

Abstract
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults. Here, we highlight important genetic alterations that contribute to tumorigenesis, clinical progression, and chemorefractoriness of CLL. All CLLs share a common gene expression profile that suggests derivation from antigen-experienced B cells, a model supported by frequent B cell receptor repertoire skewing and stereotypy. Many CLL patients carry mutated immunoglobulin heavy-chain variable genes, while approximately 35% harbor unmutated IgV genes, which are associated with an inferior outcome. Deletion of chromosome 13q14, which is the most common genetic mutation at diagnosis, is considered an initiating lesion that frequently results in disruption of the tumor suppressor locus DLEU2/MIR15A/MIR16A. Next-generation sequencing has revealed additional recurrent genetic lesions that are implicated in CLL pathogenesis. These advancements in the molecular genetics of CLL have important implications for stratifying treatment based on molecular prognosticators and for targeted therapy.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in adults (1, 2). Historically, CLL was viewed as a tumor caused by the accumulation of long-lived but mainly resting lymphocytes with a very low proliferation index (3). However, this model has been challenged in recent years. Heavy-water experiments have shown that CLL contains a small fraction of actively proliferating cells, with approximately 2% of cells newly generated each day (4). Though lymphocytes in the peripheral blood (PB) are predominantly resting (5), specific structures known as proliferation centers, which are localized in the lymph nodes and in the bone marrow, replenish the CLL cell population (1, 6, 7). The small proportion of CLL cells in the PB with a proliferative phenotype likely represent cells that have recirculated through the lymph node microenvironment and have exited the proliferation centers before becoming quiescent again (1, 6).
The natural history of many CLLs involves progression toward a more malignant disease. Most, if not all, cases of CLL are preceded by monoclonal B cell lymphocytosis (MBL), a very indolent cell expansion defined by less than 5,000 monoclonal B cells in the PB (8–12). MBL is detectable in approximately 5% of the elderly population and carries a risk of evolving into CLL that approximates 1% per year (11, 13, 14). Clinically, CLL is characterized by a marked degree of heterogeneity, ranging from patients that harbor highly stable disease with a nearly normal life expectancy to patients with rapidly progressive disease who are destined to succumb in a short time (15, 16). The variable course of CLL is driven, at least in part, by heterogeneity in the disease biology. Over time, a small fraction of CLL cases transform into a very aggressive form known as Richter syndrome (RS), which morphologically mimics diffuse large B cell lymphoma (DLBCL) (1, 17, 18).
This review focuses on recent advances in the analysis of the genome of CLL. Genome-wide analysis have allowed the characterization of the spectrum of genetic lesions present in the CLL coding genome, thus providing further insights into CLL pathogenesis and disease progression.
Phenotype and putative cell of origin
CLL expresses a distinct immunophenotype, characterized by co-expression of CD19, CD5, and CD23 coupled with low levels of surface immunoglobulins (1). This phenotypic profile is different from any normal B cell subsets, concealing any indication of a normal counterpart. Although the precise cell of origin of CLL is still under investigation (19), immunogenetic studies and gene expression profiling (GEP) analyses have provided important information regarding the putative CLL progenitor (Figure 1).
Figure 1. A model for the cellular origin of CLL.

Encounter of naive B cells with antigen may proceed either through a T cell–dependent reaction occurring in the GC and leading to the generation of memory B cells that have undergone somatic hypermutation of IGHV genes, or in T cell–independent immune responses that may lead to the formation of antigen-experienced B cells harboring unmutated IGHV genes. CLL, and the preceding MBL phase, may originate from both of these subsets of antigen-experienced B cells. CLL originating from B cells that have experienced somatic hypermutation carry mutated IGHV genes and are defined as M-CLL. Conversely, CLL originating from B cells that have been involved in T cell–independent immune reactions harbor germline IGHV genes and are defined as U-CLL. The emergence and growth of a CLL (or MBL) clone is due to the accumulation of genetic lesions in the neoplastic population as well as interactions of the leukemic cells with antigen through the BCR and with microenvironmental components that promote cell proliferation and inhibit apoptosis.
Extensive molecular investigations of the B cell receptor (BCR) indicate that 60%–65% of CLLs carry immunoglobulin heavy-chain variable (IGHV) genes with evidence of somatic hypermutation in their variable regions, a process that occurs in the germinal center (GC) and may modify BCR affinity for antigens (refs. 20–23 and Figure 1). Conversely, 35%–40% of CLLs are devoid of IGHV somatic mutations (20–22). The association with IGHV gene mutations suggests that a fraction of cases (CLL with mutated IGHV genes [M-CLL]) derive from GC-experienced B cells, while the remaining cases (CLL with unmutated IGHV genes [U-CLL]) derive from B cells that have undergone differentiation in a GC-independent fashion (19, 24, 25).
Both M-CLL and U-CLL share a largely similar GEP, with few genes, such as ZAP70, differentiating the two categories (26, 27). This observation suggests that M-CLL and U-CLL derive from progenitors that are reminiscent of antigen-experienced B cells (26). In humans, antigen-experienced B cells include memory B cells and marginal zone B cells, whose IGHV genes can be somatically mutated or unmutated (28–30). Both these B cell subsets have been proposed to be the cell of origin of CLL (19). The definition of antigen-experienced B cells includes both GC-experienced cells as well as memory-like B cells generated in a T cell–independent reaction not requiring the GC microenvironment and not necessarily involving IGHV somatic hypermutation (31). This notion accounts for the fact that lymphocytes from U-CLLs resemble antigen-experienced B cells even though their IGHV genes have not undergone somatic hypermutation in the GC (26, 27).
The role of antigen stimulation is corroborated by the skewed BCR repertoire expressed by many CLL patients and by the association of approximately 30% of cases with a stereotyped BCR. Certain IGHV genes are utilized at a significantly higher frequency than expected based on the representation of these same IGHV genes among normal B cells (20, 32, 33). In addition to preferential usage of specific IGHV genes, the BCRs of approximately 30% of CLLs are clustered into stereotyped subsets, each of which is characterized by a high degree of homology of the CDR3 region (34–38). By chance alone, the possibility is extremely low (<1 × 10–12) that the same BCR rearrangement is shared by different CLL patients, suggesting that stereotyped BCRs have been positively selected for their interactions with specific antigens (34–38). Importantly, stereotyped BCRs have also been detected in B cells of normal individuals, though with features distinct from those associated with CLL (39). Although the antigens involved in CLL have not been fully elucidated, a potential role of autoantigens such as non-muscle myosin has been proposed (40, 41).
The distinction between M-CLL and U-CLL is clinically relevant because germline IGHV genes predict poor outcome (21, 22). The worse prognosis predicted by unmutated IGHV genes is conceivably due to the enrichment of some genetic lesions that confer higher aggressiveness (e.g., mutations of NOTCH1) among U-CLLs, as well as to the greater predisposition of U-CLLs, compared with M-CLLs, to undergo clonal evolution (42–47).
In contrast with the model suggesting a B cell origin of the entire CLL pathogenesis, recent data suggest that the initiating event may occur in a hematopoietic stem cell (48). Hematopoietic stem cells obtained from CLL patients and transplanted into xenograft models have a propensity to generate clonal B cells with a CLL-like phenotype and IGHV usage similar to that of CLL. Nonetheless, the lymphoid expansions that occur in mice are clonally unrelated to those from the original patient, indicating de novo generation, presumably initiated by genetic lesions that are present in the donor stem cells. Although the nature of the initiating event(s) in the hematopoietic precursors are unknown, and the above Results require independent confirmation, these findings challenge current theories on the nature of the cell of origin of CLL and its pathogenesis.
The genome of CLL
Karyotypic investigations revealed the association of CLL with del13q14, trisomy 12, del11q22-q23, and del17p13 (refs. 1, 2, 49, 50, and Figure 2). The advent of next-generation sequencing (NGS) technologies, coupled with gene copy number analysis, has enabled exploration of the CLL genome (45, 46, 51, 52), uncovering genetic lesions that recurrently target this leukemia (Figure 2).
Figure 2. Genetic lesions of CLL at different phases of the disease.
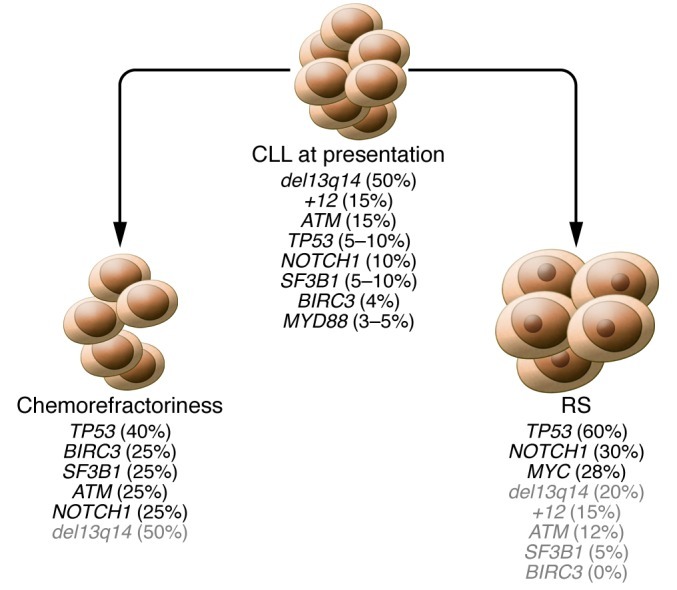
The frequency (in parentheses) of genetic lesions is shown for CLL at presentation and for two different types of CLL progression: chemorefractoriness without evidence of histologic transformation, and histologic transformation to RS. The two types of CLL progression follow distinct molecular pathways in terms of type and frequency of genetic lesions. Genetic lesions that occur at CLL presentation but are not enriched at CLL progression are indicated in gray.
The CLL genome is largely devoid of the chromosomal translocations and aberrant somatic hypermutations that are involved in several B cell non-Hodgkin lymphomas (B-NHLs) (1, 2, 49, 50, 53). These observation are consistent with a post-GC or GC-independent derivation of CLL because B-NHL–associated translocations are caused by errors during somatic hypermutation and class switch recombination, two mechanisms that are active in GC B cells (23).
NGS studies have further elucidated the genomic complexity of CLLs and have shown that the average number of non-silent mutations (i.e., mutations that alter the protein sequence) per case is 10–12 at diagnosis, whereas the average number of copy number abnormalities is approximately two (45, 46, 51, 52). The order of magnitude of lesions detected in the coding genome of CLL appears considerably lower than in common epithelial cancers (54). Among hematologic malignancies, the complexity of the CLL genome is also on the lower side, markedly smaller than DLBCL and multiple myeloma, and comparable to some acute leukemias (55–58).
Del13q14 is the most frequent alteration and occurs in 50%–60% of cases (2, 49, 50). Because this lesion is found at a similar frequency in MBL and is often detectable as a single lesion (11, 13), this alteration may represent an early event in the disease. The minimal deleted region (MDR) of del13q14 contains the deleted in leukemia-1 (DLEU1) and DLEU2 genes, which code for noncoding transcripts, as well as the MIR15A/MIR16A microRNAs, which map within intron 4 of DLEU2 (59, 60). Expression of DLEU2 and MIR15A/MIR16A is also downregulated in CLL in the absence of the del13q14 deletion, possibly as a consequence of epigenetic mechanisms (61, 62). The DLEU2 locus generates a long non-coding RNA that has only one known function, to serve as the primary RNA from which the mature miR-15a/miR-16a microRNAs are processed (59–61, 63). Based on animal models, MIR15A/MIR16A are considered the main candidate tumor suppressor genes involved with del13q14, although a role is likely for additional genetic elements including DLEU2 (refs. 64, 65, and see below). In normal cells, these two microRNAs appear to inhibit expression of multiple genes, including BCL2, the cyclins CCND1 and CCND3, and cyclin-dependent kinase 6 (CDK6) (66–71). Consequently, deletion of MIR15A/MIR16A abrogates this inhibitory effect and favors the constitutive cycling of B cells. In addition, miR-15a/miR-16a may also participate in a microRNA/tumor protein p53 (microRNA/TP53) feedback circuitry associated with CLL pathogenesis and prognosis (72).
Trisomy 12 occurs in approximately 15% of CLL cases and is thought to alter the gene dosage of one or more proto-oncogenes, although the precise molecular mechanism of this alteration is not currently understood (2, 49). Del11q22-q23 in most cases affects the ataxia telangiectasia mutated (ATM) gene, the deficiency of which causes genomic instability and, if inherited in the germline, a predisposition for lymphoid malignancies (73). In CLL, ATM is also affected by mutations that disrupt its function (74–77). In a fraction of cases carrying del11q22-q23, the ATM gene appears to be intact and the deletion targets the BIRC3 gene, which is in close proximity to ATM (ref. 78 and see below).
In approximately 5%–10% of untreated CLL patients, del17p13 disrupts the TP53 tumor suppressor gene (49, 79–87). Many cases of CLL with del17p13 display inactivation of the second TP53 allele by point mutation (80–87). Altered TP53 function due to 17p deletion and/or TP53 gene mutation is an important predictor of chemorefractoriness (see below) and is associated with reduced survival (81–87). From a diagnostic standpoint, it should be noted that a fraction of CLLs display TP53 mutations in the absence of del17p13. Such cases are not recognized by FISH analysis, and their identification requires mutation analysis of the TP53 gene (88).
Recently, whole exome sequencing studies have revealed recurrent genetic lesions that affect genes implicated in different biological pathways of potential pathogenetic relevance for CLL (45, 46, 51, 52, 89). These genes include NOTCH1, splicing factor 3b, subunit 1 (SF3B1), BIRC3, and myeloid differentiation primary response gene 88 (MYD88).
Mutational activation of the Notch pathway.
NOTCH1 encodes a ligand-activated transcription factor that regulates several downstream pathways important for cell growth control and is affected by activating mutations in 60% of T-lineage acute lymphoblastic leukemias (90). In CLL, the frequency of NOTCH1 mutations at the time of diagnosis is approximately 10% (45–47, 91). Conversely, mutations of NOTCH1 are very rare in MBL (92). NOTCH1 mutations at CLL diagnosis preferentially occur among U-CLLs and cluster with trisomy 12 (45–47, 93, 94). NOTCH1 mutations identify a high-risk subgroup of patients showing poor survival comparable to that associated with TP53 abnormalities and exert a prognostic role that is independent of widely accepted risk factors, as confirmed in multiple consecutive series from different institutions (45–47).
NOTCH1 mutations in CLL are highly consistent among cases and disrupt the C-terminal PEST domain that normally limits the intensity and duration of NOTCH1 signaling (45–47). Removal of the PEST domain Results in impaired NOTCH1 degradation and accumulation of an active NOTCH1 isoform (90). One recurrent mutation (c.7544_7545delCT) accounts for approximately 80% of all NOTCH1 mutations (45–47) and can be rapidly detected by a simple PCR-based strategy, providing a potential approach for a first-level screening of NOTCH1 alterations (47). Although the precise role of NOTCH1 activation in CLL pathogenesis is still under scrutiny, the relevance of these mutations is highlighted by the distinctive GEP signature of CLL carrying NOTCH1 mutations (46, 94).
Mutations of the SF3B1 gene.
SF3B1 is a critical component of both major (U2-like) and minor (U12-like) spliceosomes, which enact the precise excision of introns from pre-mRNA (95). At diagnosis, SF3B1 is mutated in 5%–10% of CLLs (51, 52, 89). Conversely, SF3B1 mutations are exceptional in MBL (96). The pathogenicity of SF3B1 mutations in CLL is supported by the clustering of these mutations in evolutionarily conserved hotspots localized within HEAT domains (51, 52, 89, 97). Although the precise mechanistic aspects of SF3B1 mutations are still under investigation, the observation that SF3B1 regulates the alternative splicing program of genes controlling cell cycle progression and apoptosis points to a potential contribution of SF3B1 mutations in modulating tumor cell proliferation and survival (51, 52). Across the spectrum of mature B cell neoplasias, mutations of SF3B1 are selectively restricted to CLL, whereas these same mutations are highly recurrent in myelodysplastic syndromes (98). At CLL diagnosis, SF3B1 mutations predict reduced survival independent of other clinical and biological risk factors (51, 52, 89).
BIRC3 gene inactivation.
BIRC3, along with TRAF2 and TRAF3, cooperates in the same protein complex that negatively regulates MAP3K14, an activator of the non-canonical pathway of NF-κB signaling (99). All BIRC3 mutations in CLL are predicted to disrupt the C-terminal RING domain, which is essential for proteasomal degradation of MAP3K14 by BIRC3 (78). Consistently, CLLs harboring BIRC3 disruption display constitutive NF-κB activation (78). At CLL diagnosis, BIRC3 disruption associates with unfavorable clinical and genetic features and predicts poor outcome independent of other risk factors (78). Among B cell neoplasms, mutations of BIRC3 appear to be specific for CLL and for splenic marginal zone lymphoma (78, 100).
Other mutations revealed by NGS studies.
Whole exome sequencing studies have revealed mutations of several other genes in CLL, albeit at low frequency (45, 46, 51, 52). Of these, MYD88 is mutated in 3%–5% of CLL at diagnosis (46). MYD88 is a critical adaptor molecule of the TLR complex that is also mutated in other B cell malignancies, namely DLBCL and marginal zone lymphoma (100, 101). In CLL, mutations of MYD88 target specific hotspots also implicated in other B cell malignancies (46, 101, 102).
Pathways to progression and chemorefractoriness
The predisposition to clinical progression of CLL is influenced by the genetic lesions of the leukemic clone. At CLL diagnosis, TP53 disruption, ATM deletion, NOTCH1 mutations, SF3B1 mutations, and inactivation of BIRC3 have all been shown to predict poor outcome (45–47, 49, 51, 52, 78, 81–89). While the prognostic values of TP53 and ATM have been assessed both in consecutive series and in the context of clinical trials, the roles of NOTCH1, SF3B1, and BIRC3 as clinical predictors have been tested only in consecutive series. Therefore, it remains to be determined whether specific treatments, such as the fludarabine/cyclophosphamide/rituximab regimen when administered to ATM-deleted CLL patients (103), are able to overcome the unfavorable prognostic value of NOTCH1, SF3B1, and BIRC3.
Molecular knowledge of the CLL genotype has significantly advanced the understanding of the genetic basis of chemorefractoriness (Figure 2). TP53 disruption predicts treatment failure with many regimens and mandates the use of alternative therapeutic strategies that are more efficacious (81–88, 103, 104). TP53 disruption, however, accounts for only 30%–40% of CLLs refractory to fludarabine, the mainstay drug for this leukemia. In the remaining cases, the molecular basis of chemorefractoriness is unknown. The newly identified genetic lesions of NOTCH1, SF3B1, and BIRC3 have clarified, at least in part, this issue. All these alterations are enriched in patients requiring therapy and refractory to fludarabine (NOTCH1 mutations: 20%; SF3B1 mutations: 17%; BIRC3 disruption: 24%) compared with CLL at diagnosis or compared with cases requiring therapy but sensitive to the drug (45–47, 78, 89). Among chemorefractory patients, alterations of NOTCH1, SF3B1, and BIRC3 appear to be largely exclusive with disruption of TP53, indicating that multiple pathways can lead to treatment failure (45, 47, 78, 89).
Pathways to Richter transformation
A fraction of CLLs develop RS, which represents the clinico-pathologic transformation of CLL into DLBCL (17, 18, 105); the cumulative incidence of RS at 5 and 10 years after CLL diagnosis is approximately 10% and 15%, respectively (106).
TP53 disruption (50%–60%), NOTCH1 activation (30%), and MYC abnormalities (20%) have emerged as the most frequent recurrent genetic lesions in RS (refs. 45, 107, and Figure 2). These abnormalities are frequently acquired at transformation, thus pointing to the pathogenetic relevance of these genetic lesions in acquiring the DLBCL phenotype (45, 107).
In RS, NOTCH1 mutations are largely mutually exclusive with MYC oncogenic activation (45). This is consistent with the observation that NOTCH1 directly stimulates MYC transcription and suggests that activation of oncogenic MYC may be a common pathway in RS transformation (45, 108). Conversely, both NOTCH1 mutational activation and MYC deregulation often co-exist with TP53 inactivation. The concurrence of TP53 inactivation with MYC activation in tumors may be selected to prevent the apoptotic effects and the response to genomic instability induced by MYC overexpression (45). Genetic lesions of TP53, NOTCH1, and MYC account for many (∼60%), though not all, cases of RS (45, 107). Additional genetic alterations acquired at the time of CLL transformation to RS are likely to be revealed by NGS studies.
The high prevalence of TP53 disruption in RS reflects the selection of a chemorefractory clone under the pressure of previous CLL treatments, thus suggesting an explanation for the poor outcome and the limited sensitivity to conventional drugs (17, 18, 105). Consistent with this hypothesis, TP53 status is an independent predictor of RS survival after transformation, segregating RS into prognostically meaningful categories (107).
Despite morphologic and phenotypic similarities with non-GC DLBCL arising de novo, the molecular profile of RS is distinct (107). Most genetic features that are recurrent in de novo DLBCL are rare or absent in RS (107). Moreover, the high frequency of unmutated IGHV genes observed in RS is in sharp contrast with the immunogenetic features of de novo DLBCL, which carry mutated IGHV genes in virtually all cases (107). Such molecular and immunogenetic differences between RS and de novo DLBCL reflect differences in the cell of origin and/or the microenvironment in which the lymphoma develops.
Early recognition of RS may be clinically useful to avoid the exposure of patients to multiple lines of therapy that are of little efficacy. A number of biological risk factors help to independently predict RS at the time of CLL diagnosis (106, 109–113). Biological predictors of RS reflect both the molecular features of the CLL clone — namely, stereotyped CDR3 of specific IGHV genes, telomere length, and mutations of NOTCH1 — as well as the genotype of the host, as exemplified by SNPs of the CD38 and LRP4 genes (109–113). CLL patients exhibiting activation of a specific IGHV gene such as IGHV4-39 in a stereotyped fashion (so-called BCR subset 8) have a very high risk of RS transformation (110). Curiously, a stereotyped IGHV4-39 BCR tends to associate with CLLs expressing surface IgG (35). The affinity of the BCR expressed by these patients for vimentin and possibly other apoptosis-associated autoantigens points to the role of BCR stimulation in RS development (41). Importantly, up to 40%–50% of CLL patients carrying NOTCH1 mutations at diagnosis are ultimately projected to develop and die of RS (47, 113). Consistent with the enrichment of NOTCH1 mutations in IGHV4-39–positive CLL, patients harboring both NOTCH1 mutations and IGHV4-39 constitute the disease category with the highest risk of RS transformation (113).
Since immunogenetic investigations have shown that the subclone from which RS emerges is already identifiable at CLL diagnosis in most patients who subsequently develop RS (114), deep sequencing studies performed in sequential samples are needed to assess whether traces of the genetic lesions manifestly acquired at RS transformation are already present at CLL diagnosis. Preliminary evidence suggests that this might be the case, at least in some patients (45).
Animal models of CLL
Mouse models have formally proven the pathogenetic role of del13q14 in CLL development. Three different lines of transgenic mice designed to mimic del13q14 develop CLL and other del13q14-associated lymphoproliferative disorders (64, 65). One transgenic line mimics the MDR common to all cases with del13q14, a second line of transgenic mice contains a deletion selectively targeting the Mir15a/16-1 cluster within intron 4 of the Dleu2 gene, and the third line carries a relatively large deletion spanning the genomic region most commonly deleted in 13q14 and including several genes (64, 65). All three models develop clonal lymphoproliferative disorders that occur late in life, are mostly indolent, and arise in a B cell–autonomous fashion. The severity of the disease appears to be proportional to the extent of the deletion, suggesting that genetic elements in addition to Mir15a/16-1 may contribute to the pathogenetic role of del13q14 (64, 65). The observed lymphoproliferations are characterized by low penetrance in all transgenic lines (40% MDR, 25% Mir15a/16-1 deletion) (64). This observation, coupled with the long latency associated with the disease phenotype, suggests that the development of a full-blown phenotype in mice might require accumulation of additional genetic alterations.
In addition to mice mimicking del13q14, other mouse models of CLL have been reported, including TCL1-transgenic mice and New Zealand black (NZB) mice (67, 115, 116). However, neither TCL1-mice nor NZB mice genetically mimic lesions in human CLL. Transgenic mice overexpressing TCL1 specifically in B cells develop lymphoproliferative disorders that follow an indolent course and display CD5 expression and stereotyped BCR, similar to mouse models of del13q14 (115, 116). Although TCL1 is highly expressed in human CLL, no structural genetic lesions are associated with this phenotype (117, 118). In NZB mice, three loci have been linked to CLL development, including a point mutation that affects the processing of Mir16-1, though this mutation is not detectable in human CLL (67).
Future mouse models combining del13q14 with mutations of NOTCH1, SF3B1, and BIRC3 might clarify the synergy of these genetic lesions in CLL pathogenesis and may more closely recapitulate the human disease.
Therapeutic implications
Advancing the knowledge of CLL molecular genetics may have important therapeutic implications including: (a) therapeutic stratification based on molecular prognosticators; and (b) targeted therapy aimed at correcting the genetic defect or its functional consequences.
The guidelines for CLL management that are currently in use consider only the prognostic value of TP53 disruption for the stratification of CLL therapy (88, 104). Because CLL patients with TP53 disruption have an extremely high likelihood of failing standard immunochemotherapy, it is recommended that these patients receive treatment with alternative regimens that are able to overcome the chemorefractoriness induced by TP53 disruption (104). Given their recent identification (45–47, 51, 52, 78, 89), it has not yet been established whether genetic alterations of NOTCH1, SF3B1, and BIRC3, all of which predict poor outcome in consecutive series, will also be useful for therapeutic stratification.
Some of the genetic lesions that have been recently identified by NGS represent attractive candidates for targeted therapy of CLL. In particular, NOTCH1 is a well-established therapeutic target with some drugs already available and others under active development (118, 119). Compounds inhibiting the enzymatic conversion of NOTCH1 to an active transcription factor have already entered clinical trials for T lineage acute lymphoblastic leukemia (119, 120). The identification of BIRC3 disruption might also bear potential therapeutic implications, because NF-κB pathway inhibitors may potentially target NF-κB activation resulting from BIRC3 abrogation (78).
Apart from the potential targeting of genetic lesions with specific inhibitors, the biological advances in CLL have revealed that the BCR plays a central role in disease pathogenesis and that, consequently, BCR signaling might represent a suitable target for therapy in many patients (121). Currently, two drugs that act as inhibitors of BCR signaling, i.e., the Bruton tyrosine kinase inhibitor ibrutinib and the PI3K-δ inhibitor GS-1101, appear to be particularly promising and are under advanced clinical investigation (121).
Improved understanding of the genetic basis of CLL continues to refine the stratification of patients, with a goal of improving CLL management through targeted therapy. The clinical heterogeneity of CLL is underscored by the variety of genetic lesions observed, from classical markers of CLL to new genetic lesions uncovered by whole genome and whole exome sequencing. Translating these molecular prognosticators into the clinic, along with continued development of targeted therapies, will be critical to improving treatment options.
Acknowledgments
Work by the authors described in this review was supported by the Italian Association for Cancer Research (AIRC) Special Program in Clinical Molecular Oncology 5 × 1000 (number 10007).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(10):3432–3438. doi:10.1172/JCI64101.
References
- 1.Swerdlow SH, et al. WHO Classification Of Tumours Of Haematopoietic And Lymphoid Tissues. 4th ed. Lyon, France: World Health Organization; 2008. [Google Scholar]
- 2.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 3.Dameshek W. Chronic lymphocytic leukemia: an accumulative disease of immunologically incompetent lymphocytes. Blood. 1967;29(4 suppl):566–584. [PubMed] [Google Scholar]
- 4.Messmer BT, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest. 2005;115(3):755–764. doi: 10.1172/JCI23409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dighiero G, et al. B-cell chronic lymphocytic leukemia: present status and future directions. French Cooperative Group on CLL. Blood. 1991;78(8):1901–1914. [PubMed] [Google Scholar]
- 6.Ponzoni M, Doglioni C, Caligaris-Cappio F. Chronic lymphocytic leukemia: the pathologist’s view of lymph node microenvironment. Semin Diagn Pathol. 2011;28(2):161–166. doi: 10.1053/j.semdp.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. 2009;114(16):3367–3375. doi: 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marti GE, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol. 2005;130(3):325–332. doi: 10.1111/j.1365-2141.2005.05550.x. [DOI] [PubMed] [Google Scholar]
- 9.Rawstron AC, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575–583. doi: 10.1056/NEJMoa075290. [DOI] [PubMed] [Google Scholar]
- 10.Landgren O, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360(7):659–667. doi: 10.1056/NEJMoa0806122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rawstron AC. Monoclonal B-cell lymphocytosis. Hematology Am Soc Hematol Educ Program. 2009. 2009 430 439 . 10.1182/asheducation-2009.1.430 [DOI] [PubMed] [Google Scholar]
- 12.Ghia P, Caligaris-Cappio F. Monoclonal B-cell lymphocytosis (MBL): right track or red herring? Blood. 2012;119(19):4358–4362. doi: 10.1182/blood-2012-01-404681. [DOI] [PubMed] [Google Scholar]
- 13.Rossi D, et al. The prognosis of clinical monoclonal B cell lymphocytosis differs from prognosis of Rai 0 chronic lymphocytic leukaemia and is recapitulated by biological risk factors. Br J Haematol. 2009;146(1):64–75. doi: 10.1111/j.1365-2141.2009.07711.x. [DOI] [PubMed] [Google Scholar]
- 14.Shanafelt TD, et al. Natural history of individuals with clinically recognized monoclonal B-cell lymphocytosis compared with patients with Rai 0 chronic lymphocytic leukemia. J Clin Oncol. 2009;27(24):3959–3963. doi: 10.1200/JCO.2008.21.2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10(1):37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- 16.Zenz T, Gribben JG, Hallek M, Döhner H, Keating MJ, Stilgenbauer S. Risk categories and refractory CLL in the era of chemoimmunotherapy. Blood. 2012;119(18):4101–4107. doi: 10.1182/blood-2011-11-312421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rossi D, Gaidano G. Richter syndrome: molecular insights and clinical perspectives. Hematol Oncol. 2009;27(1):1–10. doi: 10.1002/hon.880. [DOI] [PubMed] [Google Scholar]
- 18.Fangazio M, De Paoli L, Rossi D, Gaidano G. Predictive markers and driving factors behind Richter syndrome development. Expert Rev Anticancer Ther. 2011;11(3):433–442. doi: 10.1586/era.10.237. [DOI] [PubMed] [Google Scholar]
- 19.Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117(6):1781–1791. doi: 10.1182/blood-2010-07-155663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kipps TJ. Immunoglobulin genes in chronic lymphocytic leukemia. Blood Cells. 1993;19(3):615–625. [PubMed] [Google Scholar]
- 21.Damle RN, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840–1847. [PubMed] [Google Scholar]
- 22.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848–1854. [PubMed] [Google Scholar]
- 23.Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol. 2008;8(1):22–33. doi: 10.1038/nri2217. [DOI] [PubMed] [Google Scholar]
- 24.Küppers R, Klein U, Hansmann ML, Rajewsky K. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341(20):1520–1529. doi: 10.1056/NEJM199911113412007. [DOI] [PubMed] [Google Scholar]
- 25.Stevenson FK, Sahota SS, Ottensmeier CH, Zhu D, Forconi F, Hamblin TJ. The occurrence and significance of V gene mutations in B cell-derived human malignancy. Adv Cancer Res. 2001;83:81–116. doi: 10.1016/s0065-230x(01)83004-9. [DOI] [PubMed] [Google Scholar]
- 26.Klein U, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenwald A, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639–1647. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein U, et al. Somatic hypermutation in normal and transformed human B cells. Immunol Rev. 1998;162:261–280. doi: 10.1111/j.1600-065X.1998.tb01447.x. [DOI] [PubMed] [Google Scholar]
- 29.Martin F, Kearney JF. Marginal zone B cells. Nat Rev Immunol. 2002;2(5):323–335. doi: 10.1038/nri799. [DOI] [PubMed] [Google Scholar]
- 30.Pillai S, Cariappa A, Moran ST. Marginal zone B cells. Annu Rev Immunol. 2005;23:161–196. doi: 10.1146/annurev.immunol.23.021704.115728. [DOI] [PubMed] [Google Scholar]
- 31.Carsetti R, Rosado MM, Wardmann H. Peripheral development of B cells in mouse and man. Immunol Rev. 2004;197:179–191. doi: 10.1111/j.0105-2896.2004.0109.x. [DOI] [PubMed] [Google Scholar]
- 32.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21:841–894. doi: 10.1146/annurev.immunol.21.120601.141018. [DOI] [PubMed] [Google Scholar]
- 33.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103(12):4389–4395. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 34.Messmer BT, et al. Multiple distinct sets of stereotyped antigen receptors indicate a role for antigen in promoting chronic lymphocytic leukemia. J Exp Med. 2004;200(4):519–525. doi: 10.1084/jem.20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamatopoulos K, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007;109(1):259–270. doi: 10.1182/blood-2006-03-012948. [DOI] [PubMed] [Google Scholar]
- 36.Murray F, et al. Stereotyped patterns of somatic hypermutation in subsets of patients with chronic lymphocytic leukemia: implications for the role of antigen selection in leukemogenesis. Blood. 2008;111(3):1524–1533. doi: 10.1182/blood-2007-07-099564. [DOI] [PubMed] [Google Scholar]
- 37.Bomben R, et al. Molecular and clinical features of chronic lymphocytic leukaemia with stereotyped B cell receptors: Results from an Italian multicentre study. Br J Haematol. 2009;144(4):492–506. doi: 10.1111/j.1365-2141.2008.07469.x. [DOI] [PubMed] [Google Scholar]
- 38.Agathangelidis A, et al. Stereotyped B-cell receptors in one third of chronic lymphocytic leukemia: towards a molecular classification with implications for targeted therapeutic interventions. Blood. 2012;119(19):4467–4475. doi: 10.1182/blood-2011-11-393694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forconi F, et al. The IGHV1-69/IGHJ3 recombinations of unmutated CLL are distinct from those of normal B cells. Blood. 2012;119(9):2106–2109. doi: 10.1182/blood-2011-08-375501. [DOI] [PubMed] [Google Scholar]
- 40.Chu CC, et al. Chronic lymphocytic leukemia antibodies with a common stereotypic rearrangement recognize nonmuscle myosin heavy chain IIA. Blood. 2008;112(13):5122–5129. doi: 10.1182/blood-2008-06-162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu CC, et al. Many chronic lymphocytic leukemia antibodies recognize apoptotic cells with exposed nonmuscle myosin heavy chain IIA: implications for patient outcome and cell of origin. Blood. 2010;115(19):3907–3915. doi: 10.1182/blood-2009-09-244251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shanafelt TD, et al. Prospective evaluation of clonal evolution during long-term follow-up of patients with untreated early-stage chronic lymphocytic leukemia. J Clin Oncol. 2006;24(28):4634–4641. doi: 10.1200/JCO.2006.06.9492. [DOI] [PubMed] [Google Scholar]
- 43.Stilgenbauer S, et al. Clonal evolution in chronic lymphocytic leukemia: acquisition of high-risk genomic aberrations associated with unmutated VH, resistance to therapy, and short survival. Haematologica. 2007;92(9):1242–1245. doi: 10.3324/haematol.10720. [DOI] [PubMed] [Google Scholar]
- 44.Guarini A, et al. BCR ligation induced by IgM stimulation Results in gene expression and functional changes only in IgV H unmutated chronic lymphocytic leukemia (CLL) cells. Blood. 2008;112(3):782–792. doi: 10.1182/blood-2007-12-127688. [DOI] [PubMed] [Google Scholar]
- 45.Fabbri G, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. 2011;208(7):1389–1401. doi: 10.1084/jem.20110921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi D, et al. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood. 2012;119(2):521–529. doi: 10.1182/blood-2011-09-379966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kikushige Y, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246–259. doi: 10.1016/j.ccr.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 49.Döhner H, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910–1916. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 50.Caporaso N, et al. Chronic lymphocytic leukaemia genetics overview. Br J Haematol. 2007;139(5):630–634. doi: 10.1111/j.1365-2141.2007.06846.x. [DOI] [PubMed] [Google Scholar]
- 51.Quesada V, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011;44(1):47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 52.Wang L, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pasqualucci L, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 54.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11(10):685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 55.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471(7339):467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasqualucci L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–837. doi: 10.1038/ng.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang J, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, et al. Cloning of two candidate tumor suppressor genes within a 10 kb region on chromosome 13q14, frequently deleted in chronic lymphocytic leukemia. Oncogene. 1997;15(20):2463–2473. doi: 10.1038/sj.onc.1201643. [DOI] [PubMed] [Google Scholar]
- 60.Migliazza A, et al. Nucleotide sequence, transcription map, and mutation analysis of the 13q14 chromosomal region deleted in B-cell chronic lymphocytic leukemia. Blood. 2001;97(7):2098–2104. doi: 10.1182/blood.V97.7.2098. [DOI] [PubMed] [Google Scholar]
- 61.Calin GA, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–15529. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mertens D, et al. Allelic silencing at the tumor-suppressor locus 13q14.3 suggests an epigenetic tumor-suppressor mechanism. Proc Natl Acad Sci U S A. 2006;103(20):7741–7746. doi: 10.1073/pnas.0600494103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 64.Klein U, et al. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17(1):28–40. doi: 10.1016/j.ccr.2009.11.019. [DOI] [PubMed] [Google Scholar]
- 65.Lia M, et al. Functional dissection of the chromosome 13q14 tumor-suppressor locus using transgenic mouse lines. Blood. 2012;119(13):2981–2990. doi: 10.1182/blood-2011-09-381814. [DOI] [PubMed] [Google Scholar]
- 66.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Proc Natl Acad Sci U S A. 2005;102(39):13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raveche ES, et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood. 2007;109(12):5079–5086. doi: 10.1182/blood-2007-02-071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Linsley PS, et al. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Mol Cell Biol. 2007;27(6):2240–2252. doi: 10.1128/MCB.02005-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bandi N, et al. miR-15a and miR-16 are implicated in cell cycle regulation in a Rb-dependent manner and are frequently deleted or down-regulated in non-small cell lung cancer. Cancer Res. 2009;69(13):5553–5559. doi: 10.1158/0008-5472.CAN-08-4277. [DOI] [PubMed] [Google Scholar]
- 70.Liu Q, et al. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008;36(16):5391–5404. doi: 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao H, Kalota A, Jin S, Gewirtz AM. The c-myb proto-oncogene and microRNA-15a comprise an active autoregulatory feedback loop in human hematopoietic cells. Blood. 2009;113(3):505–516. doi: 10.1182/blood-2008-01-136218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fabbri M, et al. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305(1):59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossi D, Gaidano G. ATM and chronic lymphocytic leukemia: mutations, and not only deletions, matter. Haematologica. 2012;97(1):5–8. doi: 10.3324/haematol.2011.057109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schaffner C, Stilgenbauer S, Rappold GA, Döhner H, Lichter P. Somatic ATM mutations indicate a pathogenetic role of ATM in B-cell chronic lymphocytic leukemia. Blood. 1999;94(2):748–753. [PubMed] [Google Scholar]
- 75.Stankovic T, et al. Inactivation of ataxia teleangiectasia mutated gene in B-cell chronic lymphocytic leukemia. Lancet. 1999;353(1):26–29. doi: 10.1016/S0140-6736(98)10117-4. [DOI] [PubMed] [Google Scholar]
- 76.Guarini A, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2011;97(1):47–55. doi: 10.3324/haematol.2011.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Skowronska A, et al. ATM germline heterozygosity does not play a role in CLL initiation but influences rapid disease progression through loss of the remaining ATM allele. Haematologica. 2011;97(1):142–146. doi: 10.3324/haematol.2011.048827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rossi D, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119(12):2854–2862. doi: 10.1182/blood-2011-12-395673. [DOI] [PubMed] [Google Scholar]
- 79.Bieging KT, Attardi LD. Deconstructing p53 transcriptional networks in tumor suppression. Trends Cell Biol. 2012;22(2):97–106. doi: 10.1016/j.tcb.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaidano G, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 1991;88(12):5413–5417. doi: 10.1073/pnas.88.12.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zenz T, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: Results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–3329. doi: 10.1182/blood-2008-04-154070. [DOI] [PubMed] [Google Scholar]
- 82.Rossi D, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15(3):995–1004. doi: 10.1158/1078-0432.CCR-08-1630. [DOI] [PubMed] [Google Scholar]
- 83.Zenz T, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–4479. doi: 10.1200/JCO.2009.27.8762. [DOI] [PubMed] [Google Scholar]
- 84.Gonzalez D, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: Results from the LRF CLL4 trial. J Clin Oncol. 2011;29(16):2223–2229. doi: 10.1200/JCO.2010.32.0838. [DOI] [PubMed] [Google Scholar]
- 85.Dicker F, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009;23(1):117–124. doi: 10.1038/leu.2008.274. [DOI] [PubMed] [Google Scholar]
- 86.Malcikova J, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114(26):5307–5314. doi: 10.1182/blood-2009-07-234708. [DOI] [PubMed] [Google Scholar]
- 87.Chiaretti S, et al. Evaluation of TP53 mutations with the AmpliChip p53 research test in chronic lymphocytic leukemia: correlation with clinical outcome and gene expression profiling. Genes Chromosomes Cancer. 2011;50(4):263–274. doi: 10.1002/gcc.20852. [DOI] [PubMed] [Google Scholar]
- 88.Pospisilova S, et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia. 2012;26(7):1458–1461. doi: 10.1038/leu.2012.25. [DOI] [PubMed] [Google Scholar]
- 89.Rossi D, et al. Mutations of the SF3B1 splicing factor in chronic lymphocytic leukemia: association with progression and fludarabine-refractoriness. Blood. 2011;118(26):6904–6908. doi: 10.1182/blood-2011-08-373159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferrando AA. The role of NOTCH1 signaling in T-ALL. Hematology Am Soc Hematol Educ Program. 2009;2009:353–361. doi: 10.1182/asheducation-2009.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sportoletti P, et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br J Haematol. 2010;151(4):404–406. doi: 10.1111/j.1365-2141.2010.08368.x. [DOI] [PubMed] [Google Scholar]
- 92.Rasi S, Monti S, Spina V, Foà R, Gaidano G, Rossi D. Analysis of NOTCH1 mutations in monoclonal B-cell lymphocytosis. Haematologica. 2012;97(1):153–154. doi: 10.3324/haematol.2011.053090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balatti V, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119(2):329–331. doi: 10.1182/blood-2011-10-386144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Del Giudice I, et al. NOTCH1 mutations in +12 chronic lymphocytic leukemia (CLL) confer an unfavorable prognosis, induce a distinctive transcriptional profiling and refine the intermediate prognosis of +12 CLL. Haematologica. 2012;97(3):437–441. doi: 10.3324/haematol.2011.060129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136(4):701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 96.Greco M, et al. Hematol Oncol. Analysis of SF3B1 mutations in monoclonal B-cell lymphocytosis [published online ahead of print March 29, 2012]. doi: 10.1002/hon.2013 . [DOI] [PubMed] [Google Scholar]
- 97.Andrade MA, Perez-Iratxeta C, Ponting CP. Protein repeats: structures, functions, and evolution. J Struct Biol. 2001;134(2–3):117–131. doi: 10.1006/jsbi.2001.4392. [DOI] [PubMed] [Google Scholar]
- 98.Hahn CN, Scott HS. Spliceosome mutations in hematopoietic malignancies. Nat Genet. 2011;44(1):9–10. doi: 10.1038/ng.1045. [DOI] [PubMed] [Google Scholar]
- 99.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 100.Rossi D, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118(18):4930–4934. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- 101.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yan Q, et al. BCR and TLR signaling pathways are recurrently targeted by genetic changes in splenic marginal zone lymphomas. Haematologica. 2012;97(4):595–598. doi: 10.3324/haematol.2011.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hallek M, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376(9747):1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- 104.Hallek M, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–5456. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tsimberidou AM, Keating MJ. Richter syndrome: biology, incidence, and therapeutic strategies. Cancer. 2005;103(2):216–228. doi: 10.1002/cncr.20773. [DOI] [PubMed] [Google Scholar]
- 106.Rossi D, et al. Biological and clinical risk factors of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2008;142(2):202–215. doi: 10.1111/j.1365-2141.2008.07166.x. [DOI] [PubMed] [Google Scholar]
- 107.Rossi D, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–3401. doi: 10.1182/blood-2010-09-302174. [DOI] [PubMed] [Google Scholar]
- 108.Weng AP, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20(15):2096–2109. doi: 10.1101/gad.1450406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aydin S, et al. CD38 gene polymorphism and chronic lymphocytic leukemia: a role in transformation to Richter syndrome? Blood. 2008;111(12):5646–5653. doi: 10.1182/blood-2008-01-129726. [DOI] [PubMed] [Google Scholar]
- 110.Rossi D, et al. Stereotyped B-cell receptor is an independent risk factor of chronic lymphocytic leukemia transformation to Richter syndrome. Clin Cancer Res. 2009;15(13):4415–4422. doi: 10.1158/1078-0432.CCR-08-3266. [DOI] [PubMed] [Google Scholar]
- 111.Rossi D, et al. Telomere length is an independent predictor of survival, treatment requirement and Richter’s syndrome transformation in chronic lymphocytic leukemia. Leukemia. 2009;23(6):1062–1072. doi: 10.1038/leu.2008.399. [DOI] [PubMed] [Google Scholar]
- 112.Rasi S, et al. A variant of the LRP4 gene affects the risk of chronic lymphocytic leukaemia transformation to Richter syndrome. Br J Haematol. 2011;152(3):284–294. doi: 10.1111/j.1365-2141.2010.08482.x. [DOI] [PubMed] [Google Scholar]
- 113.Rossi D, et al. Different impact of NOTCH1 and SF3B1 mutations on the risk of chronic lymphocytic leukemia transformation to Richter syndrome. Br J Haematol. 2012;158(3):426–429. doi: 10.1111/j.1365-2141.2012.09155.x. [DOI] [PubMed] [Google Scholar]
- 114.Rossi D, et al. Molecular history of Richter syndrome: origin from a cell already present at the time of chronic lymphocytic leukemia diagnosis. Int J Cancer. 2012;130(12):3006–3010. doi: 10.1002/ijc.26322. [DOI] [PubMed] [Google Scholar]
- 115.Bichi R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99(10):6955–60. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yan XJ, et al. B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2006;103(31):11713–11718. doi: 10.1073/pnas.0604564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Herling M, et al. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia. 2006;20(2):280–285. doi: 10.1038/sj.leu.2404017. [DOI] [PubMed] [Google Scholar]
- 118.Pekarsky Y, et al. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res. 2006;66(24):11590–11593. doi: 10.1158/0008-5472.CAN-06-3613. [DOI] [PubMed] [Google Scholar]
- 119.Palomero T, Ferrando A. Therapeutic targeting of NOTCH1 signaling in T-cell acute lymphoblastic leukemia. Clin Lymphoma Myeloma. 2009;9(suppl):S205–S210. doi: 10.3816/CLM.2009.s.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sarmento LM, Barata JT. Therapeutic potential of Notch inhibition in T-cell acute lymphoblastic leukemia: rationale, caveats and promises. Expert Rev Anticancer Ther. 2011;11(9):1403–1415. doi: 10.1586/era.11.73. [DOI] [PubMed] [Google Scholar]
- 121.Wojach JA, Johnson AJ, Byrd JC. The B-cell receptor signaling pathway as a therapeutic target in CLL. Blood. 2012;120(6):1175–1184. doi: 10.1182/blood-2012-02-362624. [DOI] [PMC free article] [PubMed] [Google Scholar]