

Abstract
Thalamocortical neurons in dorsal lateral geniculate nucleus (dLGN) dynamically convey visual information from retina to the neocortex. Activation of metabotropic glutamate receptors (mGluRs) exerts multiple effects on neural integration in dLGN; however, their direct influence on the primary sensory input, namely retinogeniculate afferents, is unknown. In the present study, we found that pharmacological or synaptic activation of type 1 mGluRs (mGluR1s) significantly depresses glutamatergic retinogeniculate excitation in rat thalamocortical neurons. Pharmacological activation of mGluR1s attenuates excitatory synaptic responses in thalamocortical neurons at a magnitude sufficient to decrease suprathreshold output of these neurons. The reduction in both NMDA and AMPA receptor-dependent synaptic responses results from a presynaptic reduction in glutamate release from retinogeniculate terminals. The suppression of retinogeniculate synaptic transmission and dampening of thalamocortical output was mimicked by tetanic activation of retinogeniculate afferent in a frequency-dependent manner that activated mGluR1s. Retinogeniculate excitatory synaptic transmission was also suppressed by the glutamate transport blocker TBOA (dl-threo-β-benzyloxyaspartic acid), suggesting that mGluR1s were activated by glutamate spillover. The data indicate that presynaptic mGluR1 contributes to an activity-dependent mechanism that regulates retinogeniculate excitation and therefore plays a significant role in the thalamic gating of visual information.
Introduction
The lateral geniculate nucleus (LGN) is the primary thalamic relay that receives excitatory inputs from both the ascending retinal fibers and descending cortical fibers from layer 6 of the visual cortex (Jones, 1985; Sherman and Guillery, 1996, 2002). Retinogeniculate synapses, although making up a small number of excitatory synapses (5–10%), are powerful and effective in driving action potentials with precise timing to dynamically relay the visual information from retina to the cortex (Sherman and Guillery, 1996; Chen and Regehr, 2000; Augustinaite and Heggelund, 2007). Synaptic transmission at the retinogeniculate synapse is mediated via ionotropic glutamate receptors consisting of both AMPA receptors (AMPARs) and NMDA receptors (NMDARs) (Salt, 1986, 2002; Scharfman et al., 1990; Chen and Regehr, 2000; Kielland and Heggelund, 2002).
Metabotropic glutamate receptors (mGluRs) are a family of G-protein-coupled receptors involved in the modulation of synaptic transmission and neuronal excitability throughout the CNS (Nakanishi, 1994; Conn and Pin, 1997; Niswender and Conn, 2010). Glutamate acts on at least eight subtypes of mGluRs that are further classified into three groups based on sequence similarity, intracellular second messenger involvement, and agonist sensitivity (Nakanishi, 1992; Conn and Pin, 1997). Group I mGluRs consisting of mGluR1 and mGluR5 exhibit distinct distribution and physiological effects within the dLGN (Godwin et al., 1996a,b; Vidnyanszky et al., 1996; von Krosigk et al., 1999; Turner and Salt, 2000; Govindaiah and Cox, 2004, 2006b; Alexander and Godwin, 2005). It has been shown that corticothalamic stimulation activates mGluR1 on thalamocortical neurons and this mechanism serves to switch firing mode from “burst” to “tonic” firing via a postsynaptic depolarization of thalamocortical neurons (McCormick and von Krosigk, 1992; Turner and Salt, 1998, 2000). In contrast, stimulation of the retinogeniculate pathway does not produce an mGluR1-mediated depolarization of thalamocortical neurons (McCormick and von Krosigk, 1992; Turner and Salt, 1998; Govindaiah and Cox, 2006a). However, high-frequency retinogeniculate stimulation activates mGluR5 on interneuron dendrites leading to increased GABAergic inhibition onto thalamocortical neurons (Govindaiah and Cox, 2006b; Govindaiah et al., 2012). Recent studies have shown that activation of both mGluR1 and mGluR5 regulates GABAA receptor-mediated tonic inhibition (Errington et al., 2011). Furthermore, in vivo studies have shown that activation of mGluR5 modulates visual responses of dorsal LGN (dLGN) neurons (de Labra et al., 2005). These findings are consistent with the anatomical evidences showing the distribution mGluR5 on dendrites of interneurons and mGluR1 on dendrites of thalamocortical neurons (Godwin et al., 1996b; Vidnyanszky et al., 1996). Studies also have shown that mGluR1s are involved in generation of intrinsic slow oscillations found during wakeful state in thalamocortical neurons in vitro with identical properties to those observed in vivo (Hughes et al., 2002). In contrast to group I mGluRs, activation of group II and group III mGluRs dampen corticogeniculate synaptic transmission via presynaptic mechanisms (Turner and Salt, 1998, 1999; Alexander and Godwin, 2005).
It is unclear how diverse actions of mGluRs at different presynaptic and postsynaptic locations shape thalamocortical output. The present study was aimed at investigating the role of mGluRs on excitatory transmission at the retinogeniculate synapse and its influence on thalamocortical output. We found that the activation of mGluR1 in an activity-dependent manner abolishes action potential output in thalamocortical neurons via reduced afferent synaptic excitation mediated by NMDAR and AMPAR currents. Thus, presynaptic mGluR1 may act as low-pass filters to narrow the frequency range of information transmitted at the retinogeniculate synapse.
Materials and Methods
All experimental procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Illinois Animal Care and Use Committee. Care was taken to use the minimal number of animals necessary to complete this series of experiments, and animals were deeply anesthetized to prevent any possible suffering.
Brain slice preparation.
Thalamic slices were prepared from Sprague Dawley rats of either sex (postnatal age, 14–21 d) as previously described (Govindaiah and Cox, 2004). Briefly, animals were deeply anesthetized with sodium pentobarbital (50 mg/kg) and decapitated, and brains placed into cold (4°C), oxygenated (95% O2/5% CO2) slicing solution containing the following (in mm): 2.5 KCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 10 MgCl2, 2 CaCl2, 234 sucrose, and 11 glucose. Parasagittal or coronal slices (250–300 μm thickness) were cut at the level of dLGN using a vibrating tissue slicer. The slices were incubated in oxygenated (95% O2/5% CO2) artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, and 10 glucose at 32°C for at least 60 min before recording.
Whole-cell recording procedures.
Whole-cell recordings were obtained from dLGN neurons, as described previously (Govindaiah and Cox, 2004). Briefly, individual brain slices were transferred to a recording chamber that was maintained at 32°C and continuously perfused with ACSF (3.0 ml/min). Neurons were visualized using a Zeiss microscope equipped with differential interference contrast optics. Recording pipettes were pulled from borosilicate glass capillaries and filled with an intracellular solution containing the following (in mm): 117 K-gluconate, 13.0 KCl, 1 MgCl2, 0.07 CaCl2, 0.1 EGTA, 10 HEPES, 2 Na-ATP, and 0.4 Na-GTP. The pH and osmolality of internal solution were adjusted to 7.3 and 290 mOsm, respectively. Recordings were obtained using a Multiclamp 700B amplifier (Molecular Devices). Access resistance (<15 MΩ) was monitored continuously throughout the experiment, and neurons in which access resistance changed by >20% were discarded. Data were filtered at 2.5 kHz, digitized at 10 kHz, and analyzed using pCLAMP 9 (Molecular Devices) or MiniAnalysis (Synaptosoft) software. A 10 mV junction potential was subtracted for all voltage recordings.
Synaptic responses were evoked by electrical stimulation using a monopolar electrode placed in the optic tract (OT). Synaptic responses were evoked with various intensities (25–400 μA) and frequencies (0.5–200 Hz) at 5–10 s interstimulus intervals (ISIs). AMPAR-dependent EPSCs were evoked at a holding potential of −70 mV in the presence of the NMDAR antagonist d(−)4-(3-phosphonopropyl)piperazine-2-carboxylic acid (d-CPP) (10 μm) and GABAA receptor (GABAAR) antagonist 4-[6-imino-3-(4-methoxyphenyl)pyridazin-1-yl]butanoic acid hydrobromide (SR95531) (10 μm). NMDAR-dependent EPSCs were evoked at a holding potential of −40 mV in the presence of AMPAR antagonist DNQX (20 μm) and SR95531 (10 μm).
Concentrated stock solutions of pharmacological agents were prepared and stored as recommended by the manufacturer. Stock solutions were diluted in ACSF to final concentration just before use. Agonists were applied via a short bolus into the input line of the recording chamber using a syringe pump. After 5 min of baseline recording, the group I mGluR agonist (R,S)-3,5-dihydroxyphenylglycine (DHPG) or nonselective mGluR agonist (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD) was applied for 20–45 s. All antagonists were bath applied at least 5–10 min before agonist application. All compounds were purchased from Tocris or Sigma-Aldrich.
Data analysis.
Data were analyzed with pCLAMP 9 (Molecular Devices) software. Acute changes induced by mGluR agonists/antagonists application and synaptic stimulation were determined by averaging three to five consecutive responses obtained before, during, and 10 min following agonist application. Data are presented as mean ± SEM. Statistical significance was assessed using Student's t test, with a significance level of 0.05.
Results
In dLGN, activation of mGluRs exerts diverse physiological effects. In general, activation of group I mGluRs (mGluR1, mGluR5) produce excitatory effects on interneurons and thalamocortical neurons, respectively (Turner and Salt, 1998; Govindaiah and Cox, 2006), whereas activation of group II mGluRs hyperpolarizes GABAergic thalamic interneurons (Cox and Sherman, 1999; Govindaiah and Cox, 2006, 2009). Activation of mGluR1 produces membrane depolarization acting on thalamocortical neurons at corticothalamic synapses, whereas activation of mGluR5 increases GABAAR-mediated inhibition acting on dendrites of GABAergic interneurons (Govindaiah and Cox, 2006). We initially examined the effects of the general mGluR agonist ACPD on synaptic responses evoked by OT stimulation in dLGN thalamocortical neurons. At resting membrane potentials, suprathreshold OT stimulation (50–350 μA, 0.1 Hz) elicited action potential discharge in dLGN neurons (Fig. 1A,B). After obtaining stable evoked responses, short bath application of ACPD (75–100 μm; 20–30 s) produced a membrane depolarization that could lead to spontaneous action potential discharge (Fig. 1A). At the peak of the depolarization, the membrane potential was manually clamped back to baseline levels by current injection. Under this condition, OT stimulation no longer produced action potential discharge (Fig. 1A). Considering a general agonist ACPD could act on multiple receptor subtypes, the selective group I mGluR agonist DHPG was tested in subsets of neurons. Exposure to DHPG (25 μm) produced a similar membrane depolarization along with suppression of action potential discharge (Fig. 1B).
Figure 1.
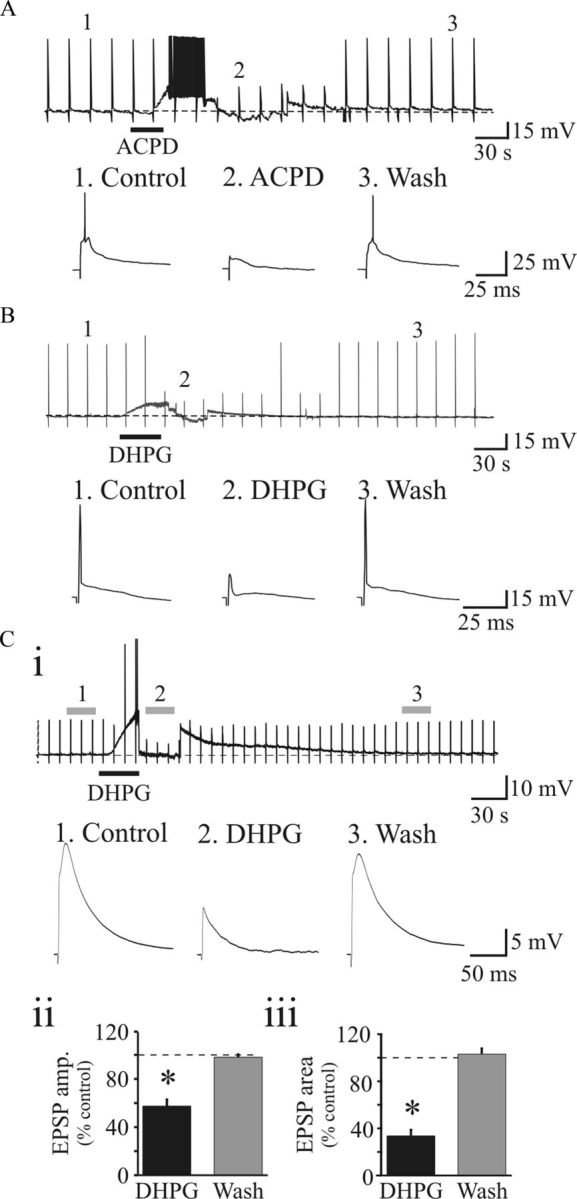
Activation of mGluRs dampens synaptic excitation of thalamocortical neurons. A, Example of current trace from dLGN thalamocortical neuron showing that suprathreshold OT stimulation (150 μA) elicits action potential discharge. After stable baseline (1), bath application of ACPD (100 μm, 25 s) produces membrane depolarization along with increase in action potential discharge. The membrane potential was clamped back to baseline levels by injecting DC current, and at this level, action potential firing is abolished (2). The suprathreshold response returns to control conditions following ACPD wash out (3). Vm = −69 mV. B, In a different neuron, DHPG (25 μm, 20 s) produces a membrane depolarization with inhibition of action potential discharge (1 vs 2) in a reversible manner (3). Vm = −69 mV. C, Activation of group I mGluRs suppresses EPSPs in dLGN neurons. Ci, Representative voltage traces from dLGN neuron showing EPSPs in response to OT stimulation (50 μA). Exposure to DHPG (25 μm, 20 s) produces a membrane depolarization along with suppression of the EPSP. Individual synaptic responses below illustrate EPSPs before DHPG (1), in DHPG clamped at resting membrane potential (2), and following DHPG wash out (3). Vm = −68 mV. Cii, Ciii, Histograms of EPSP amplitude (Cii) and area (Ciii) following DHPG exposure and washout. *p < 0.002. Error bars indicate SEM.
Considering the reduction in action potential output, we next investigated the actions of mGluRs on EPSPs evoked by subthreshold OT stimulation. Lower stimulus intensities (50–125 μA, 0.1 Hz) were used to evoke EPSPs in thalamocortical neurons (Fig. 1C). DHPG (25 μm) significantly reduced EPSP amplitude and area in 9 of 11 cells in a reversible manner that recovered to baseline levels (Fig. 1Ci). Overall, the EPSP amplitude was significantly reduced by 43.6 ± 5.7% (Fig. 1Cii; control, 17.0 ± 1.9 mV; DHPG, 9.7 ± 1.7 mV; n = 9; p < 0.001, paired t test), and EPSP area significantly reduced by 66.3 ± 5.2% (Fig. 1Ciii; n = 9; p < 0.002, paired t test).
Retinogeniculate transmission is mediated by both AMPAR and NMDARs (Salt, 1986; Scharfman et al., 1990; Chen et al., 2002). To examine whether activation of mGluRs differentially affects NMDAR and AMPAR currents, we tested the effects of mGluR agonists on pharmacologically isolated components of the synaptic response. AMPAR-mediated EPSCs were evoked at a holding potential of −70 mV in the presence of NMDAR antagonist d-CPP (10 μm) and GABAAR antagonist SR95531 (10 μm). The general mGluR agonist ACPD (100 μm) markedly depressed the AMPAR EPSC in a reversible manner (Fig. 2Ai). Overall, the AMPAR-dependent EPSC was reduced by 40.8 ± 4.3% (Fig. 2Aiii; n = 6; p < 0.001, paired t test). We next tested the effects of ACPD on NMDAR EPSCs, which were evoked at a holding potential of −40 mV in the presence of AMPAR antagonist DNQX (20 μm) and SR95531 (10 μm). Similar to the AMPAR EPSC, the NMDAR ESPC was significantly reduced by 47.6 ± 2.0% following ACPD application (Fig. 2Aii,iii; n = 8; p < 0.001, paired t test). Together, these data indicate that both AMPAR- and NMDAR-dependent synaptic currents are reduced by mGluR activation.
Figure 2.
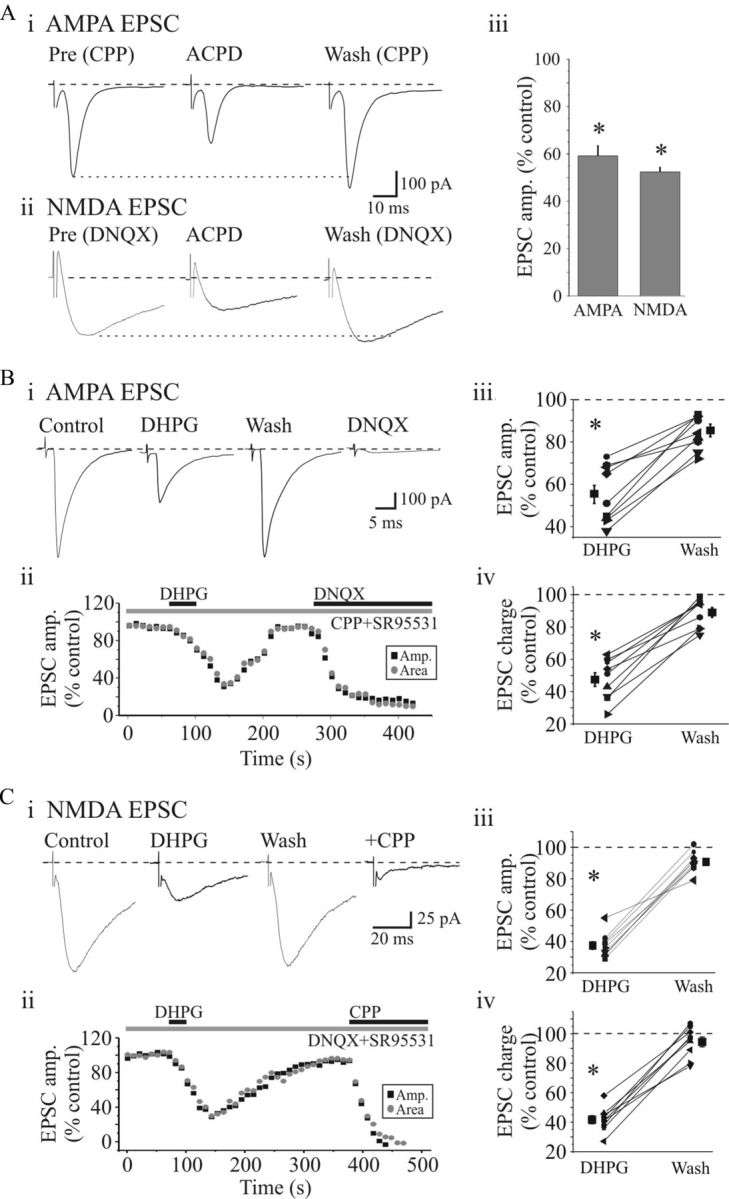
Activation of group I mGluRs suppress EPSCs in dLGN neurons. A, ACPD (100 μm, 20 s) markedly reduced the AMPAR (Ai) and NMDAR (Aii)-mediated EPSCs evoked by OT stimulation (0.1 Hz, 180 μA) in a reversible manner. In this and subsequent figures, individual traces are averages of five consecutive responses. Aiii, Histogram of population data showing that ACPD significantly attenuated both AMPAR- and NMDAR-mediated synaptic responses. *p < 0.001. Error bars indicate SEM. B, Group I mGluR agonist DHPG suppresses excitatory synaptic currents. Bi, Representative AMPAR-dependent EPSCs recorded before and following DHPG application. Bii, Time course of DHPG-induced attenuation of AMPAR EPSCs. DHPG produced a similar attenuation of EPSC amplitude (■) and charge ( ). Population data reveal a significant suppression of AMPAR EPSC amplitude (Biii) and charge (Biv) by DHPG that recovers near baseline level following washout. Ci, Representative NMDAR-dependent EPSCs recorded in presence of DNQX and SR95531. DHPG (25 μm) attenuates the NMDAR EPSC similar to that of the AMPAR EPSC. Cii, Time course of DHPG-mediated attenuation of NMDAR EPSC amplitude (■) and charge () that recovers to baseline levels following washout. NMDAR EPSCs were attenuated by d-CPP (10 μm). Population data show a significant attenuation of NMDAR EPSC amplitude (Ciii) and charge (Civ) by DHPG. *p < 0.001.
). Population data reveal a significant suppression of AMPAR EPSC amplitude (Biii) and charge (Biv) by DHPG that recovers near baseline level following washout. Ci, Representative NMDAR-dependent EPSCs recorded in presence of DNQX and SR95531. DHPG (25 μm) attenuates the NMDAR EPSC similar to that of the AMPAR EPSC. Cii, Time course of DHPG-mediated attenuation of NMDAR EPSC amplitude (■) and charge () that recovers to baseline levels following washout. NMDAR EPSCs were attenuated by d-CPP (10 μm). Population data show a significant attenuation of NMDAR EPSC amplitude (Ciii) and charge (Civ) by DHPG. *p < 0.001.
Activation of mGluR1 suppresses retinogeniculate transmission
Our initial data indicated that the group I mGluR agonist DHPG significantly suppressed excitatory synaptic responses (Fig. 1B,C). We next tested the actions of DHPG on isolated AMPAR- and NMDAR-mediated EPSCs evoked by OT stimulation. DHPG (25 μm) reversibly suppressed the AMPAR EPSC (Fig. 2Bi,ii). In our sample of cells (n = 9), DHPG significantly attenuated the AMPAR EPSC amplitude by 44.4 ± 4.1% (Fig. 2Biii; p < 0.0001, paired t test) and EPSC charge by 52.6 ± 3.2% (Fig. 2Biv; n = 9; p < 0.0001, paired t test). As with the AMPAR-dependent EPSC, DHPG also produced a strong depression of the NMDAR EPSC (Fig. 2Ci,ii). DHPG significantly reduced the NMDAR EPSC amplitude by 60.8 ± 1.6% (Fig. 2Ciii; n = 7; p < 0.0001, paired t test) and EPSC area by 58.3 ± 1.2% (Fig. 2Civ; n = 7; p < 0.0001, paired t test).
We have previously shown that activation of mGluRs can lead to increased GABAAR-mediated activity in thalamocortical neurons via presynaptic dendrites of intrinsic interneurons (Cox and Sherman, 2000; Govindaiah and Cox, 2006b). To examine whether the suppression of the excitatory synaptic response involves this inhibitory pathway, we tested the effect of DHPG in the presence of the GABAAR antagonist SR95531 on NMDAR EPSCs. In control conditions, DHPG (25 μm) produced a reversible suppression of the NMDAR EPSC. Subsequently, in the presence of SR95531 (10 μm), DHPG application still produced a similar suppression of the EPSC. Overall, the NMDAR ESPC was significantly suppressed by 47.3 ± 3.1% (n = 8; p < 0.0002, paired t test) in control conditions, which persisted in SR95531 (46.6 ± 4.3%; n = 8; p < 0.0002, paired t test).
Selective antagonists were used to determine whether the DHPG-mediated depression of ESPCs is via mGluR1 and/or mGluR5 receptor subtypes. Pharmacologically isolated NMDA EPSCs (see Materials and Methods) were evoked in the presence of the selective mGluR5 receptor antagonist 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP) (50 μm). Under these conditions, DHPG significantly depressed the NMDAR EPSC amplitude in a reversible manner (Fig. 3A; 50 ± 2.9% of control; n = 5). Following recovery, the selective mGluR1 antagonist (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385) (100 μm) was bath applied. Subsequent application of DHPG did not alter the NMDA EPSC (Fig. 3Ai–iii; 106 ± 4.4% of control; n = 5; p < 0.001, paired t test). In a different subset of cells, we tested the antagonistic effects of the mGluR5 antagonist MPEP. The DHPG-induced attenuation of NMDAR EPSC was unaltered in the presence of MPEP (Fig. 3Aiv; DHPG, 46.6 ± 2.6%; MPEP plus DHPG, 49.3 ± 4.2%; n = 6; p > 0.04, paired t test), further supporting the role of mGluR1 in suppressing retinogeniculate synaptic excitation. In addition, we tested the effect of the noncompetitive mGluR1 antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a- carboxylate ethyl ester (CPCCOEt) on DHPG-induced suppression of NMDAR EPSC. In control conditions, DHPG significantly suppressed NMDAR EPSC to 50.5 ± 1.6% (n = 5; p < 0.0001, paired t test). In the presence of CPCCOEt (150 μm), subsequent application of DHPG did not significantly alter the NMDA EPSC (Fig. 3B; 93 ± 4.1% of control; n = 5; p > 0.5, paired t test).
Figure 3.

Activation of mGluR1, but not mGluR5, depresses EPSCs in dLGN neurons. Ai, Sample current traces revealing that the DHPG-induced suppression of NMDAR EPSC is attenuated in the presence of selective mGluR1 antagonist LY367385 (100 μm). Aii, Time course of DHPG-induced depression of EPSC and antagonistic effect of LY367385. Aiii, Histogram of population data showing that the DHPG attenuates the NMDAR EPSC, which is completely blocked by LY367385 (n = 7). Aiv, Summary data indicating that suppression of NMDAR EPSC by DHPG is unaltered by the selective mGluR5 antagonist MPEP (50 μm; n = 5). *p > 0.001. Bi, Representative current traces showing suppression of NMDAR EPSCs by DHPG and antagonistic effect of a noncompetitive mGluR1 antagonist CPCCOEt (150 μm). Bii, Graph illustrating the time course of DHPG-induced depression and antagonistic effect of CPCCOEt. Biii, Histogram of population data depicting a significant suppression of NMDAR EPSC by DHPG; this effect is blocked in the presence of CPCCOEt (n = 5). *p > 0.5. Error bars indicate SEM.
We also tested the effect of LY367385 on the isolated AMPAR EPSC. Similar to that illustrated in Figure 2A, DHPG produced a reversible suppression of the AMPAR EPSC. Following recovery, LY367385 (100 μm) was bath applied, and subsequent DHPG application did not alter the EPSC amplitude. In the presence of LY367385, the DHPG-mediated suppression of the AMPAR EPSC amplitude was blocked (DHPG, 51 ± 4.3% of control; plus LY367385/DHPG, 102 ± 2.5% of control; n = 7; p < 0.0001, paired t test). To test the possible contribution of mGluR5, we tested whether the selective mGluR5 antagonist MPEP could alter the DHPG-mediated suppression. In control conditions, DHPG reduced the AMPA EPSC by 50.8 ± 4.5% (n = 5). In MPEP (50–75 μm), DHPG reduced the AMPA EPSC by 43.6 ± 2.4% (n = 5), which did not significantly differ from control conditions (p > 0.1, paired t test).
We initially found that DHPG could abolish suprathreshold retinogeniculate excitation of thalamocortical neurons (Fig. 1). Using the same stimulation paradigm, we found that DHPG dampened action potential output from OT stimulation in thalamocortical neurons (Fig. 4). Following recovery, the mGluR1 antagonist LY367385 (100 μm) was bath applied, and subsequent application of DHPG did not alter the suprathreshold excitation of the thalamocortical neurons (Fig. 4; n = 4).
Figure 4.

Activation of mGluR1 inhibits synaptic responses evoked by suprathreshold stimulation of OT. Top, Representative voltage trace recorded from dLGN neuron. Transient in trace are responses to suprathreshold OT stimulation (200 μA, 0.1 Hz). DHPG induces membrane depolarization along with suppression of synaptic responses. Following recovery, mGluR1 antagonist LY367385 (100 μm) was bath applied for 10 min, and subsequent DHPG application produces membrane depolarization but did not alter synaptic responses. Below are examples of individual synaptic responses before (1), during (2), and after DHPG exposure (3), and during DHPG in LY367385 (4).
Presynaptic mGluR1 regulates retinogeniculate transmission
To further determine whether the suppressive actions via mGluR1 activation results from presynaptic and/or postsynaptic mechanisms, we used multiple approaches: paired-pulse stimulation and recording of postsynaptic NMDA responses. The synaptic responses to paired-pulse optic tract stimulation were used to determine a paired-pulse ratio (PPR: EPSC2/EPSC1) (Zucker and Regehr, 2002; Chen and Regehr, 2003). AMPAR EPSCs were pharmacologically isolated and paired-pulse OT stimulation (50 ms ISI) resulted in paired-pulse depression (PPD) of the AMPAR EPSCs (Fig. 5A) (Turner and Salt, 1998). DHPG (25 μm) differentially suppressed EPSC1 and EPSC2 leading to an increase in PPR (Fig. 5Aii). In our sample of cells (n = 8), the PPR was significantly increased from 0.31 ± 0.01 to 0.54 ± 0.05 (Fig. 5Aiii; p < 0.002, paired t test). In a different subset of neurons, NMDAR EPSCs were evoked with 75 ms ISI, which produced robust paired-pulse facilitation (Fig. 5Bi). Subsequent application of DHPG strongly depressed EPSC1 and EPSC2 differentially resulting in a significant increase in PPR (Fig. 5Bii,iii; control, 1.61 ± 0.16; DHPG, 2.58 ± 0.25; n = 7; p < 0.001, paired t test).
Figure 5.

mGluR1-mediated depression of synaptic transmission is a presynaptic phenomenon. Ai, Paired-pulse stimulation of OT (50 ms ISI) resulted in paired-pulse depression of the AMPAR EPSCs in thalamocortical neurons. DHPG (25 μm) attenuates EPSC1 and EPSC2 to differing degrees. Aii, Time course of DHPG-induced depression on EPSC1 (■) and EPSC2 (). Aiii, Population data reveal that paired-pulse ratio (PPR = EPSC2/EPSC1) is significantly increased after DHPG application. Bi, In a different neuron, paired-pulse stimulation of OT (75 ms ISI) produces a facilitation of NMDAR EPSCs. DHPG (25 μm) depresses EPSC1 and EPSC2 to a different degree. Bii, Time course of DHPG-induced depression on EPSC1 (■) and EPSC2 (). Biii, Population data reveal that DHPG significantly increases the PPR. * p < 0.01.
Our findings indicate that mGluR1-induced suppression of retinogeniculate fast synaptic transmission results from presynaptic mechanisms. To directly test a potential postsynaptic mechanism, NMDA currents were elicited by focal application of NMDA (200 μm) via pressure ejection (2 psi, 10 ms) in the presence of 1 μm TTX. When applied at 10 s intervals, NMDA produced repeatable, consistent-amplitude, transient inward currents (Fig. 6A). DHPG (25 μm) application produced an inward current but did not attenuate the NMDAR currents. In fact, DHPG produced a significant facilitation of the NMDAR currents (Fig. 6B; control, 261 ± 52 pA; DHPG, 326 ± 66 pA; n = 6; p < 0.008, paired t test). Overall, our data indicate that the mGluR1-mediated suppression of retinogeniculate synaptic transmission occurs via presynaptic mechanisms.
Figure 6.
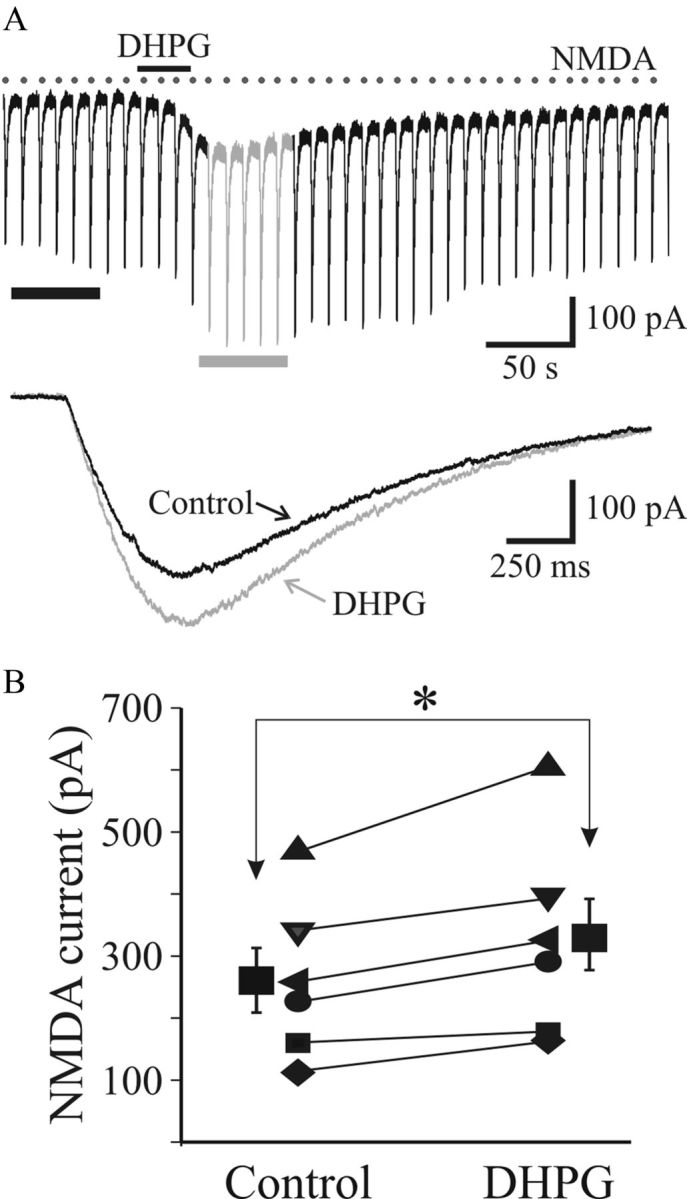
DHPG does not alter postsynaptic NMDA currents. A, Current trace revealing NMDAR-mediated currents elicited by repeated focal application of NMDA (200 μm) using pressure ejection via glass pipettes (2 psi, 10 ms, 0.1 Hz) in TTX (1 μm). Bottom, Average traces of five consecutive responses before (■) and following DHPG exposure (▩). B, Histogram illustrating that DHPG produces a significant increase in NMDA currents (n = 6). *p < 0.001.
Activity dependence of mGluR1-mediated suppression of retinogeniculate transmission
We next tested whether the mGluR1-mediated suppression of retinogeniculate excitation could be produced endogenously. By using trains of stimuli that closely resemble reported activity of retinal ganglion cell discharge, we determined whether this could engage the mGluR1-dependent suppression. After obtaining a stable synaptic response with single shock stimulation (0.05 Hz), tetanic stimulation was then applied to the OT (10–200 Hz, 10 pulses, 3–5 trains, 1 s interval, 100–350 μA), and we examined their effect on subsequent single shocks thereafter. Low-frequency tetanic stimulation (10 Hz) did not alter the synaptic responses (Fig. 7Ai). In contrast, high-frequency tetanic stimulation (50–200 Hz) resulted in a reversible suppression of the synaptic responses similar to the mGluR agonist (compare Figs. 1B, 7Aii). We next examined whether high-frequency tetanic stimulation could attenuate NMDAR EPSCs. After obtaining a stable baseline with single shock stimulation (0.1 Hz), tetanic stimulation of different frequencies (10–200 Hz, 10 pulses, 3–5 trains, 1 s interval) was applied. NMDAR EPSCs were attenuated in a frequency-dependent manner (Fig. 7B,C). During 10 Hz stimulation, the NMDAR EPSC charge was not significantly altered (92 ± 2.9% control; n = 4; p > 0.1, paired t test). However, at higher stimulus frequencies, the NMDAR EPSC was significantly attenuated (Fig. 7B,C; 50 Hz: 65.2 ± 3.4%, n = 5, p < 0.01; 200 Hz: 53.6 ± 5%, n = 8, p < 0.008, paired t tests). Considering that this tetanic stimulation-induced suppression of the EPSC could arise from multiple possible mechanisms, we next tested whether the suppression was dependent on mGluR1 activation. High-frequency tetanic stimulation (200 Hz, 10 pulses, 3–5 trains, 1 s interval) significantly suppressed the NMDAR EPSC (Fig. 7D; 61.7 ± 4.6% of control; n = 4; p < 0.008, paired t test). In the presence of selective mGluR1 antagonist CPCCOEt (150 μm), subsequent tetanic stimulation did not significantly alter the NMDAR EPSC (Fig. 7D,E; 91 ± 3.8% of control; n = 4; p > 0.05, paired t test). These data indicate that the high-frequency tetanic stimulation of OT could activate mGluR1 and thereby depresses AMPAR- and NMDAR-mediated synaptic transmission.
Figure 7.

Activation of mGluR1 regulates retinogeniculate transmission in a frequency-dependent manner. Ai, Representative voltage trace from relay neuron showing suprathreshold synaptic responses to OT stimulation (0.1 Hz, 300 μA). After obtaining a stable synaptic response with single shock stimulation (0.1 Hz), low-frequency trains were applied (300 μA, 10 Hz, 10 pulses, 3 trains, 1 s interval). Examples of traces before (control), immediately after 10 Hz stimulation (posttrain, 10 s after train), and 90 s after stimulation (recovery, 120 s after train) are shown. Low-frequency OT stimulation does not inhibit synaptic responses. Aii, Representative voltage traces before (control), immediately after high-frequency stimulation (200 Hz, 10 pulses, 3 trains, 1 s interval, posttrain, 10 s after train), and after recovery (recovery, 120 s posttrain). Note the suppression of suprathreshold output following the high-frequency OT stimulation. Bi, Examples of NMDAR-dependent EPSCs before and after high-frequency tetanic OT stimulation (200 Hz, 300 μA, 10 pulses, 3–5 trains, 1 s interval). Bii, Time course illustrating that high-frequency tetanic stimulation attenuates the NMDAR EPSC in a reversible manner. S, Single; T, tetanus. C, Population data indicating the frequency-dependent suppression of NMDAR EPSC (n = 8). D, Attenuation of NMDAR EPSC by high-frequency tetanic stimulation is mediated by mGluR1. Representative NMDAR EPSCs (Di) and plot of time course of the experiment (Dii) indicate that the mGluR1 antagonist CPCCOEt (150 μm) blocks the high-frequency (200 Hz) tetanic stimulation-induced depression of EPSC. E, Histogram of population data depicting the sensitivity of the tetanic-induced suppression of the NMDAR ESPC to the selective mGluR1 antagonist CPCCOEt (n = 4). *p < 0.05. Error bars indicate SEM.
We further hypothesized that the reduction of postsynaptic currents could be attributable to an activity-dependent activation of presynaptic mGluR1. Thus, we examined whether mGluR1 may be activated during high-frequency OT stimulation and determined the possible effects of blockade of mGluR1 on the short-term plasticity of AMPAR-mediated EPSCs at retinogeniculate synapses. AMPAR EPSCs were recorded by stimulating OT at different frequencies (5, 20, 100 Hz, 10 pulses) in control condition and in the presence of LY367385 (100 μm; n = 4) or CPCCOEt (150 μm; n = 1). After obtaining a stable baseline response with different frequencies, mGluR1 antagonist LY367385 or CPCCOEt was bath applied for 5–7 min. We found that the rate of synaptic depression at higher frequency (100 Hz) was markedly relieved by mGluR1 antagonists as the PPD was decreased in a frequency-dependent manner compared with control (Fig. 8Bi,iii). It is also evident that, at higher stimulus frequencies, there is an endogenous influence of mGluR1 activation of the frequency depression (Fig. 8, compare Aii, Bii). The PPD was not significantly altered during low-frequency stimulation (20 Hz, 10 pulses) (Fig. 8Ai,ii). Analysis of PPR (EPSC2/EPSC1) reveals that there was no significant alterations in PPR by mGluR1 antagonists at 20 Hz trains (control, 0.45 ± 0.12; LY/CPCCOEt, 0.50 ± 0.10; p > 0.3; n = 5; paired t test; Fig. 8Aiii). However, analysis of PPR revealed a significant increase in PPD in the presence of mGluR1 antagonists at 100 Hz (control, 0.36 ± 0.09; LY/CPCCOEt, 0.48 ± 0.10; p < 0.02; n = 5; paired t test; Fig. 8Biii). These data suggest that the activation of presynaptic mGluR1 decreases the probability of neurotransmitter release and limits the relative amount of neurotransmitter available for subsequent release.
Figure 8.

Blockade of mGluR1 reduces PPD during high-frequency stimulation. Ai, Representative current traces reveal marked reduction in second pulse and subsequent leveling of the EPSC amplitude with tetanic stimulation of OT (20 Hz, 75 μA, 10 pulses) in control (black) and in presence of mGluR1 antagonist LY367385 (100 μm, gray). Right, Examples of first two synaptic responses of the tetanus (EPSC1 and EPSC2) at expanded timescale. Synaptic responses were evoked in the presence of SR95531. Aii, Graph revealing frequency-dependent suppression of the EPSC (normalized to initial EPSC) before (control, black) and in the presence of LY367385 (gray). Aiii, Population data reveal no significant alterations in PPR by mGluR1 antagonists (n = 5; paired t test). Bi, Representative current traces in response to high-frequency stimulation (100 Hz, 75 μA, 10 pulses) before (black) and in presence of LY367385 (100 μm; gray). Expanded EPSC1 and EPSC2 are shown at right. Note the marked increase in PPR in LY367385. Bii, Graph illustrating the frequency-dependent suppression before (control, black) and in the presence of LY367385. Biii, Population data reveal a significant increase in PPR by mGluR1 antagonists (n = 5; paired t test). *p < 0.02.
Glutamate spillover activates mGluR1
Our results suggest that glutamate spillover might occur during higher frequency synaptic activity and thereby enable activation of extrasynaptic receptors. Spillover from synapses is usually restricted by the presence of powerful glutamate reuptake mechanisms (Arnth-Jensen et al., 2002). If the mGluRs are activated by accumulation of glutamate spillover, we speculate that blocking glutamate reuptake should mimic the actions of mGluR1 agonist by suppressing the synaptic response. We investigated whether blocking reuptake of glutamate by the inhibitor dl-threo-β-benzyloxyaspartic acid (dl-TBOA) would lead to activation of presumably extrasynaptic mGluR1. To test this, we examined the effects of DHPG and TBOA on the amplitudes of AMPA EPSC in the same neurons. DHPG (25 μm) reduced the EPSC amplitude to 59.8 ± 3.8% (p < 0.0008; n = 4; Fig. 9A) from the baseline in a reversible manner. After recovery, bath perfusion of TBOA (30 μm) suppressed the EPSC amplitude to 52.5 ± 3.3% (p < 0.0002; n = 4; Fig. 9A). We further examined whether the effects of TBOA are blocked in the presence of mGluR1 antagonists. In control conditions, TBOA reduced EPSC amplitude to 62 ± 3.2% (Fig. 9B; p < 0.0004; n = 9) and partially recovered (91 ± 2.7%; n = 6). In the presence of LY367385, the suppressive actions of TBOA on EPSC were partially but significantly blocked (87 ± 4.2%; p < 0.01; n = 5; Fig. 9B,C). These results indicate that blocking glutamate transporters increases spillover to activate extrasynaptic mGluR1 on retinal terminals.
Figure 9.

Glutamate spillover activates extrasynaptic mGluR1 at retinogeniculate synapses. Ai, Representative AMPAR-mediated EPSCs evoked by OT stimulation (0.1 Hz, 125 pA). Exposure to DHPG results in significant suppression of the EPSC that recovers following washout. Subsequent application of the glutamate uptake inhibitor dl-TBOA (30 μm) also reduces the EPSC in the same neurons. Aii, Histogram of population data illustrating that both DHPG (25 μm) and TBOA (30 μm) suppress the AMPAR EPSC amplitudes. *p < 0.0002. B, LY367385 blocks the suppressive actions of TBOA. Bi, Representative synaptic responses showing TBOA-induced suppression of EPSC that is subsequently blocked by mGluR1 antagonist LY367385. Bii, Time course of TBOA effects and partial antagonistic effects of LY367385 on the TBOA-induced suppression of the EPSC. C, Histogram of population data reveals that the suppressive actions of TBOA on EPSC is significantly reduced in the presence of LY367385 (n = 5). *p < 0.01. Error bars indicate SEM.
Discussion
In this study, we provide novel evidence that presynaptic mGluR1 regulates retinogeniculate excitation, which in turn strongly influences thalamocortical neuron output. Considering the retinogeniculate synapse serves as the primary visual input to the thalamus and subsequently primary visual cortex, presynaptic regulation of this afferent pathway can significantly shape visual information transfer. Our data demonstrate that pharmacological or endogenous activation of mGluR1 regulates fast glutamatergic synaptic excitation mediated by both NMDAR and AMPARs. While activation of mGluRs in thalamocortical circuits is more readily associated with top–down (corticothalamic) modulation, this presynaptic mechanism at the primary sensory input could significantly regulate thalamic gating.
Multiple mGluR subtypes are localized in the CNS and activation of these receptors is associated with multiple physiological effects (Nakanishi, 1992, 1994; Conn and Pin, 1997; Schoepp et al., 1999; Niswender and Conn, 2010). In thalamus, group I mGluRs (mGluR1 and mGluR5) are typically associated with excitation (Turner and Salt, 2000; Govindaiah and Cox, 2006b, 2009), whereas group II (mGluR2, mGluR3)/group III (mGluR4, mGluR7, mGluR8) mGluRs are associated with inhibitory processes (Salt and Eaton, 1991, 1994; Cox and Sherman, 1999; Turner and Salt, 1999; Alexander and Godwin, 2005; Govindaiah and Cox, 2006b). Furthermore, localization of specific mGluRs can lead to differential actions as well. For example, mGluR1 and mGluR5 are differentially distributed and exert distinct physiological effects in visual thalamus (Godwin et al., 1996a,b; Vidnyanszky et al., 1996; Rivadulla et al., 2002; de Labra et al., 2005; Govindaiah and Cox, 2006b). Previous anatomical studies have indicated that mGluR1s are localized postsynaptically on thalamocortical neurons and mGluR5 on intrinsic interneurons (Godwin et al., 1996b; Vidnyanszky et al., 1996; Govindaiah et al., 2012). Activation of mGluR1 elicits long-lasting depolarization in thalamocortical neurons resulting from corticothalamic excitation, but not retinogeniculate afferent stimulation (von Krosigk et al., 1993, 1999; Turner and Salt, 2000). In contrast, tetanic stimulation of retinogeniculate afferents leads to an increase in GABAergic inhibition via mGluR5 activation of presynaptic dendrites of intrinsic interneurons that subsequently innervate the thalamocortical neurons (Govindaiah and Cox, 2006b). In addition, activation of mGluR5, but not mGluR1, elicits membrane depolarizations in interneurons (Govindaiah and Cox, 2006b).
Our present data provide a novel role of mGluR1 in thalamic circuitry in that activation of these receptors dampens retinogeniculate excitation and subsequently influences output of thalamocortical neurons. An interesting and significant issue related to this finding is the source of glutamate that leads to this presynaptic mGluR1 activation. The suppressive actions of mGluR1 are mediated via decreased glutamate release from retinogeniculate afferents as revealed by paired-pulse stimulation paradigm. In the present study, the agonist DHPG significantly altered the paired-pulse ratio, consistent with the role of presynaptic mGluR1 in regulation of glutamate release from retinal terminals. In addition, DHPG did not suppress postsynaptic NMDA currents evoked by agonist application. Although group I mGluRs are usually associated with facilitation of glutamate release, activation of group I mGluRs has also been shown to decrease glutamate release in multiple brain regions (Gereau and Conn, 1995; Rodríguez-Moreno et al., 1998; Mannaioni et al., 2001; Watabe et al., 2002; White et al., 2003). Direct ultrastructural evidence of mGluR1 on presynaptic retinogeniculate terminals is lacking, but these cells are positive for mGluR1; however, it is not clear whether the receptors are only within the dendritic arbor and not in axonal terminals (Hartveit et al., 1995; Peng et al., 1995; Tehrani et al., 2000). The mechanisms underlying the presynaptic actions of mGluRs in inhibiting glutamate release and physiological implications are unclear. It has been hypothesized that the mGluRs are coupled to inhibitory pathway in which activation of mGluR1s inhibits glutamate release following desensitization (Rodríguez-Moreno et al., 1998). In this scheme, the mGluR is phosphorylated by protein kinase C to become coupled to the inhibitory pathway. As the mGluR becomes phosphorylated, the production of diacylglycerol decreases and results in subsequent dampening of presynaptic Ca2+ influx. Alternatively, it has been suggested that phosphorylated mGluRs become coupled to a pertussis toxin-resistant G-protein whose activation conveys the inhibition of glutamate release, probably by a block of presynaptic Ca2+ channels.
In the present study, we found suppression of glutamatergic transmission associated with high-frequency activation of retinogeniculate afferents. We speculate that the high-frequency afferent activity leads to spillover of glutamate that may in turn act on extrasynaptic mGluRs on retinal terminals. Our present data revealed the blockade of glutamate transporters with TBOA suppresses retinogeniculate signaling, supporting the notion that mGluR1s are activated by glutamate spillover. At hippocampal mossy fibers synapses, presynaptic mGluRs are not activated by low levels of glutamate release during low-frequency activity; however, when glutamate concentrations are increased by higher-frequency activity, these receptors become activated, leading to a rapid inhibition of transmitter release (Scanziani et al., 1997). Thus, the activity-dependent suppression of excitation found in the present study, at least in part, is due to the activation of extrasynaptic mGluR1 via spillover following strong afferent activation.
How presynaptic mGluR1 modulation contributes to the transfer of information from the retina to the cortex is an important issue. We hypothesize that the presynaptic mGluR1s are engaged during high-frequency activation of retinal afferents and may play crucial role in faithful information transfer to, and through the thalamus. Our data indicate that mGluR1 antagonists significantly relieved the paired-pulse depression during high-frequency stimulation of retinal afferents; indicate that these receptors are activated in an activity-dependent manner. Retinal ganglion cells in rodents have been shown to fire at rates ranging from 100 to 500 Hz in response to photostimulation (Stone and Pinto, 1993; Nirenberg and Meister, 1997). By effectively limiting the relay of visual information to those retinal ganglion cells that respond to a change in light stimuli with a robust discharge of action potentials, presynaptic mGluR1 modulation may be one of the mechanisms that dampens overexcitation of thalamocortical neurons. The result would be more faithful information transfer through the thalamus. This novel regulation of primary sensory information could have a significant influence on thalamic gating. Furthermore, given the complex actions of mGluRs within thalamic circuitry, it is important to understand the physiological conditions that give rise to differential mGluR-mediated actions and their subsequent influence on thalamocortical processing. Our data indicate that these mGluR1s represent efficient sensors of extracellular glutamate. Thus, mGluR1s are able to accommodate glutamate release to changes in the ambient glutamate concentration through a feedback mechanism. The latter effect may be particularly important in preventing excessive accumulation of extracellular glutamate as a result of repetitive retinogeniculate activity. Future studies at the circuit and systems level will be required to gain a complete understanding of the overall impact of mGluR1 activation in corticothalamic and retinogeniculate circuits.
Footnotes
This work was supported by National Institutes of Health Grants EY014024, MH085324, and HL086870.
References
- Alexander GM, Godwin DW. Presynaptic inhibition of corticothalamic feedback by metabotropic glutamate receptors. J Neurophysiol. 2005;94:163–175. doi: 10.1152/jn.01198.2004. [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- Augustinaite S, Heggelund P. Changes in firing pattern of lateral geniculate neurons caused by membrane potential dependent modulation of retinal input through NMDA receptors. J Physiol. 2007;582:297–315. doi: 10.1113/jphysiol.2007.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron. 2000;28:955–966. doi: 10.1016/s0896-6273(00)00166-5. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Presynaptic modulation of the retinogeniculate synapse. J Neurosci. 2003;23:3130–3135. doi: 10.1523/JNEUROSCI.23-08-03130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron. 2002;33:779–788. doi: 10.1016/s0896-6273(02)00611-6. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Cox CL, Sherman SM. Glutamate inhibits thalamic reticular neurons. J Neurosci. 1999;19:6694–6699. doi: 10.1523/JNEUROSCI.19-15-06694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Sherman SM. Control of dendritic outputs of inhibitory interneurons in the lateral geniculate nucleus. Neuron. 2000;27:597–610. doi: 10.1016/s0896-6273(00)00069-6. [DOI] [PubMed] [Google Scholar]
- de Labra C, Rivadulla C, Cudeiro J. Modulatory effects mediated by metabotropic glutamate receptor 5 on lateral geniculate nucleus relay cells. Eur J Neurosci. 2005;21:403–410. doi: 10.1111/j.1460-9568.2005.03847.x. [DOI] [PubMed] [Google Scholar]
- Errington AC, Di Giovanni G, Crunelli V, Cope DW. mGluR control of interneuron output regulates feedforward tonic GABAA inhibition in the visual thalamus. J Neurosci. 2011;31:8669–8680. doi: 10.1523/JNEUROSCI.0317-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gereau RW, 4th, Conn PJ. Multiple presynaptic metabotropic glutamate receptors modulate excitatory and inhibitory synaptic transmission in hippocampal area CA1. J Neurosci. 1995;15:6879–6889. doi: 10.1523/JNEUROSCI.15-10-06879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin DW, Vaughan JW, Sherman SM. Metabotropic glutamate receptors switch visual response mode of lateral geniculate nucleus cells from burst to tonic. J Neurophysiol. 1996a;76:1800–1816. doi: 10.1152/jn.1996.76.3.1800. [DOI] [PubMed] [Google Scholar]
- Godwin DW, Van Horn SC, Eriir A, Sesma M, Romano C, Sherman SM. Ultrastructural localization suggests that retinal and cortical inputs access different metabotropic glutamate receptors in the lateral geniculate nucleus. J Neurosci. 1996b;16:8181–8192. doi: 10.1523/JNEUROSCI.16-24-08181.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindaiah, Cox CL. Synaptic activation of metabotropic glutamate receptors regulates dendritic outputs of thalamic interneurons. Neuron. 2004;41:611–623. doi: 10.1016/s0896-6273(04)00013-3. [DOI] [PubMed] [Google Scholar]
- Govindaiah G, Cox CL. Excitatory actions of synaptically released catecholamines in the rat lateral geniculate nucleus. Neuroscience. 2006a;137:671–683. doi: 10.1016/j.neuroscience.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Govindaiah G, Cox CL. Metabotropic glutamate receptors differentially regulate GABAergic inhibition in thalamus. J Neurosci. 2006b;26:13443–13453. doi: 10.1523/JNEUROSCI.3578-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindaiah G, Cox CL. Distinct roles of metabotropic glutamate receptor activation on inhibitory signaling in the ventral lateral geniculate nucleus. J Neurophysiol. 2009;101:1761–1773. doi: 10.1152/jn.91107.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindaiah G, Venkitaramani DV, Chaki S, Cox CL. Spatially distinct actions of metabotropic glutamate receptor activation in dorsal lateral geniculate nucleus. J Neurophysiol. 2012;107:1157–1163. doi: 10.1152/jn.00401.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartveit E, Brandstätter JH, Enz R, Wässle H. Expression of the mRNA of seven metabotropic glutamate receptors (mGluR1 to 7) in the rat retina. An in situ hybridization study on tissue sections and isolated cells. Eur J Neurosci. 1995;7:1472–1483. doi: 10.1111/j.1460-9568.1995.tb01142.x. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Blethyn KL, Crunelli V. Cellular mechanisms of the slow (<1 Hz) oscillation in thalamocortical neurons in vitro. Neuron. 2002;33:947–958. doi: 10.1016/s0896-6273(02)00623-2. [DOI] [PubMed] [Google Scholar]
- Jones EG. The thalamus. New York: Plenum; 1985. [Google Scholar]
- Kielland A, Heggelund P. AMPA and NMDA currents show different short-term depression in the dorsal lateral geniculate nucleus of the rat. J Physiol. 2002;542:99–106. doi: 10.1113/jphysiol.2002.019240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci. 2001;21:5925–5934. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, von Krosigk M. Corticothalamic activation modulates thalamic firing through glutamate “metabotropic” receptors. Proc Natl Acad Sci U S A. 1992;89:2774–2778. doi: 10.1073/pnas.89.7.2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Nirenberg S, Meister M. The light response of retinal ganglion cells is truncated by a displaced amacrine circuit. Neuron. 1997;18:637–650. doi: 10.1016/s0896-6273(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YW, Blackstone CD, Huganir RL, Yau KW. Distribution of glutamate receptor subtypes in the vertebrate retina. Neuroscience. 1995;66:483–497. doi: 10.1016/0306-4522(94)00569-q. [DOI] [PubMed] [Google Scholar]
- Rivadulla C, Martínez LM, Varela C, Cudeiro J. Completing the corticofugal loop: a visual role for the corticogeniculate type 1 metabotropic glutamate receptor. J Neurosci. 2002;22:2956–2962. doi: 10.1523/JNEUROSCI.22-07-02956.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Sistiaga A, Lerma J, Sánchez-Prieto J. Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron. 1998;21:1477–1486. doi: 10.1016/s0896-6273(00)80665-0. [DOI] [PubMed] [Google Scholar]
- Salt TE. Mediation of thalamic sensory input by both NMDA receptors and non-NMDA receptors. Nature. 1986;322:263–265. doi: 10.1038/322263a0. [DOI] [PubMed] [Google Scholar]
- Salt TE. Glutamate receptor functions in sensory relay in the thalamus. Philos Trans R Soc Lond B Biol Sci. 2002;357:1759–1766. doi: 10.1098/rstb.2002.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salt TE, Eaton SA. Excitatory actions of the metabotropic excitatory amino acid receptor agonist, trans-(+/−)-1-amino-cyclopentane-1,3-dicarboxylate (t-ACPD), on rat thalamic neurons in vivo. Eur J Neurosci. 1991;3:1104–1111. doi: 10.1111/j.1460-9568.1991.tb00045.x. [DOI] [PubMed] [Google Scholar]
- Salt TE, Eaton SA. The function of metabotropic excitatory amino-acid receptors in synaptic transmission in the thalamus—studies with novel phenylglycine antagonists. Neurochem Int. 1994;24:451–458. doi: 10.1016/0197-0186(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Lu SM, Guido W, Adams PR, Sherman SM. N-Methyl-d-aspartate receptors contribute to excitatory postsynaptic potentials of cat lateral geniculate neurons recorded in thalamic slices. Proc Natl Acad Sci U S A. 1990;87:4548–4552. doi: 10.1073/pnas.87.12.4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. Functional organization of thalamocortical relays. J Neurophysiol. 1996;76:1367–1395. doi: 10.1152/jn.1996.76.3.1367. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery RW. The role of the thalamus in the flow of information to the cortex. Philos Trans R Soc Lond B Biol Sci. 2002;357:1695–1708. doi: 10.1098/rstb.2002.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone C, Pinto LH. Response properties of ganglion cells in the isolated mouse retina. Vis Neurosci. 1993;10:31–39. doi: 10.1017/s0952523800003205. [DOI] [PubMed] [Google Scholar]
- Tehrani A, Wheeler-Schilling TH, Guenther E. Coexpression patterns of mGluR mRNAs in rat retinal ganglion cells: a single-cell RT-PCR study. Invest Ophthal Vis Sci. 2000;41:314–319. [PubMed] [Google Scholar]
- Turner JP, Salt TE. Characterization of sensory and corticothalamic excitatory inputs to rat thalamocortical neurones in vitro. J Physiol. 1998;510:829–843. doi: 10.1111/j.1469-7793.1998.829bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol. 1999;519:481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Synaptic activation of the group I metabotropic glutamate receptor mGlu1 on the thalamocortical neurons of the rat dorsal lateral geniculate nucleus in vitro. Neuroscience. 2000;100:493–505. doi: 10.1016/s0306-4522(00)00280-3. [DOI] [PubMed] [Google Scholar]
- Vidnyanszky Z, Gorcs TJ, Negyessy L, Borostyankio Z, Knopfel T, Hamori J. Immunocytochemical visualization of the mGluR1a metabotropic glutamate receptor at synapses of corticothalamic terminals originating from area 17 of the rat. Eur J Neurosci. 1996;8:1061–1071. doi: 10.1111/j.1460-9568.1996.tb01273.x. [DOI] [PubMed] [Google Scholar]
- von Krosigk M, Bal T, McCormick DA. Cellular mechanisms of a synchronized oscillation in the thalamus. Science. 1993;261:361–364. doi: 10.1126/science.8392750. [DOI] [PubMed] [Google Scholar]
- von Krosigk M, Monckton JE, Reiner PB, McCormick DA. Dynamic properties of corticothalamic excitatory postsynaptic potentials and thalamic reticular inhibitory postsynaptic potentials in thalamocortical neurons of the guinea-pig dorsal lateral geniculate nucleus. Neuroscience. 1999;91:7–20. doi: 10.1016/s0306-4522(98)00557-0. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Carlisle HJ, O'Dell TJ. Postsynaptic induction and presynaptic expression of group 1 mGluR-dependent LTD in the hippocampal CA1 region. J Neurophysiol. 2002;87:1395–1403. doi: 10.1152/jn.00723.2001. [DOI] [PubMed] [Google Scholar]
- White AM, Kylänpää RA, Christie LA, McIntosh SJ, Irving AJ, Platt B. Presynaptic group I metabotropic glutamate receptors modulate synaptic transmission in the rat superior colliculus via 4-AP sensitive K+ channels. Br J Pharmacol. 2003;140:1421–1433. doi: 10.1038/sj.bjp.0705570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]