

Abstract
Cadmium has been widely used in industry and is known to be carcinogenic to humans. Although it is widely accepted that chronic exposure to cadmium increases the incidence of cancer, the mechanisms underlying cadmium-induced carcinogenesis are unclear. The main aim of this study was to investigate the role of reactive oxygen species (ROS) in cadmium-induced carcinogenesis and the signal transduction pathways involved. Chronic exposure of human bronchial epithelial BEAS-2B cells to cadmium induced cell transformation, as evidenced by anchorage-independent growth in soft agar and clonogenic assays. Chronic cadmium treatment also increased the potential of these cells to invade and migrate. Injection of cadmium-stimulated cells into nude mice resulted in the formation of tumors. In contrast, the cadmium-mediated increases in colony formation, cell invasion and migration were prevented by transfection with catalase, superoxide dismutase-1 (SOD1), or SOD2. In particular, chronic cadmium exposure led to activation of signaling cascades involving PI3K, AKT, GSK-3β, and β-catenin and transfection with each of the above antioxidant enzymes markedly inhibited cadmium-mediated activation of these signaling proteins. Inhibitors specific for AKT or β-catenin almost completely suppressed the cadmium-mediated increase in total and active β-catenin proteins and colony formation. Moreover, there was a marked induction of AKT, GSK-3β, β-catenin, and carcinogenic markers in tumor tissues formed in mice after injection with cadmium-stimulated cells. Collectively, our findings suggest a direct involvement of ROS in cadmium-induced carcinogenesis and implicate a role of AKT/GSK-3β/β-catenin signaling in this process.
Keywords: cadmium, carcinogenesis, ROS, GSK-3β, β-catenin
Introduction
Cadmium is widely distributed in the earth’s crust, air, and water and is categorized as a human and animal carcinogen (IARC, 1993). Major sources of cadmium exposure are food, cigarette smoke, and cadmium-related industries (Jarup, 2003). Cadmium has a very long biological half-life, resulting in accumulative toxic and carcinogenic effects. The lung is a primary target organ of exposure to cadmium because this metal is mainly absorbed through inhalation.
Carcinogenesis is a multistep process of tumor initiation, promotion, and progression, in which cancer cells arise from normal cells following the accumulation of mutations that result in the activation of proto-oncogenes and inactivation of tumor suppressor genes (Harris, 1991). Cadmium has the potential to affect all stages of the carcinogenic processes. Cadmium alters the cellular homeostasis of secondary messengers, such as reactive oxygen species (ROS) and Ca2+, and induces overexpression of proto-oncogenes. These properties are considered major factors in cadmium-induced cell transformation and tumorigenesis. Cadmium also activates gene expression of c-myc and c-Jun, inhibits tumor suppressor genes, such as p53 and p27, and accelerates the proliferation of cells that are stimulated with organic carcinogens (Fang et al., 2002; Jin and Ringertz, 1990).
A few studies suggest that cadmium-induced toxicity and carcinogenesis are associated with its potential to induce ROS production (Bagchi et al., 2000; Szuster-Ciesielska et al., 2000) but there is no direct evidence of a correlation between ROS and cadmium-induced carcinogenesis. Although cadmium is considered a redox-inactive metal that does not catalyze a Fenton-type reaction (Ercal et al., 2001; Stohs and Bagchi, 1995), it increases the generation of ROS by depleting cellular glutathione (GSH) and antioxidant enzymes, such as superoxide dismutase (SOD) and catalase (CAT) (Bagchi et al., 2000; Shaikh et al., 1999). Cadmium also induces ROS production by inhibiting the mitochondrial electron transfer chain (Wang et al., 2004). These findings suggest that cadmium induces carcinogenesis through the accumulation of intracellular ROS by disrupting cellular redox regulatory systems rather than by inducing ROS generation directly.
β-catenin signaling plays important roles in embryonic development and tissue homoeostasis, and also in carcinogenesis (Clevers, 2006; Lustig and Behrens, 2003). Phosphorylated β-catenin becomes multi-ubiquitinated and subsequently degraded in proteasomes (Lustig and Behrens, 2003). During this process, the tumor suppressor protein Axin acts as a scaffold by directly interacting with adenomatous polyposis coli (APC), glycogen synthase kinase-3β (GSK-3β), casein kinase 1-α (CK1-α), and β-catenin (Behrens, 2005). The serine/threonine kinase GSK-3β is constitutively active in unstimulated cells (Cohen and Frame, 2001). GSK-3 is a downstream effector of the PI3K/AKT pathway and its activity is inhibited by AKT-mediated phosphorylation at residue Ser 9 (Cross et al., 1995). GSK-3β also tightly regulates β-catenin signaling: phosphorylation of β-catenin by GSK-3β leads to ubiquitin-mediated degradation of β-catenin in proteasomes (MacDonald et al., 2009). Cadmium can induce β-catenin signaling (Chakraborty et al., 2010). Because β-catenin signaling is regulated by ROS in various types of cell (Heo and Lee, 2011; Ladelfa et al., 2011), it is likely that cadmium exerts its transformative and carcinogenic effects by increasing cellular ROS levels and activating β-catenin signaling. However, the mechanisms by which cadmium induces cell transformation and carcinogenesis remain unclear.
In this study, we examined the transformative and carcinogenic effects of cadmium in the human bronchial epithelial cell line BEAS-2B and an animal xenograft model. We also investigated the roles of ROS in cadmium-induced carcinogenesis and the signal transduction pathways involved. We demonstrate that cadmium exerts transformative and carcinogenic effects in epithelial cells through ROS-dependent signaling cascades of AKT/GSK-3 β/β-catenin.
Materials and methods
Chemicals and supplies
Unless specified otherwise, all chemicals and laboratory wares were purchased from Sigma Chemical Co. (St. Louis, MO) and Falcon Labware (Becton-Dickinson, Franklin Lakes, NJ). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), gentamicin, and L-glutamine were purchased from Gibco Co. (Gibco BRL, NY). The PI3 kinase inhibitor LY294002 was obtained from Cell Signaling (Beverly, MA). Inhibitors specific for GSK-3 (SB216763) and β-catenin (FH535) were purchased from Calbiochem (San Diego, CA).
Cell culture and treatment
The human bronchial epithelial cell line BEAS-2B was obtained from the American Type Culture Collection (Rockville, MD). Cells were maintained in DMEM supplemented with 10% heat-inactivated FBS and 1% penicillin-streptomycin. The cells were exposed continuously to cadmium (0–2 μM) in the growing medium. Cells were sub-cultured every week for 2 months and then processed for experiments.
Plasmids and transfection
CAT-Myc-DDK- and SOD1-Myc-DDK-tagged plasmids were purchased from Origene (Rockville, MD). SOD2-EGFP-tagged plasmid was from Addgene (Cambridge, MA). Transfections were performed using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Briefly, BEAS-2B cells were seeded in 6-well culture plates and transfected with 4 μg plasmid at approximately 50% confluency. Expression of CAT, SOD1, and SOD2 protein was measured by immunoblotting. The pooled stable transfectants cell lines were maintained using G418.
Anchorage-independent colony growth assays
Soft agar colony formation assay was performed as described previously (Huang et al., 1999). Briefly, 2.5 ml of 0.5% agar in DMEM supplemented with 10% FBS was spread onto each well of a 6-well culture plate. A 1 ml suspension containing 1 × 104 cadmium-exposed BEAS-2B cells was mixed with 2 ml of 0.5% agar-DMEM and layered on the top of the 0.5% agar layer. The plates were incubated at 37°C in 5% CO2 for 2 months and colonies larger than 50 μm in diameter were counted under a light microscope.
Clonal assay
The clonal assay was performed as described elsewhere (Plaisant et al., 2011). Cadmium-exposed cells were plated in 6-well plates at a density of 1 × 103 cells/well and incubated for 7 days with two media changes. The cells were fixed with 2% formalin for 10 min and stained with 0.5% crystal violet for 5 min prior to observation by light microscopy.
Cell migration assay
Cell migration was measured using transwell chambers (8-μm pore size, BD Biosciences) according to methods described previously (Meng et al., 2009). The top chambers of the transwells were loaded with 0.2 ml cells (5 × 105 cells) and the bottom chambers contained 0.7 ml complement medium. After incubation at 37°C and 5% CO2 for 12 h, the migrated cells were fixed, stained with hematoxylin solution, and counted under a microscope.
Cell invasion assay
The cell invasion assay was performed according to methods described previously (Qian et al., 2003). Cadmium-stimulated BEAS-2B cells (5 × 105 cells) were loaded into 24-well invasion chambers (BD Biosciences) that were pre-coated with 100 μl Matrigel solution. Medium containing 10% fetal calf serum was added to the plates (0.7 ml/well). The Matrigel invasion chambers were incubated for 3 days and the invaded cells were stained with hematoxylin and observed by microscopy.
Tumorigenicity assay
Athymic nude mice (NU/NU, 6–8 weeks old; Charles River) were housed in a pathogen-free room in the animal facilities at the Chandler Medical Center, University of Kentucky. All animals were handled according to the Institutional Animal Care and Use (IACUC). Cadmium-stimulated cells (1 × 106 cells) were resuspended in serum-free medium with Matrigel basement membrane matrix (BD Biosciences) at a 1:1 ratio (total volume = 100 μl) and subcutaneously injected into the flanks of nude mice (two sites/mouse). Body weight and tumor mass were measured every week for 4 months. Tumor volumes were determined using a caliper and calculated according to the following formula: (width2 × length)/2. Finally, tumor tissues were dissected, weighed, and fixed with formalin for immunohistochemical staining. The implantation site containing BD Matrigel plug, subcutaneous tissue, peritoneum, and skin was isolated to use as control tissue corresponding to the tumor tissue.
Western blot analyses
Cell lysates were prepared in lysis buffer (50 mM Tris-Cl, pH 7.4, 1 mM EDTA, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate, and 1 μg/ml aprotinin, leupeptin, and pepstatin). Equal amounts of protein (30 μg/sample) were separated by the NuPAGE Bis-Tris electrophoresis system (Invitrogen, Carlsbad, CA) and blotted onto nitrocellulose membrane (Whatman, Dassel, Germany). Blots were probed with primary and secondary antibodies before exposure to Hyperfilm (Amersham Pharmacia Biotech), and bands were quantified with ImageJ densitometry software (NIH, Bethesda, MD). Antibodies used were as follows: β-catenin (SC-70509), GSK-3β (SC-9166), AKT (SC-5298), and β-actin (SC-47778) from Santa Cruz Biotechnology (Santa Cruz, CA); phospho-GSK-3β (ser 9) (#9323) and phospho-AKT (ser 473) (#9271) from Cell Signaling (Beverly, MA); active-β-catenin (#05-665) from Millipore (Temecula, CA). Secondary antibodies and enhanced chemiluminescence substrate were from Pierce (Rockford, IL).
Immunohistochemical staining
Tumor tissues were fixed with 4% paraformaldehyde at room temperature for 24 h, paraffin-embedded, and sectioned (10 μm thickness). The slides were deparaffinized and processed for immunohistochemical staining according to the VECTASTAIN ABC KIT protocol (Vector Laboratories, Burlingame, CA). Briefly, the sections were incubated with 3% H2O2 in distilled water to block endogenous peroxidase activity. After antigen retrieval, the sections were blocked with normal serum for 20 min and then incubated with primary antibodies for 1 h. A negative control was provided by incubating sections with nonspecific mouse or rabbit serum IgG at the same dilution as for the primary antibodies. After washing with PBS, the sections were incubated with biotinylated-secondary antibodies for 30 min. The sections were then washed twice with PBS, incubated with ABC reagent for 30 min, and developed in DAB solution until the desired staining intensity was achieved.
Statistical analysis
All data are expressed as mean ± standard error (SE). One-way analysis of variance (ANOVA) using SPSS ver. 10.0 software was used for multiple comparisons. A value of P < 0.05 was considered statistically significant.
Results
Chronic exposure to cadmium induces carcinogenic properties in BEAS-2B cells
The cell transformation assay is used as a predictive tool for carcinogenicity (Barrett et al., 1984). An anchorage-independent colony formation assay was performed 2 months after stimulation with cadmium. Continuous exposure of BEAS-2B cells to cadmium induced dose-dependent transformation of these cells, as shown by the marked increase in size and number of colonies compared with the vehicle control (Figs. 1A and 1B). Continuous exposure to 1 μM and 2 μM cadmium increased the number of colonies to approximately 300/104 cells (P < 0.01) and 800/104 cells (P < 0.001), respectively, whereas few colonies were observed in the untreated control (Fig. 1B). Cadmium-stimulated clonogenicity was also demonstrated by a clonal assay in which cadmium treatment significantly increased the number of colonies in a dose-dependent manner (Figs. 2A and 2B).
Fig. 1.
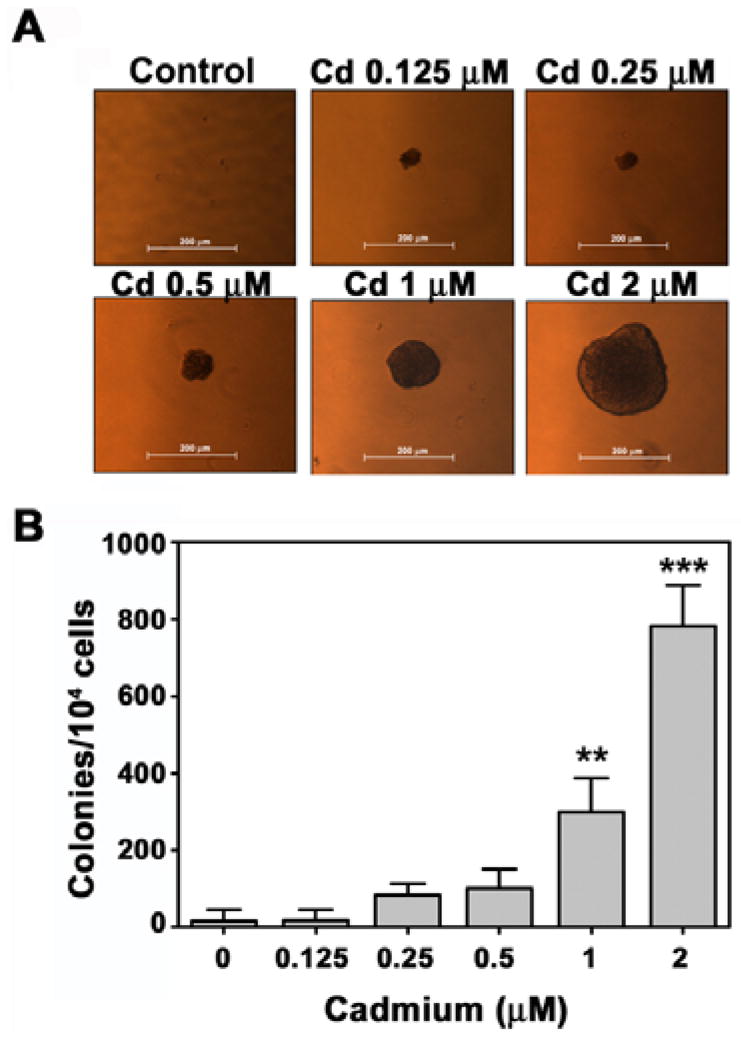
Cadmium induces cell transformation in BEAS-2B cells in a dose-dependent manner. After exposure of cells to increasing concentrations (0–2 μM) of cadmium for 2 months, the soft agar assay was performed as described in ‘Materials and Methods’. Representative images from three independent experiments are shown (A) and colonies larger than 50 μm were counted (B). **P < 0.01 and ***P < 0.001 vs. vehicle control.
Fig. 2.

Cadmium induces colony formation in BEAS-2B cells. Cells were stimulated with cadmium at the indicated concentrations for 2 months and then plated at clonal density and maintained in medium supplemented with10% FBS for 1 week. (A) Photographs of colony formation of cadmium-stimulated cells. (B) Graphs showing the number of colony-forming cells from three independent experiments. ***P < 0.001 vs. vehicle control.
Chronic cadmium stimulation increases cell migration and invasion in BEAS-2B cells
Cell migration is an important physiological process in tumor metastasis. We used transwell chambers to examine the effect of cadmium on migration of BEAS-2B cells. As shown in Fig. 3A, continuous cadmium stimulation stimulated migration of these cells in a dose-dependent manner. Treatment with 2 μM cadmium increased cell migration up to 3.5-fold, compared with the vehicle control (Fig. 3B). These results were consistent with those of a wound-healing assay (data not shown). Subsequently, we performed a Matrigel invasion assay to determine whether cadmium stimulation also increases the invasiveness of BEAS-2B cells. Microscopic observation of cadmium-stimulated cells revealed a dose-dependent increase in the level of cell invasion (Fig. 4A). Treatment with cadmium at 0.5, 1, and 2 μM concentrations increased cell invasiveness approximately 6-, 10-, and 17-fold respectively, compared with the vehicle control (Fig. 4B).
Fig. 3.

Cadmium increases the potential of cells to migrate. A cell migration assay was performed using transwell chambers as described in ‘Materials and Methods’. Panel ‘A’ shows a representative result from three independent experiments. The number of hematoxylin-stained cells was also determined (B). **P < 0.01 vs. vehicle control.
Fig. 4.

Cadmium increases cell invasion ability in BEAS-2B cells. A cell invasion assay was performed using transwell chambers. Cadmium-stimulated cells were loaded in the upper chambers and the lower chambers contained 0.7 ml of complement medium. After incubation for 3 days the invaded cells were stained with hematoxylin and visualized by microscopy (A). The number of stained cells was also counted (B). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control.
Cadmium increases tumor growth in a mouse xenograft model
The carcinogenic potential of cadmium in vivo was investigated by subcutaneous injection of nude mice with control cells or cadmium-stimulated cells. To avoid any effect of cell density on tumor formation, we injected each site with 1 × 106 cells. Over a 1-month post injection period there was no visible tumor formation in the vehicle control and there was no significant increase in tumor volume in the controls throughout the entire experimental period (Fig. 5A). However, mice injected with cadmium-stimulated cells showed obvious formation of tumors that increased in size according to the duration after injection. Fig. 5B indicates that gained body weight during the experimental period was not different between the control and experimental animal groups.
Fig. 5.

Cadmium increases tumor growth in a xenograft animal model. Cadmium-exposed BEAS-2B cells (1 × 106 cells/site) were injected subcutaneously into 6-week-old male athymic nude mice. Mice were checked daily for tumors, and tumor volume and body weight were measured every week. Tumor volume of animals is shown in (A) and body weight in (B).
Overexpression of antioxidant enzymes attenuates cadmium-induced carcinogenic potential in BEAS-2B cells
To investigate whether intracellular ROS are involved in cadmium-induced carcinogenesis, we generated BEAS-2B cells that overexpress CAT, SOD1, or SOD2 (Fig. 6A). The results from the soft agar assay showed that cadmium-stimulated formation of colonies was inhibited significantly by transfection with each of the antioxidant-specific vectors (Fig. 6B). This was in part consistent with results from the cell migration assay, in which transfection with SOD2, but not CAT or SOD1, significantly diminished the cadmium-stimulated increase in cell migration (Fig. 6C). Similarly, transfection with SOD1 or SOD2 attenuated the cadmium-mediated cell invasion: a greater than 60% reduction in the level of cell invasion was observed in the transfected cells compared with the positive controls (Fig. 6D). Overexpression of CAT did not suppress cadmium-stimulated cell invasion at a significant level.
Fig. 6.

Involvement of ROS in cadmium-induced tumorigenicity of BEAS-2B cells. CAT-Myc-DDK-, SOD1-Myc-DDK-, and SOD2-EGFP-tagged plasmids were transfected into BEAS-2B cells. (A) Overexpression of the proteins CAT, SOD1, and SOD2 was confirmed by Western blotting. The pooled stable transfectants were stimulated with cadmium for 2 months and soft agar (B), cell migration (C), and cell invasion (D) assays were performed in three independent experiments. Fold changes were calculated by the each stable transfected cells without cadmium exposure versus the same cells with cadmium exposure. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control. #P < 0.05 and ##P < 0.01 vs. cadmium-exposed vector control.
Cadmium activates ROS-dependent AKT/GSK-3β/β-catenin-mediated signaling in BEAS-2B cells
To elucidate the molecular mechanisms involved in cadmium-induced carcinogenesis we measured the protein levels of several protein kinases using Western blot analysis. The levels of phosphorylated PI3K and its downstream target AKT increased after stimulation with cadmium in a dose-dependent manner (Figs. 7A and B). The expression level of p-GSK-3β (Ser 9) also increased markedly in cadmium-exposed cells. Overall, the cellular levels of active and total β-catenin proteins increased in treated cells, and the highest level of these proteins was observed after treatment with 1 μM cadmium. The levels of p-PI3K, p-AKT, p-GSK-3β and active and total β-catenin were reduced by transfection with either CAT, SOD1, or SOD2 (Figs. 7C and D). Treatment with an AKT inhibitor, LY294002, or a β-catenin inhibitor, FH535, attenuated the cadmium-induced increase in the levels of active and total β-catenins (Figs. 7E and F). In contrast, addition of the GSK-3β inhibitor SB216763 facilitated the cadmium-stimulated induction of β-catenins. These findings were consistent with the results of the soft agar assay, which showed that inhibitors specific to AKT and β-catenin almost completely prevented formation of colonies whereas GSK-3β inhibitor accelerated colony formation (Fig. 7G). Western blot analysis also revealed that the levels of p-PI3K, p-AKT, p-GSK-3β and active β-catenin were higher in the tumor tissues formed at the injection site of the xenograft mice than in the corresponding tissues of the control mice (Fig. 8A). Similarly, high expression levels of carcinogenesis marker proteins, such as c-myc, cyclooxygenase-2 (COX-2), hypoxia-inducible factor-1α (HIF-1α, matrix metalloproteinase (MMPs), and vascular endothelial growth factor (VEGF), were detected in the tumor tissues compared with flank tissues of the control mice (Fig. 8B). Immunohistochemical staining confirmed that the levels of total and active β-catenin proteins were markedly higher in the tumor tissues than in the controls (Fig. 8C).
Fig. 7.

Cadmium leads to ROS-dependent activation of PI3K, AKT, GSK-3β and β-catenin signaling in BEAS-2B cells. Western blot analysis of protein levels of PI3K, AKT, GSK-3β and β-catenin in cells stimulated with increasing concentrations of cadmium (0–2 μM) (A), or after treatment of cells transfected with CAT, SOD1, or SOD2 with 2 μM cadmium for 2 months (C). Cadmium-stimulated BEAS-2B cells for 2 months were exposed to 2 μM cadmium in the presence of LY294002 (20 μM), SB216763 (10 μM), or FH535 (20 μM) for 48 h and the protein levels of total and active β-catenin forms were analyzed by Western blotting (E). In addition, colony formation ability of these stimulated cells was analyzed by soft agar assay with additional treatment of the inhibitors or cadmium (G).The band intensity of each Western results was expressed as the mean ± SE relative to the control of triplicate experiments and represented in (B), (D), and (F). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. vehicle control. #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. cadmium treatment alone.
Fig. 8.

Increased protein levels of carcinogenic markers in tumor tissues formed in mice injected with cadmium-stimulated BEAS-2B cells. Cells that had been exposed to cadmium for 2 months were subcutaneously injected into the flanks of nude mice (1 × 106 cells/site). After 4 months each tumor was dissected and tumor tissues were lysed for Western blot analysis (A and B) or fixed with paraformaldehyde for immunohistochemistry (C).
Discussion
Cadmium is a heavy toxic metal and group 1 human carcinogen (IARC, 1993) that causes human cancer through occupational and environmental exposure (Waalkes, 2000). The carcinogenic potential of cadmium and the mechanisms involved have been widely studied using in vitro and in vivo systems (Waalkes, 2000; Waalkes et al., 1999). However, although the carcinogenic property of cadmium is well recognized, the precise mechanisms underlying its carcinogenic activity are barely understood. In the present study, we demonstrate that cadmium induces cell transformation and carcinogenesis in human bronchial epithelial cells via ROS-dependent activation of PI3K/AKT/GSK-3β/β-catenin signaling.
Epidemiological studies suggest that occupational exposure to cadmium induces lung cancer (IARC, 1993; Waalkes, 2000). Animal studies have shown that chronic inhalation of cadmium causes pulmonary adenocarcinomas (Waalkes, 2000, 2003). However, most previous studies described the potential of cadmium to cause carcinogenic effects using short-term exposure to this toxic metal at high concentrations. To mimic conditions more similar to occupational and biologically relevant cadmium exposure (Glahn et al., 2008; Henson and Chedrese, 2004; Jin and Ringertz, 1990), this study exposed human bronchial epithelial cells to cadmium at concentrations of 0–2 μM for 2 months. Long-term exposure to low doses of cadmium seems to induce pre-neoplastic cells: the number of colonies formed in soft agar was significantly higher in cadmium-stimulated cells than in unstimulated controls (Fig. 1). This was consistent with the results of the clonal assay, in which chronic exposure of cells to cadmium increased the number of colonies in a dose-dependent manner (Fig. 2). Exposure of BEAS-2B cells to cadmium at concentrations greater than 2 μM exerted a cytotoxic effect such that viable cells were rarely found during the culture period (data not shown). Moreover, there was no prominent increase in colony formation in the soft agar assay unless the cells were exposed to cadmium for more than 1 month. Based on these observations, we used a maximum cadmium concentration of 2 αM and an exposure period of up to 2 months.
Cell migration and invasion are hallmark characteristics of tumor metastasis. Chronic stimulation of BEAS-2B cells with cadmium resulted in a marked increase in cell migration and invasion compared with unstimulated cells (Figs. 3 and 4). We also found that the expression level of MMPs and VEGF in tumor tissues formed in the xenograft mice were higher than in tissues of control mice. In addition to these proteins, expression of other markers of carcinogenesis including c-myc, COX-2, and HIF-1α was markedly induced in the tumor tissues. These data suggest that cadmium has a potential to induce metastasis. Injection of mice with cadmium-stimulated cells induced formation of tumors that gradually increased in volume, in contrast to the control mice (Fig. 5). Additional experiments revealed that these tumors were not aggressive, compared with the tumors xenografts generated by injection of transformed cells (data not shown). Notwithstanding, our present findings suggest that cadmium not only induces malignant transformation in vitro, but also induces tumor formation in vivo.
Although cadmium is not a direct mutagenic inducer, proposed mechanisms of cadmium-induced carcinogenesis include formation of ROS, alteration of antioxidant enzymes, inhibition of DNA repair enzymes, and an imbalance between pro- and anti-apoptotic proteins (Arita and Costa, 2009; Waisberg et al., 2003). Over the last decade many investigators have highlighted the involvement of ROS-stimulated signaling in metal-induced carcinogenesis. Unlike general concepts on the relationship between metals and ROS, a direct role of ROS is not considered likely in cadmium-induced carcinogenesis because cadmium does not participate in Fenton-type chemical reactions. However, metals can cause oxidative stress by interaction with cellular reductants or by inhibiting the activity of cellular antioxidants, and there is considerable evidence that cadmium induces the generation of hydroxyl radicals (•OH), superoxide anion (O2•−), nitric oxide (NO), and hydrogen peroxide (H2O2) (Galan et al., 2001; O’Brien and Salacinski, 1998; Son et al., 2010; Stohs et al., 2001). To verify the involvement of ROS in cadmium-induced carcinogenesis, we constructed cells that overexpress antioxidant enzymes (CAT, SOD1, or SOD2) (Fig. 6). As evidenced by the inhibition of colony formation and cell migration and invasion, overexpression of each of the antioxidant enzymes prevented cadmium-stimulated carcinogenesis. In particular, transfection with SOD2 showed the most prominent suppression of cadmium-induced carcinogenic effects. These results provide the first direct evidence of the involvement of ROS in cadmium-induced carcinogenesis. Interestingly, however, injection of cells that overexpress antioxidant enzymes also stimulated formation of tumors in mice (data not shown). Although we cannot fully explain the mechanisms involved, this appears to be related to the difference in ROS formation between in vitro and in vivo conditions or the nature of the cells; i.e., normal vs. tumor. It is likely that ROS are involved in initiation, rather than other stages in the process of cadmium-induced carcinogenesis. Our research group recently reported that ROS levels are lower in metal-transformed cells than in normal cells (Chang et al., 2010). Although it is commonly accepted that intracellular ROS levels are higher in cancer cells than in normal cells (Trachootham et al., 2009), some studies report the opposite phenomenon where ROS levels seem to be lower in cancer cells than in normal cells (Diehn et al., 2009; Jang and Sharkis, 2007). This led us to postulate that ROS are required for the initiation step of carcinogenesis in cadmium-exposed cells, but not for the processes of promotion or progression. Further detailed studies are needed to elucidate the molecular mechanisms involved in cadmium-induced carcinogenesis.
To determine the signal transduction pathways underlying cadmium-induced cell transformation and carcinogenesis, we examined the role of the Wnt/β-catenin pathway. The Wnt/β-catenin pathway is required for the development of leukemia stem cells (Wang et al., 2010), colon carcinomas (Behrens, 2005; Lustig and Behrens, 2003; Zhang et al., 2011), and other cancer types (Klaus and Birchmeier, 2008; Lustig and Behrens, 2003). It has also been reported that β-catenin regulates proto-oncogenes including c-myc, cyclin D1, and ABCB1 (Chakraborty et al., 2010), VEGF (Zhang et al., 2001), COX-2 (Howe et al., 2001), and MMPs (Marchenko et al., 2002). β-catenin is tightly regulated by the Axin-APC-GSK-3β complex; phosphorylation of β-catenin by GSK-3β leads to its degradation through a ubiquitin-proteasome pathway (Lustig and Behrens, 2003) (Nakamura et al., 1998). Our present findings revealed that levels of both total and active β-catenin (dephosphorylated β-catenin) were markedly increased in cadmium-stimulated cells, as well as in the tumor tissues of mice injected with cadmium-stimulated cells (Figs. 7 and 8). These results suggest that β-catenin regulates the expression of target genes involved in cell transformation and carcinogenesis. Further, β-catenin stabilization seems to be regulated by GSK-3β in cadmium-induced carcinogenesis, based on the increase in p-GSK-3β levels in cadmium-stimulated cells and in xenograft mice injected with these cells. This is consistent with the increased phosphorylation of PI3K and AKT in cadmium-stimulated cells. It is likely that phosphorylation of GSK-3β at Ser 9 by AKT inhibits GSK-3β activity and leads to β-catenin stabilization during cadmium-induced carcinogenesis.
To clarify the roles of ROS in signal transduction pathways involved in cadmium-induced carcinogenesis, cells expressing antioxidant enzymes were stimulated with 2 μM cadmium. Transfection with CAT, SOD1, or SOD2 suppressed the phosphorylation levels of signaling molecules, and also reduced total and active forms of β-catenin proteins. Similarly, pharmacological inhibitors of AKT and β-catenin not only attenuated the induction of total and active β-catenin forms, but also prevented the increase in the number of colonies after cadmium exposure. In contrast, blockage of GSK-3β activation by SB216763 enhanced colony formation in cadmium-exposed cells. These results strongly suggest that ROS activate PI3K/AKT/GSK-3β/β-catenin signaling in cadmium-induced carcinogenesis; thus, efficient regulation of this signaling may control the carcinogenic processes. Our results also indicate that cadmium-mediated activation of β-catenin signaling leads to increased expression of c-myc, COX-2, HIF-1α, MMPs, and VEGF during carcinogenesis. Our findings are partially consistent with Jing et al.’s results (Jing et al., 2012). They observed that chronic exposure of human lung epithelial cells to cadmium increased HIF-1α and VEGF expression through an ROS dependent pathway and induced malignant transformation. However, in the present study, we have provided further evidence of the involvement of ROS signaling and its down steam cascades in cadmium-induced carcinogenesis. Taken together, the current findings demonstrate that PI3K/AKT/GSK-3β/β-catenin signaling is a critical signaling pathway in cadmium-induced carcinogenesis, and that intracellular ROS play important roles in the activation of this pathway (Fig. 9).
Fig. 9.
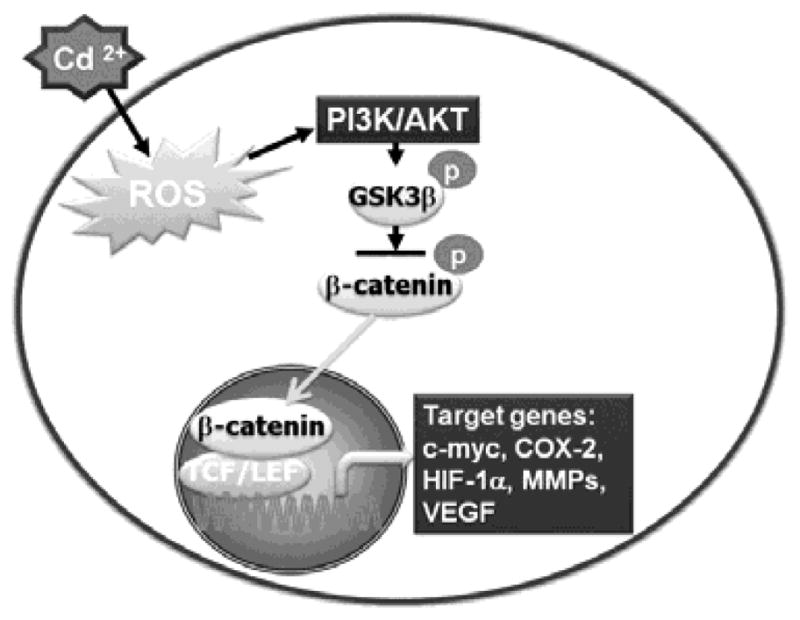
Proposed model of cadmium-induced carcinogenesis. Cadmium treatment of BEAS-2B cells induces ROS that activate PI3K/AKT signaling, leading to phosphorylation of GSK-3β. Phosphorylated GSK-3β dephosphorylates β-catenin and prevents its degradation. This results in translocation of β-catenin into the nucleus, leading to expression of target genes such as c-myc, COX-2, HIF-1α, MMPs, and VEGF.
In summary, the results obtained from the present study demonstrate for the first time that ROS play important roles in the activation of signaling cascades involving PI3K/AKT/GSK-3β/β-catenin in cadmium-mediated transformation of epithelial cells. Increases in total and active forms of β-catenin stimulate the expression of target genes, such as c-myc, COX-2, HIF-1α, MMPs, and VEGF, which might be critical events in cadmium-induced carcinogenesis. Collectively, our results implicate ROS and ROS-dependent signaling molecules as potential therapeutic targets in cadmium-stimulated carcinogenesis.
Highlights.
Chronic exposure to cadmium induces carcinogenic properties in BEAS-2B cells.
ROS involved in cadmium-induced tumorigenicity of BEAS-2B cells.
Cadmium activates ROS-dependent AKT/GSK-3β/β-catenin-mediated signaling in BEAS-2B cells.
ROS and ROS-dependent signaling molecules as potential therapeutic targets in cadmium-stimulated carcinogenesis.
Acknowledgments
This work was supported by grants from National Institutes of Health (R01ES015518, 1R01CA119028, and 1R01CA116697).
Abbreviations
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- CAT
catalase
- GSH
glutathione
- APC
adenomatous polyposis coli
- GSK-3β
glycogen synthase kinase-3β
- CK1-α
casein kinase 1-α
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- IACUC
Institutional Animal Care and Use
- COX-2
cyclooxygenase-2
- HIF-1α
hypoxia-inducible factor-1α
- MMPs
matrix metalloproteinase
- VEGF
vascular endothelial growth factor
- •OH
hydroxyl radicals
- O2•−
superoxide anion
- NO
nitric oxide
- H2O2
hydrogen peroxide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arita A, Costa M. Epigenetics in metal carcinogenesis: nickel, arsenic, chromium and cadmium. Metallomics. 2009;1:222–228. doi: 10.1039/b903049b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagchi D, Joshi SS, Bagchi M, Balmoori J, Benner EJ, Kuszynski CA, Stohs SJ. Cadmium- and chromium-induced oxidative stress, DNA damage, and apoptotic cell death in cultured human chronic myelogenous leukemic K562 cells, promyelocytic leukemic HL-60 cells, and normal human peripheral blood mononuclear cells. J Biochem Mol Toxicol. 2000;14:33–41. doi: 10.1002/(sici)1099-0461(2000)14:1<33::aid-jbt5>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Hesterberg TW, Thomassen DG. Use of cell transformation systems for carcinogenicity testing and mechanistic studies of carcinogenesis. Pharmacol Rev. 1984;36:53S–70S. [PubMed] [Google Scholar]
- Behrens J. The role of the Wnt signalling pathway in colorectal tumorigenesis. Biochem Soc Trans. 2005;33:672–675. doi: 10.1042/BST0330672. [DOI] [PubMed] [Google Scholar]
- Chakraborty PK, Lee WK, Molitor M, Wolff NA, Thevenod F. Cadmium induces Wnt signaling to upregulate proliferation and survival genes in sub-confluent kidney proximal tubule cells. Mol Cancer. 2010;9:102. doi: 10.1186/1476-4598-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Pan J, Wang X, Zhang Z, Chen F, Shi X. Reduced reactive oxygen species-generating capacity contributes to the enhanced cell growth of arsenic-transformed epithelial cells. Cancer Res. 2010;70:5127–5135. doi: 10.1158/0008-5472.CAN-10-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercal N, Gurer-Orhan H, Aykin-Burns N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Curr Top Med Chem. 2001;1:529–539. doi: 10.2174/1568026013394831. [DOI] [PubMed] [Google Scholar]
- Fang MZ, Mar W, Cho MH. Cadmium affects genes involved in growth regulation during two-stage transformation of Balb/3T3 cells. Toxicology. 2002;177:253–265. doi: 10.1016/s0300-483x(02)00229-9. [DOI] [PubMed] [Google Scholar]
- Galan A, Garcia-Bermejo L, Troyano A, Vilaboa NE, Fernandez C, de Blas E, Aller P. The role of intracellular oxidation in death induction (apoptosis and necrosis) in human promonocytic cells treated with stress inducers (cadmium, heat, X-rays) Eur J Cell Biol. 2001;80:312–320. doi: 10.1078/0171-9335-00159. [DOI] [PubMed] [Google Scholar]
- Glahn F, Schmidt-Heck W, Zellmer S, Guthke R, Wiese J, Golka K, Hergenroder R, Degen GH, Lehmann T, Hermes M, Schormann W, Brulport M, Bauer A, Bedawy E, Gebhardt R, Hengstler JG, Foth H. Cadmium, cobalt and lead cause stress response, cell cycle deregulation and increased steroid as well as xenobiotic metabolism in primary normal human bronchial epithelial cells which is coordinated by at least nine transcription factors. Arch Toxicol. 2008;82:513–524. doi: 10.1007/s00204-008-0331-9. [DOI] [PubMed] [Google Scholar]
- Harris CC. Chemical and physical carcinogenesis: advances and perspectives for the 1990s. Cancer Res. 1991;51:5023s–5044s. [PubMed] [Google Scholar]
- Henson MC, Chedrese PJ. Endocrine disruption by cadmium, a common environmental toxicant with paradoxical effects on reproduction. Exp Biol Med (Maywood) 2004;229:383–392. doi: 10.1177/153537020422900506. [DOI] [PubMed] [Google Scholar]
- Heo JS, Lee JC. beta-Catenin mediates cyclic strain-stimulated cardiomyogenesis in mouse embryonic stem cells through ROS-dependent and integrin-mediated PI3K/Akt pathways. J Cell Biochem. 2011;112:1880–1889. doi: 10.1002/jcb.23108. [DOI] [PubMed] [Google Scholar]
- Howe LR, Crawford HC, Subbaramaiah K, Hassell JA, Dannenberg AJ, Brown AM. PEA3 is up-regulated in response to Wnt1 and activates the expression of cyclooxygenase-2. J Biol Chem. 2001;276:20108–20115. doi: 10.1074/jbc.M010692200. [DOI] [PubMed] [Google Scholar]
- Huang C, Li J, Ma WY, Dong Z. JNK activation is required for JB6 cell transformation induced by tumor necrosis factor-alpha but not by 12-O-tetradecanoylphorbol-13-acetate. J Biol Chem. 1999;274:29672–29676. doi: 10.1074/jbc.274.42.29672. [DOI] [PubMed] [Google Scholar]
- IARC. Beyllium, cadmium, Hg, and exposure in the glass manufacturing industry. IARC Monogr Eval Carcinogen Risks Hum. 1993;58:119–237. [PMC free article] [PubMed] [Google Scholar]
- Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110:3056–3063. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarup L. Hazards of heavy metal contamination. Br Med Bull. 2003;68:167–182. doi: 10.1093/bmb/ldg032. [DOI] [PubMed] [Google Scholar]
- Jin P, Ringertz NR. Cadmium induces transcription of proto-oncogenes c-jun and c-myc in rat L6 myoblasts. J Biol Chem. 1990;265:14061–14064. [PubMed] [Google Scholar]
- Jing Y, Liu LZ, Jiang Y, Zhu Y, Guo NL, Barnett J, Rojanasakul Y, Agani F, Jiang BH. Cadmium increases HIF-1 and VEGF expression through ROS, ERK, and AKT signaling pathways and induces malignant transformation of human bronchial epithelial cells. Toxicol Sci. 2012;125:10–19. doi: 10.1093/toxsci/kfr256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Ladelfa MF, Toledo MF, Laiseca JE, Monte M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid Redox Signal. 2011;15:1749–1761. doi: 10.1089/ars.2010.3652. [DOI] [PubMed] [Google Scholar]
- Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko GN, Marchenko ND, Leng J, Strongin AY. Promoter characterization of the novel human matrix metalloproteinase-26 gene: regulation by the T-cell factor-4 implies specific expression of the gene in cancer cells of epithelial origin. Biochem J. 2002;363:253–262. doi: 10.1042/0264-6021:3630253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng D, Lv DD, Zhuang X, Sun H, Fan L, Shi XL, Fang J. Benzo[a]pyrene induces expression of matrix metalloproteinases and cell migration and invasion of vascular smooth muscle cells. Toxicol Lett. 2009;184:44–49. doi: 10.1016/j.toxlet.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Hamada F, Ishidate T, Anai K, Kawahara K, Toyoshima K, Akiyama T. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3:395–403. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- O’Brien P, Salacinski HJ. Evidence that the reactions of cadmium in the presence of metallothionein can produce hydroxyl radicals. Arch Toxicol. 1998;72:690–700. doi: 10.1007/s002040050562. [DOI] [PubMed] [Google Scholar]
- Plaisant M, Giorgetti-Peraldi S, Gabrielson M, Loubat A, Dani C, Peraldi P. Inhibition of hedgehog signaling decreases proliferation and clonogenicity of human mesenchymal stem cells. PLoS One. 2011;6:e16798. doi: 10.1371/journal.pone.0016798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Luo J, Leonard SS, Harris GK, Millecchia L, Flynn DC, Shi X. Hydrogen peroxide formation and actin filament reorganization by Cdc42 are essential for ethanol-induced in vitro angiogenesis. J Biol Chem. 2003;278:16189–16197. doi: 10.1074/jbc.M207517200. [DOI] [PubMed] [Google Scholar]
- Shaikh ZA, Vu TT, Zaman K. Oxidative stress as a mechanism of chronic cadmium-induced hepatotoxicity and renal toxicity and protection by antioxidants. Toxicol Appl Pharmacol. 1999;154:256–263. doi: 10.1006/taap.1998.8586. [DOI] [PubMed] [Google Scholar]
- Son YO, Lee JC, Hitron JA, Pan J, Zhang Z, Shi X. Cadmium induces intracellular Ca2+- and H2O2-dependent apoptosis through JNK- and p53-mediated pathways in skin epidermal cell line. Toxicol Sci. 2010;113:127–137. doi: 10.1093/toxsci/kfp259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radic Biol Med. 1995;18:321–336. doi: 10.1016/0891-5849(94)00159-h. [DOI] [PubMed] [Google Scholar]
- Stohs SJ, Bagchi D, Hassoun E, Bagchi M. Oxidative mechanisms in the toxicity of chromium and cadmium ions. J Environ Pathol Toxicol Oncol. 2001;20:77–88. [PubMed] [Google Scholar]
- Szuster-Ciesielska A, Stachura A, Slotwinska M, Kaminska T, Sniezko R, Paduch R, Abramczyk D, Filar J, Kandefer-Szerszen M. The inhibitory effect of zinc on cadmium-induced cell apoptosis and reactive oxygen species (ROS) production in cell cultures. Toxicology. 2000;145:159–171. doi: 10.1016/s0300-483x(00)00144-x. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis in review. J Inorg Biochem. 2000;79:241–244. doi: 10.1016/s0162-0134(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533:107–120. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Anver MR, Diwan BA. Chronic toxic and carcinogenic effects of oral cadmium in the Noble (NBL/Cr) rat: induction of neoplastic and proliferative lesions of the adrenal, kidney, prostate, and testes. J Toxicol Environ Health A. 1999;58:199–214. doi: 10.1080/009841099157296. [DOI] [PubMed] [Google Scholar]
- Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Fang J, Leonard SS, Rao KM. Cadmium inhibits the electron transfer chain and induces reactive oxygen species. Free Radic Biol Med. 2004;36:1434–1443. doi: 10.1016/j.freeradbiomed.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001;61:6050–6054. [PubMed] [Google Scholar]
- Zhang Z, Wang X, Cheng S, Sun L, Son YO, Yao H, Li W, Budhraja A, Li L, Shelton BJ, Tucker T, Arnold SM, Shi X. Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/beta-catenin pathway in human colorectal adenocarcinoma DLD1 cells. Toxicol Appl Pharmacol. 2011;256:114–121. doi: 10.1016/j.taap.2011.07.016. [DOI] [PubMed] [Google Scholar]