

Abstract
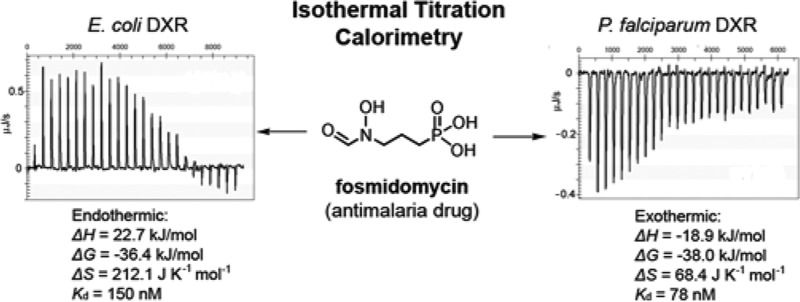
Isothermal titration calorimetry (ITC) was used to investigate the binding of six inhibitors to 1-deoxy-d-xylulose-5-phosphate reductoisomerase (DXR), a target for developing novel anti-infectives. The binding of hydroxamate inhibitors to Escherichia coli DXR is Mg2+-dependent, highly endothermic (ΔH, 22.7–24.3 kJ/mol), and entropy-driven, while that of nonhydroxamate compounds is metal ion-independent and exothermic (ΔH, −19.4 to −13.8 kJ/mol), showing that hydration/dehydration of the enzyme metal ion binding pocket account for the drastic ΔH change. However, for DXRs from Plasmodium falciparum and Mycobacterium tuberculosis, the binding of all inhibitors is exothermic (ΔH, −24.9 to −9.2 kJ/mol), suggesting that the metal ion binding sites of these two enzymes are considerably less hydrated. The dissociation constants measured by ITC are well correlated with those obtained by enzyme inhibition assays (R2 = 0.75). Given the rapid rise of antibiotic resistance, this work is of interest since it provides novel structural implications for rational development of potent DXR inhibitors.
Keywords: 1-deoxy-d-xylulose-5-phosphate reductoisomerase, isothermal titration calorimetry, enthalpy/entropy driven inhibitor binding, anti-infective
Isoprenoid biosynthesis, producing isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP), is essential for all organisms. IPP and DMAPP are the only two starting compounds for biosynthesizing over 55000 isoprenoid/terpenoid natural products,1 including important substances such as steroids and long-chain (C15–C55) isoprenyl diphosphates for protein anchoring to cell membranes. Different from humans and animals, the vast majority of bacteria (except for Gram-positive cocci) as well as apicomplexan parasites exclusively use the nonmevalonate isoprene biosynthesis pathway2 [or 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway] to produce IPP and DMAPP. These include important human pathogens such as Mycobacterium tuberculosis, Escherichia coli, and Plasmodium falciparum (malaria parasite), which cause millions of human mortalities each year. Particularly problematic is the quickly rising antibiotic resistance during the past few decades.3
1-Deoxy-d-xylulose-5-phosphate reductoisomerase (DXR), the second enzyme in the nonmevalonate pathway, catalyzes the isomerization and reduction of 1-deoxy-d-xylulose-5-phosphate (DXP) to MEP with Mg2+ (or Mn2+) and NADPH being the enzyme cofactors (Figure 1). It has been found to be an important target for developing novel anti-infective drugs.4−6 Fosmidomycin (1, Chart 1), a potent DXR inhibitor,7 possesses strong antimalarial activity.8 In combination with clindamycin, fosmidomycin was found to be able to achieve 100% cure for uncomplicated P. falciparum malaria in clinical trials.9 Great interest has therefore been generated to develop more lipophilic DXR inhibitors that possess broad and enhanced anti-infective activity.10−20
Figure 1.
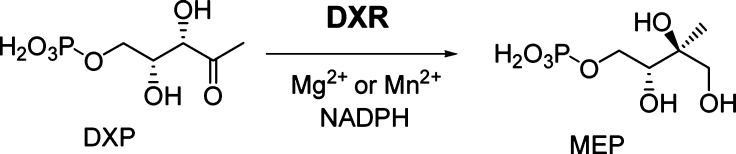
Reaction catalyzed by DXR.
Chart 1. Structures of DXR Inhibitors.

Our early work18−20 as well as that from other groups10,11,14,21−24 in X-ray crystallography and medicinal chemistry have shown the importance of metal ion chelation, van der Waals, and π–π stacking interactions between the DXR inhibitors and the protein. However, these models have not taken into account the solvation effects during ligand binding to DXR, which may cause significant enthalpy and entropy changes and therefore affect the overall binding affinity. In addition, knowledge of whether inhibitor binding is enthalpy- or entropy-driven could be helpful in drug development, since several recent isothermal titration calorimetry (ITC) studies show the best drugs generally exhibit the greatest ΔH contributions (i.e., enthalpy-driven inhibitors).25−27 Here, we use ITC to investigate the thermodynamic interactions of six selected inhibitors shown in Chart 1 with the DXR enzymes from E. coli, M. tuberculosis, and P. falciparum.
These compounds represent a broad chemical diversity as well as interesting biological activity, making them particularly suited for the study. Fosmidomycin (1) and FR900098 (2) are highly polar phosphono-hydroxamic acids. Fosmidomycin derivative 3 contains a hydrophobic α-3,4-dichlorophenyl substituent and possesses an enhanced antimalarial activity,14 presumably due to the increased lipophilicity. Nonhydroxamate inhibitors 4 and 5 do not occupy the DXR metal ion binding site.19 Compound 6 with a high lipophilicity is the only potent DXR inhibitor without a phosphonate group, showing a broad antibacterial activity.18
First, we investigated thermodynamics of the inhibitor binding to E. coli DXR (EcDXR) in the presence of 2 mM NADPH and 150 mM NaCl at 279 K (DXR is more stable at this temperature) and pH 7.6, and the results are summarized in Table 1 and representatively are shown in Figure 2. Without a metal ion (Mg2+ or Mn2+), fosmidomycin does not bind to EcDXR (data not shown), which is in agreement with the X-ray crystal structures showing its hydroxamate group chelates the metal ion.21−24 In the presence of 2 mM Mg2+, the binding of fosmidomycin was found, surprisingly, to be strongly endothermic (ΔH = 22.7 kJ/mol) with a Ka (affinity constant) value of 6.67 × 106 M–1, which equals a ΔG value of −36.4 kJ/mol (Figure 2a). With a calculated ΔS = 212.1 J K–1 mol–1, the experiment shows that fosmidomycin binding to EcDXR is entropy-driven. This is a rather unexpected result since fosmidomycin binding involves the formation of multiple H-bonds and strong electrostatic interactions for its phosphonate as well as chelation to the metal ion for the hydroxamate group,21−24 all of which are expected to be highly exothermic. An ITC experiment using 2 mM Mn2+, another commonly used metal ion for the reaction, yielded a similar result with ΔH = 27.7 kJ/mol, ΔG = −36.7 kJ/mol, and ΔS = 230.7 J K–1 mol–1. These results suggest that the apo-EcDXR is extensively hydrated at the active site. The newly formed H-bond, electrostatic, and chelation interactions between fosmidomycin and EcDXR cannot fully compensate for the loss of those between the hydrates and the protein. The binding is therefore driven by considerably enhanced entropy, including contributions from the originally ordered water molecules at the active site as well as those around fosmidomycin. Structurally similar FR900098 (2) has an almost identical thermodynamic profile, showing entropy-driven binding with ΔH = 23.5 kJ/mol, ΔG = −36.6 kJ/mol, and ΔS = 215.4 J K–1 mol–1. With a bulky, hydrophobic 3,4-dichlorophenyl group, fosmidomycin derivative 3 was also found to be an entropy-driven ligand, having ΔH = 24.3 kJ/mol, ΔG = −34.6 kJ/mol, and ΔS = 211.1 J K–1 mol–1 (Figure 2b).
Table 1. Thermodynamic Parameters for Inhibitor Binding to E. coli DXRa.
| compd | ΔH (kJ/mol) | Ka (M–1) | ΔG (kJ/mol) | ΔS (J K–1 mol–1) | Ki (μM)c |
|---|---|---|---|---|---|
| 1 (Mg2+) | 22.7 | 6.67 × 106 | –36.4 | 212.1 | 0.027 |
| 1 (Mn2+) | 27.7 | 7.35 × 106 | –36.7 | 230.7 | NTd |
| 1 (Mg2+, pH 8.5) | 20.0 | 5.60 × 106 | –36.0 | 202.9 | NTd |
| 2 (Mg2+) | 23.5 | 7.14 × 106 | –36.6 | 215.4 | 0.019 |
| 3 (Mg2+) | 24.3 | 3.03 × 106 | –34.6 | 211.1 | 0.058 |
| 4 (Mg2+) | –19.4 | 4.93 × 105 | –30.4 | 39.3 | 2.3 |
| 4b | –18.7 | 4.31 × 105 | –30.1 | 40.8 | NTd |
| 4 (pH 8.5)b | –16.1 | 4.89 × 105 | –30.4 | 51.2 | NTd |
| 5 (Mg2+) | –14.2 | 4.83 × 105 | –30.4 | 58.0 | 0.42 |
| 6 (Mg2+) | –13.8 | 1.72 × 105 | –28.0 | 50.7 | 0.7 |
| 6b | –12.1 | 2.87 × 105 | –29.1 | 61.1 | NTd |
| 6 (pH 8.5)b | –10.1 | 2.55 × 105 | –28.9 | 67.4 | NTd |
At pH 7.6 unless indicated.
No Mg2+ (or Mn2+).
Ki values were measured using enzyme inhibition assay.
Not tested.
Figure 2.

Representative ITC results and fitting curves for inhibitor binding to DXR.
Interestingly, titration of EcDXR with nonhydroxamate compounds 4 and 5 in the presence of 2 mM Mg2+ was observed to be exothermic (Figure 2c). The binding of the pyridine phosphonate inhibitor 4 gave a ΔH of −19.4 kJ/mol and a Ka of 4.93 × 105 M–1, which equals a ΔG value of −30.4 kJ/mol. The result shows both enthalpy and entropy (−TΔS = −11.0 kJ/mol) contribute considerably to binding. As compared to 4, compound 5 possesses an additional phenyl group. The binding of 5 demonstrates a generally similar thermodynamic feature, with ΔH = −14.2 kJ/mol, ΔG = −30.3 kJ/mol, and ΔS = 58.0 J K–1 mol–1. Likely due to the extra hydrophobic ring of 5, a larger entropy contribution (−TΔS = −16.2 kJ/mol) was observed. We next attempted to understand what factors lead to the drastic thermodynamic change from an average ΔH of 23.5 kJ/mol for 1–3 to that of −16.8 kJ/mol for 4 and 5. As shown in the Supporting Information, Figure S1a, our previous X-ray crystal structures of EcDXR in complex with 4 and 5 show that the phosphonate group of these two molecules is located in the same position as that of fosmidomycin, while the EcDXR Mg2+ binding site is not occupied.19 We titrated EcDXR with compound 4 in the absence of Mg2+ (or Mn2+) and found, unlike fosmidomycin, that the binding of 4 to EcDXR is indeed independent of the metal ion, with a set of similar thermodynamic parameters (ΔH = −18.7 kJ/mol, ΔG = −30.1 kJ/mol, and ΔS = 40.8 J K–1 mol–1) (Figure 2f and Table 1). In addition, our previous modeling studies further suggested the aromatic rings of compounds 3–5 are located in the same EcDXR pocket (Figure S1b in the Supporting Information).19 The distinct thermodynamic profiles between 3 and 4 clearly demonstrate that hydration/dehydration of the metal ion binding pocket of EcDXR as well as the hydroxamate group of compound 3 (and that of 1 and 2) are mainly responsible for the drastic ΔH differences. In addition, fosmidomycin is a slow, tight-binding inhibitor of EcDXR,28 and formation of the more tightly bound EcDXR:inhibitor complex could have additional effects on thermodynamics. This feature could also contribute to the difference between the binding of fosmidomycin (as well as structurally similar 2 and 3) and that of other competitive inhibitors 4 and 5.
For the structurally distinct inhibitor 6,18 binding to EcDXR in the presence of Mg2+ was found to be exothermic (ΔH = −13.8 kJ/mol), with almost equal contributions from enthalpy and entropy (−TΔS = −14.1 kJ/mol). Moreover, we found titration without Mg2+ yielded a similar result (ΔH = −12.1 kJ/mol, ΔG = −29.1 kJ/mol, and ΔS = 61.1 J K–1 mol–1), indicating the binding of compound 6 also does not require Mg2+. In our early work,18 both O atoms of 6 were found to be important for the activity, and the docking study suggested they chelate Mg2+. However, the ITC experiments demonstrate that no metal ion is needed for the binding of 6. Therefore, its 1-hydroxypyridin-2-one group could be involved in H-bond and/or other (e.g., electrostatic) interactions with the protein.
Binding thermodynamics of three representative inhibitors, 1, 4, and 6, were also measured at pH 8.5, at which DXR still has good enzyme activity. ITC experiments gave similar thermodynamic parameters for each of these compounds (Table 1). This is not surprising since from pH 7.6 to 8.5, there is little change in the protonation status of these ligands, given the pKa values of the phosphonate (Ka2), hydroxamate, and 1-hydroxypyridin-2-one being ∼7, 9.5, and 6, respectively.
Next, we performed ITC studies of inhibitor binding to recombinant P. falciparum and M. tuberculosis DXR (PfDXR and MtDXR, respectively) to find out whether DXRs from the eukaryotic and Gram-positive species have any different thermodynamic properties. Despite the high number of human deaths (∼3 million annually) caused by these two pathogens, the vast majority of DXR studies to date have used the E. coli enzyme. We cloned the PfDXR gene, inserted into pQE30 vector, expressed in E. coli (M15), and purified the enzyme using a standard protocol for His6-tagged protein (Supporting Information, Experimental Section). The ITC experiments for compounds 1–6 binding to PfDXR were conducted in the presence of 2 mM Mn2+ (previously used in ref (8)) and 2 mM NADPH at 279 K and pH 7.6, and the results are summarized in Table 2. Use of Mn2+ is also due to a higher enzyme activity and stability. Different from E. coli DXR, the binding of fosmidomycin (1) to PfDXR was, however, found to be exothermic (ΔH = −18.9 kJ/mol), with a Ka value of 1.28 × 107 M–1, which equals a ΔG value of −38.0 kJ/mol (Figure 2d). This result shows enthalpy and entropy (−TΔS = −19.1 kJ/mol) equally contribute to the high affinity binding of this ligand to PfDXR. In addition, titrations of PfDXR with the two other hydroxamate inhibitors 2 and 3 are also exothermic (ΔH = −21.1 kJ/mol for 2 and ΔH = −19.0 kJ/mol for 3). These thermodynamic experiments suggest a different state of hydration at the metal ion binding site between the two apo-DXR enzymes with PfDXR being considerably less hydrated. Upon binding of a hydroxamate ligand, relatively more heat is therefore produced together with less increase of entropy from liberated water molecules. For nonhydroxamate inhibitors 4–6, which do not need a metal ion for binding, ITC experiments showed similar thermodynamic parameters to those of EcDXR. Titrations of PfDXR with these compounds are exothermic (ΔH = −9.2 kJ/mol for 4, −12.4 kJ/mol for 5, and −15.7 kJ/mol for 6), and the binding is driven by both enthalpy and entropy. Also, as can be seen in Table 2, the binding of compounds 1–6 to M. tuberculosis DXR in general possesses similar thermodynamic features as observed for PfDXR. The ITC experiments using hydroxamate inhibitors 1–3 gave negative enthalpy change (ΔH = −24.9 kJ/mol for 1, −22.5 kJ/mol for 2, and −17.4 kJ/mol for 3), suggesting, as compared to EcDXR, the metal ion binding site of apo-MtDXR is also less hydrated. In addition, the binding of compounds 4–6 to MtDXR was also observed to be exothermic (ΔH = −19.3 kJ/mol for 4, −15.2 kJ/mol for 5, and −17.6 kJ/mol for 6).
Table 2. Thermodynamic Parameters for Inhibitor Binding to PfDXR and MtDXRa.
| enzyme | compd | ΔH (kJ/mol) | Ka (M–1) | ΔG (kJ/mol) | ΔS (J K–1 mol–1) | Ki (μM)b |
|---|---|---|---|---|---|---|
| PfDXR | 1 | –18.9 | 1.28 × 107 | –38.0 | 68.4 | 0.021 |
| 2 | –21.1 | 1.28 × 107 | –38.0 | 60.5 | 0.011 | |
| 3 | –19.0 | 3.13 × 106 | –34.7 | 56.2 | 0.015 | |
| 4 | –9.2 | 8.77 × 105 | –31.7 | 81.0 | 3.3 | |
| 5 | –12.4 | 4.67 × 105 | –30.3 | 64.2 | 1.1 | |
| 6 | –15.7 | 2.69 × 105 | –29.0 | 47.8 | 14.6 | |
| MtDXR | 1 | –24.9 | 3.45 × 106 | –34.9 | 36.0 | 0.14 |
| 2 | –22.5 | 1.79 × 106 | –33.4 | 39.0 | 0.13 | |
| 3 | –17.4 | 2.08 × 106 | –33.7 | 58.5 | 1.2 | |
| 4 | –19.3 | 6.29 × 105 | –31.0 | 41.9 | 1.6 | |
| 5 | –15.2 | 4.44 × 105 | –30.2 | 53.6 | 3.2 | |
| 6 | –17.6 | 3.42 × 105 | –29.6 | 42.7 | 21.8 |
With 2 mM Mn2+.
Ki values were measured using the enzyme activity inhibition assay.
Finally, we measured the enzyme inhibitory activities of compounds 1–6 against PfDXR. The Ki (inhibition constant) values as well as those against EcDXR and MtDXR from our previous work20 are shown in Tables 1 and 2. As illustrated in Figure 3, the Kd (dissociation constant, the inverse of Ka) values of these compounds for the three DXR enzymes measured by ITC are well correlated to the inhibition data (R2 = 0.75).
Figure 3.

Correlation between the Kd measured by ITC and the Ki obtained by enzyme inhibition assays.
In summary, this work represents the first thermodynamic investigation of the interactions between a series of structurally diverse DXR inhibitors and their target from three important human pathogens. First, the binding of the three hydroxamate inhibitors to E. coli DXR requires a divalent metal ion (Mg2+ or Mn2+) and is strongly endothermic and entropy-driven, while the binding of the three nonhydroxamate compounds is metal ion independent and exothermic. The unexpected thermodynamics of the hydroxamate ligands could be due to the hydration/dehydration of the metal ion pocket of EcDXR as well as the hydroxamate group of the inhibitors. Second, drastically different from EcDXR, the binding of the three hydroxamates to P. falciparum and M. tuberculosis DXRs is exothermic, suggesting a different hydration status between these enzymes, with PfDXR and MtDXR being considerably less hydrated at the metal ion binding site. Third, the binding affinities of these compounds measured by ITC are well correlated to the results from the enzyme kinetic studies.
Glossary
Abbreviations
- ITC
isothermal titration calorimetry
- DXR
1-deoxy-d-xylulose-5-phosphate reductoisomerase
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- EcDXR
Escherichia coli DXR
- PfDXR
Plasmodium falciparum DXR
- MtDXR
Mycobacterium tuberculosis DXR
Supporting Information Available
Supplementary Figure S1 and experimental section. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Parasitology, Wuhan University School of Basic Medical Science, Wuhan 430071, China.
This work was supported by a grant (R21AI088123) from the National Institute of Health (NIH) to Y.S. and a grant (R01AI32956) from NIH to T.P.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Thulasiram H. V.; Erickson H. K.; Poulter C. D. Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science 2007, 316, 73–76. [DOI] [PubMed] [Google Scholar]
- Hunter W. N. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J. Biol. Chem. 2007, 282, 21573–21577. [DOI] [PubMed] [Google Scholar]
- Nathan C. Antibiotics at the crossroads. Nature 2004, 431, 899–902. [DOI] [PubMed] [Google Scholar]
- Singh N.; Cheve G.; Avery M. A.; McCurdy C. R. Targeting the methyl erythritol phosphate (MEP) pathway for novel antimalarial, antibacterial and herbicidal drug discovery: Inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) enzyme. Curr. Pharm. Des. 2007, 13, 1161–1177. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Concepcion M. The MEP pathway: A new target for the development of herbicides, antibiotics and antimalarial drugs. Curr. Pharm. Des. 2004, 10, 2391–2400. [DOI] [PubMed] [Google Scholar]
- Testa C. A.; Brown M. J. The methylerythritol phosphate pathway and its significance as a novel drug target. Curr. Pharm. Biotechnol. 2003, 4, 248–259. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Shimizu T.; Takahashi S.; Seto H. Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. [Google Scholar]
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Turbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999, 285, 1573–1576. [DOI] [PubMed] [Google Scholar]
- Borrmann S.; Lundgren I.; Oyakhirome S.; Impouma B.; Matsiegui P. B.; Adegnika A. A.; Issifou S.; Kun J. F.; Hutchinson D.; Wiesner J.; Jomaa H.; Kremsner P. G. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob. Agents Chemother. 2006, 50, 2713–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima S.; Hara K.; Sanders J. M.; Yin F.; Ohsawa K.; Wiesner J.; Jomaa H.; Oldfield E. Crystallographic structures of two bisphosphonate:1-deoxyxylulose-5-phosphate reductoisomerase complexes. J. Am. Chem. Soc. 2004, 126, 10824–10825. [DOI] [PubMed] [Google Scholar]
- Silber K.; Heidler P.; Kurz T.; Klebe G. AFMoC enhances predictivity of 3D QSAR: A case study with DOXP-reductoisomerase. J. Med. Chem. 2005, 48, 3547–3563. [DOI] [PubMed] [Google Scholar]
- Merckle L.; de Andres-Gomez A.; Dick B.; Cox R. J.; Godfrey C. R. A fragment-based approach to understanding inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase. ChemBioChem 2005, 6, 1866–1874. [DOI] [PubMed] [Google Scholar]
- Kuntz L.; Tritsch D.; Grosdemange-Billiard C.; Hemmerlin A.; Willem A.; Bach T. J.; Rohmer M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: Phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reductoisomerase. Biochem. J. 2005, 386, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemers T.; Wiesner J.; Van Poecke S.; Goeman J.; Henschker D.; Beck E.; Jomaa H.; Van Calenbergh S. Synthesis of α-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1888–1891. [DOI] [PubMed] [Google Scholar]
- Woo Y. H.; Fernandes R. P.; Proteau P. J. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem. 2006, 14, 2375–2385. [DOI] [PubMed] [Google Scholar]
- Kurz T.; Schlüter K.; Pein M.; Behrendt C.; Bergmann B.; Walter R. D. Synthesis and antimalarial activity of chain substituted pivaloyloxymethyl ester analogues of Fosmidomycin and FR900098. Arch. Pharm. Chem. Life Sci. 2007, 340, 339–344. [Google Scholar]
- Munos J. W.; Pu X.; Liu H. W. Synthesis and analysis of a fluorinated product analogue as an inhibitor for 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem. Lett. 2008, 18, 3090–3094. [DOI] [PubMed] [Google Scholar]
- Deng L.; Sundriyal S.; Rubio V.; Shi Z.; Song Y. Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-d-xylulose-5-phosphate reductoisomerase. J. Med. Chem. 2009, 52, 6539–6542. [DOI] [PubMed] [Google Scholar]
- Deng L.; Endo K.; Kato M.; Cheng G.; Yajima S.; Song Y. Structures of 1-deoxy-d-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes. ACS Med. Chem. Lett. 2011, 2, 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Diao J.; Chen P.; Pujari V.; Yao Y.; Cheng G.; Crick D. C.; Prasad B. V. V; Song Y. Inhibition of 1-deoxy-d-xylulose-5-phosphate reductoisomerase by lipophilic phosphonates: SAR, QSAR, and crystallographic studies. J. Med. Chem. 2011, 54, 4721–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima S.; Nonaka T.; Kuzuyama T.; Seto H.; Ohsawa K. Crystal structure of 1-deoxy-D-xylulose 5-phosphate reductoisomerase complexed with cofactors: Implications of a flexible loop movement upon substrate binding. J. Biochem. 2002, 131, 313–317. [DOI] [PubMed] [Google Scholar]
- Steinbacher S.; Kaiser J.; Eisenreich W.; Huber R.; Bacher A.; Rohdich F. Structural Basis of Fosmidomycin Action Revealed by the Complex with 2-C-Methyl-D-erythritol 4-phosphate Synthase (IspC). J. Biol. Chem. 2003, 278, 18401–18407. [DOI] [PubMed] [Google Scholar]
- Mac Sweeney A.; Lange R.; Fernandes R. P.; Schulz H.; Dale G. E.; Douangamath A.; Proteau P. J.; Oefner C. The crystal structure of E.coli 1-deoxy-D-xylulose-5-phosphate reductoisomerase in a ternary complex with the antimalarial compound fosmidomycin and NADPH reveals a tight-binding closed enzyme conformation. J. Mol. Biol. 2005, 345, 115–127. [DOI] [PubMed] [Google Scholar]
- Yajima S.; Hara K.; Iino D.; Sasaki Y.; Kuzuyama T.; Ohsawa K.; Seto H. Structure of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in a quaternary complex with a magnesium ion, NADPH and the antimalarial drug fosmidomycin. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2007, 63, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladbury J. E.; Klebe G.; Freire E. Adding calorimetric data to decision making in lead discovery: A hot tip. Nat. Rev. Drug Discovery 2010, 9, 23–27. [DOI] [PubMed] [Google Scholar]
- Yin F.; Cao R.; Goddard A.; Zhang Y.; Oldfield E. Enthalpy versus entropy-driven binding of bisphosphonates to farnesyl diphosphate synthase. J. Am. Chem. Soc. 2006, 128, 3524–3525. [DOI] [PubMed] [Google Scholar]
- Freire E. Do enthalpy and entropy distinguish first in class from best in class?. Drug Discovery Today 2008, 13, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisch A. T.; Fox D. T.; Blagg B. S.; Poulter C. D. E. coli MEP synthase: Steady-state kinetic analysis and substrate binding. Biochemistry 2002, 41, 236–243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.