 1989;261(18):2653–2655. doi: 10.1001/jama.1989.03420180077034. [DOI] [PubMed] [Google Scholar]
- 2.Bassett JH, Williams GR. The skeletal phenotypes of TRalpha and TRbeta mutant mice. J Mol Endocrinol. 2009;42(4):269–282. doi: 10.1677/JME-08-0142. [DOI] [PubMed] [Google Scholar]
- 3.Földes J, Tarján G, Szathmari M, Varga F, Krasznai I, Horvath C. Bone mineral density in patients with endogenous subclinical hyperthyroidism: is thyroid status a risk factor for osteoporosis? Clin Endocrinol (Oxf). 1993;39(5):521–527. doi: 10.1111/j.1365-2265.1993.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 4.Abe E, et al. TSH is a negative regulator of skeletal remodeling. Cell. 2003;115(2):151–162. doi: 10.1016/S0092-8674(03)00771-2. [DOI] [PubMed] [Google Scholar]
- 5.Sampath TK, et al. Thyroid-stimulating hormone restores bone volume, microarchitecture, and strength in aged ovariectomized rats. J Bone Miner Res. 2007;22(6):849–859. doi: 10.1359/jbmr.070302. [DOI] [PubMed] [Google Scholar]
- 6.Sun L, et al. Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci U S A. 2008;105(11):4289–4294. doi: 10.1073/pnas.0712395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazziotti G, et al. Recombinant human TSH modulates in vivo C-telopeptides of type-1 collagen and bone alkaline phosphatase, but not osteoprotegerin production in postmenopausal women monitored for differentiated thyroid carcinoma. J Bone Miner Res. 2005;20(3):480–486. doi: 10.1359/JBMR.041126. [DOI] [PubMed] [Google Scholar]
- 8.Baliram R, et al. Thyroid-stimulating hormone induces a Wnt-dependent, feed-forward loop for osteoblastogenesis in embryonic stem cell cultures. Proc Natl Acad Sci U S A. 2011;108(39):16277–16282. doi: 10.1073/pnas.1110286108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martini G, et al. The effects of recombinant TSH on bone turnover markers and serum osteoprotegerin and RANKL levels. Thyroid. 2008;18(4):455–460. doi: 10.1089/thy.2007.0166. [DOI] [PubMed] [Google Scholar]
- 10.Bauer DC, Ettinger B, Nevitt MC, Stone KL. Risk for fracture in women with low serum levels of thyroid-stimulating hormone. Ann Intern Med. 2001;134(7):561–568. doi: 10.7326/0003-4819-134-7-200104030-00009. [DOI] [PubMed] [Google Scholar]
- 11.Zofkova I, Hill M. Biochemical markers of bone remodeling correlate negatively with circulating TSH in postmenopausal women. Endocr Regul. 2008;42(4):121–127. [PubMed] [Google Scholar]
- 12.La Vignera S, et al. L-thyroxin treatment and post-menopausal osteoporosis: relevance of the risk profile present in clinical history. Minerva Ginecol. 2008;60(6):475–484. [PubMed] [Google Scholar]
- 13.Flynn RW, et al. Serum thyroid-stimulating hormone concentration and morbidity from cardiovascular disease and fractures in patients on long-term thyroxine therapy. J Clin Endocrinol Metab. 2010;95(1):186–193. doi: 10.1210/jc.2009-1625. [DOI] [PubMed] [Google Scholar]
- 14.Baqi L, et al. The level of TSH appeared favourable in maintaining bone mineral density in postmenopausal women. Endocr Regul. 2010;44(1):9–15. doi: 10.4149/endo_2010_01_9. [DOI] [PubMed] [Google Scholar]
- 15.Grimnes G, Emaus N, Joakimsen RM, Figenschau Y, Jorde R. The relationship between serum TSH and bone mineral density in men and postmenopausal women: the Tromso study. Thyroid. 2008;18(11):1147–1155. doi: 10.1089/thy.2008.0158. [DOI] [PubMed] [Google Scholar]
- 16.Mikosch P, et al. High cathepsin K levels in men with differentiated thyroid cancer on suppressive L-thyroxine therapy. Thyroid. 2008;18(1):27–33. doi: 10.1089/thy.2007.0186. [DOI] [PubMed] [Google Scholar]
- 17.Svare A, et al. Hyperthyroid levels of TSH correlate with low bone mineral density: the HUNT 2 study. Eur J Endocrinol. 2009;161(5):779–786. doi: 10.1530/EJE-09-0139. [DOI] [PubMed] [Google Scholar]
- 18.Morris MS. The association between serum thyroid-stimulating hormone in its reference range and bone status in postmenopausal American women. Bone. 2007;40(4):1128–1134. doi: 10.1016/j.bone.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 19.Mazziotti G, Porcelli T, Patelli I, Vescovi PP, Giustina A. Serum TSH values and risk of vertebral fractures in euthyroid post-menopausal women with low bone mineral density. Bone. 2010;46(3):747–751. doi: 10.1016/j.bone.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 20.Heemstra KA, et al. Thyroid hormone independent associations between serum TSH levels and indicators of bone turnover in cured patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2008;159(1):69–76. doi: 10.1530/EJE-08-0038. [DOI] [PubMed] [Google Scholar]
- 21.van der Deure WM, et al. Effects of serum TSH and FT4 levels and the TSHR-Asp727Glu polymorphism on bone: the Rotterdam Study. Clin Endocrinol (Oxf). 2008;68(2):175–181. doi: 10.1111/j.1365-2265.2007.03016.x. [DOI] [PubMed] [Google Scholar]
- 22.Albagha OME, Natrajan R, Reid DM, Ralston SH. The D727 polymorphism of the human thyroid stimulating hormone receptor is associated with bone mineral density and bone loss in women from the UK. J Bone Min Res. 2005;20(supp 1):S341. doi: 10.1359/JBMR.041107. [DOI] [Google Scholar]
- 23. Onigata K, Kowasi T, Nishiyama S, Micuno H, Morikawa A. Bone mineral density in human cases with TSH receptor gene mutations. In: Zaidi M, ed. Proceedings of the New York Academy of Sciences 1st Conference on Skeletal Development and Remodeling in Health, Disease, and Aging. Vol. 1. New York, New York, USA: New York Academy of Sciences; 2005: 11
- 24.El Hadidy el HM, Ghonaim M, El Gawad SSh, El Atta MA. Impact of severity, duration, and etiology of hyperthyroidism on bone turnover markers and bone mineral density in men. BMC Endocr Disord. 2011;11:15. doi: 10.1186/1472-6823-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma R, Morshed S, Latif R, Zaidi M, Davies TF. The influence of thyroid stimulating hormone and thyroid stimulating hormone receptor antibodies on osteoclastogenesis. Thyroid. 2011;21(8):897–906. doi: 10.1089/thy.2010.0457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vestergaard P, Mosekilde L. Fractures in patients with hyperthyroidism and hypothyroidism: a nationwide follow-up study in 16,249 patients. Thyroid. 2002;12(5):411–419. doi: 10.1089/105072502760043503. [DOI] [PubMed] [Google Scholar]
- 27.Vincent BH, et al. Bone marrow cells produce a novel TSHbeta splice variant that is upregulated in the thyroid following systemic virus infection. Genes Immun. 2009;10(1):18–26. doi: 10.1038/gene.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isales CM, Zaidi M, Blair HC. ACTH is a novel regulator of bone mass. Ann N Y Acad Sci. 2010;1192:110–116. doi: 10.1111/j.1749-6632.2009.05231.x. [DOI] [PubMed] [Google Scholar]
- 29.Colaianni G, et al. Regulated production of the pituitary hormone oxytocin from murine and human osteoblasts. Biochem Biophys Res Commun. 2011;411(3):512–515. doi: 10.1016/j.bbrc.2011.06.158. [DOI] [PubMed] [Google Scholar]
- 30.Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13(7):791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
- 31.Blair HC, et al. Skeletal receptors for steroid-family regulating glycoprotein hormones: a multilevel, integrated physiological control system. Ann N Y Acad Sci. 2011;1240:26–31. doi: 10.1111/j.1749-6632.2011.06287.x. [DOI] [PubMed] [Google Scholar]
- 32.Sun L, et al. FSH directly regulates bone mass. Cell. 2006;125(2):247–260. doi: 10.1016/j.cell.2006.01.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.