

Abstract
Amphiphilic macromolecules (AMs) based on carbohydrate domains functionalized with poly(ethylene glycol) can inhibit the uptake of oxidized low density lipoprotein (oxLDL) mediated by scavenger receptor A (SR-A) and counteract foam cell formation, the characteristic “atherosclerotic” phenotype. A series of AMs were prepared by altering the carbohydrate chemistry to evaluate the influence of backbone architecture on the physicochemical and biological properties. Upon evaluating the degree of polymer-based inhibition of oxLDL uptake in human embryonic kidney cells expressing SR-A, two AMs (2a and 2c) were found to have the most efficacy. Molecular modeling and docking studies show that these same AMs have the most favorable binding energies and most close interactions with the molecular model of SR-A collagen-like domain. Thus, minor changes in the AMs architecture can significantly affect the physicochemical properties and inhibition of oxLDL uptake. These insights can be critical for designing optimal AM-based therapeutics for management of cardiovascular disease.
Keywords: Amphiphilic polymer, atherosclerosis, self-assembled micelle, oxLDL inhibition
1. INTRODUCTION
Atherosclerosis, the occlusive artery disease, is triggered by the build-up of oxidized low density lipoprotein (oxLDL) in blood vessel walls.[1] The oxLDL accumulation generates an inflammatory response which results in the recruitment of circulating monocytes followed by their differentiation to macrophages and upregulation of macrophage membrane-based scavenger receptors.[2] The major scavenger receptors, namely scavenger receptor A (SR-A) and CD36, mediate the uptake of oxLDL,[3–5] leading to unregulated cholesterol accumulation and foam cell formation, a key characteristic of the onset of atherogenesis.[6,7]
Aside from lifestyle changes, cholesterol lowering therapies (i.e., statins) are the most common methods for atherosclerosis treatment. Such drugs, however, indirectly mediate atherosclerosis by decreasing cholesterol synthesis which ultimately results in decreased oxLDL in the blood vessel walls. A more direct and promising approach in the treatment and prevention of atherosclerosis involves designing functional inhibitors against scavenger receptors to abrogate uncontrolled oxLDL uptake.[8–11] Our research group has previously presented carbohydrate-based, nanoscale amphiphilic macromolecules (AMs) that competitively inhibit scavenger receptor-mediated oxLDL uptake.[12,13] Comprised of a hydrophobic domain based on the sugar, mucic acid, acylated with four aliphatic chains, and a hydrophilic poly(ethylene glycol) (PEG) tail, the AMs exhibit high biocompatibility and stability.[14] Further, the AMs self-assemble into micelles in aqueous media at relatively low (10−7 M) concentrations, enabling drug encapsulation within the micellar core. To understand the key structural features relevant to oxLDL inhibition, systematic variations to the AM structure were performed, including the PEG chain length, PEG architecture and aliphatic chain length as well as type, charge, number, and rotational motion of anionic charges. These studies also tested whether amphiphilicity was a significant attribute in preventing oxLDL internalization.[15,16] The structure-function studies demonstrated that while amphiphilicity was necessary, it alone was insufficient; charge and hydrophobicity better contributed to the polymer’s ability to inhibit oxLDL uptake. Overall, anionic charge and a rigid, hydrophobic carboxylic acid presentation significantly enhanced the inhibitory activity of AMs.
The AM hydrophobic domain compositions have not been optimized to date, however, and the impact of carbohydrate stereochemistry and conformation remain to be elucidated. Minute changes in carbohydrate stereochemistry and conformation can potentially have far-reaching consequences on polymer physicochemical and biological properties.[17–20] For example, Wang et al. demonstrated that the L configuration of poly(N-acryloyl-L(D)-valine) films promote better cytocompatibility (i.e., adhesion, growth, assembly and proliferation) compared to the D configuration.[19] Additionally, the introduction of aromaticity can significantly affect the properties of the resultant polymers.[21] To this end, several AMs were synthesized with incremental backbone modifications to investigate the influence of carbohydrate stereochemistry, aromatic groups and cyclic carbohydrates that are incorporated within the polymer backbone on biological activity, specifically, oxLDL inhibition (Figure 1). Molecular modeling and docking studies were employed to quantify the structure-activity relationship between the polymers and SR-A scavenger receptors. Previous experiments performed with macrophages (both mouse and human) indicated that the polymers were acting, in part, through interactions with SR-A.[12, 15,16,22,23] Although polymer interactions with CD36 and other surface proteins were observed, the goal of the studies outlined in this manuscript was to isolate the interactions of the polymers with SR-A only. To confirm the extent of backbone influence on scavenger receptor binding, a functional assay was developed. The AMs were evaluated for their ability to inhibit SR-A mediated oxLDL uptake in human embryonic kidney cells expressing the SR-A scavenger receptor (HEK-SRA). These cells have a well-established history of use for studying various SR-A functions due to their over expression of the SR-A construct.[23–26] AM binding to the cells was then determined by quantifying the amount of a competitor molecule (oxLDL) bound and internalized by the cells after 24 hours incubation in the presence of the polymers.
Figure 1.

Structures of charged and uncharged AMs where R = 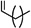 .
.
2. MATERIAL AND METHODS
2.1. Synthetic Materials
All reagents and solvents were purchased from Sigma-Aldrich and used as received unless otherwise noted. 4-(dimethylamino)pyridinium p-toluene-sulfonate (DPTS) was prepared as described by Moore and Stupp.[27] Monomethoxy-poly(ethylene glycol) (mPEG, Mn = 5000 Da) was azeotropically distilled with toluene prior to use. The synthesis of 2a and 2d have been previously reported.[14,28]
2.2 Instrumentation
1H-NMR spectra were obtained using a Varian 400 MHz or 500 MHz spectrophotometer with TMS as internal reference. Samples were dissolved in CDCl3, or CDCl3 with a few drops of DMSO-d6 if necessary. Molecular weights (MW) were determined using gel permeation chromatography (GPC) with respect to PEG standards (Sigma-Aldrich) on a Waters Stryagel® HR 3 THF column (7.8 × 300 mm). The Waters LC system (Milford, MA) was equipped with a 2414 refractive index detector, a 1515 isocratic HPLC pump, and 717plus autosampler. Samples (10 mg/mL) were dissolved in THF and filtered using 0.45 µm pore size nylon or PTFE syringe filters (Fisher Scientific). Dynamic light scattering (DLS) analysis was carried out on a Zetasizer nanoseries ZS90 (Malvern instruments). CMC studies were carried out on a Spex fluoromax-3 spectrofluorometer (Jobin Yvon Horiba) at 25 °C. Melting points were determined by DSC on a TA DSC Q200. TA universal analysis 2000 software was used for data collection on a Dell dimension 3000 computer. Samples (4–8 mg) were heated under dry nitrogen gas. Data were collected at heating and cooling rates of 10 °C min−1 with a two-cycle minimum.
2.3. Polymer synthesis (Figure 2)
Figure 2.

General scheme for the AM synthesis where BB represents the “building block” of the hydrophobic domain.
2.3.1. Synthesis of 2b
D-Saccharic acid (2.0 g, 8.1 mmol) and zinc chloride (0.26 g, 1.9 mmol) were stirred with lauroyl chloride (26 mL, 12 mmol). The solution was heated to 95 °C in a temperature-controlled oil bath and stirred under argon overnight. Diethyl ether (100 mL) and water (30 mL) were added and the solution was allowed to stir for 45 minutes. The organic phase was washed with water (5 × 100 mL) and then concentrated via rotary evaporation. The concentrated solution was precipitated from hexanes (2.5 L) yielding intermediate 1b as a white solid (4.2 g, 56%) 1H NMR (CDCl3): δ = 0.82–0.91 (t, 12H, CH3), 1.21–1.40 (m, 64H, CH2), 1.61–1.71 (m, 8H, CH2), 2.40–2.50 (m, 8H, CH2), 5.85 (s, 1H, CH), 5.10–5.12 (1H, d, CH), 5.35–5.39 (d, 1H, CH), 5.49–5.51 (d, 1H, CH); 13C NMR (CDCl3): 13.09, 21.67, 23.58, 23.68, 27.90, 28.05, 28.13, 28.16, 28.23, 28.32, 28.36, 28.38, 28.43, 28.55, 28.57, 30.89, 32.12, 32.31, 66.12, 71.90, 71.93, 73.47, 77.88, 167.98, 168.12, 171.82, 172.32; IR (NaCl cm−1): 3440, 2918, 2849, 1803, 1755 (C=O), 1298, 1196; Tm = 75 °C.
Intermediate 1b (1.5 g, 1.6 mmol) and DPTS (0.15 g 0.48 mmol) were dissolved in anhydrous CH2Cl2 (15 mL). mPEG was cooled to room temperature under argon and added to the 1b/DPTS solution. Once the PEG had dissolved, N,N’-dicyclohexylcarbodiimide (DCC) (1.7 mL, 1.7 mmol) was added dropwise. The reaction mixture was stirred under argon for 48 hours, cooled and the resulting white solid precipitate (dicyclohexylurea) was removed by vacuum filtration. The filtrate was washed with 0.1N HCl (20 mL), followed by 50:50 brine:water (2 × 20 mL), dried over MgSO4 and concentrated via rotary evaporation. The product was precipitated from diethyl ether yielding 2b as a white solid (2.6 g, 83%). 1H NMR (CDCl3): δ = 0.88 (t, 12H, CH3), 1.30 (m, 64H, CH2),1.65 (m, 8H, CH2), 2.38 (m, 4H, CH2), 2.60 (m, 4H, CH2), 3.63 (m, ~0.4kH, CH2), 5.42 (m, 1H, CH), 5.50 (m, 1H, CH), 5.59 (m, 2H,CH); IR (NaCl cm−1): 3478 (O-H), 2883 (C-H), 1749 (C=O), 1241, 1113 (C-O); Tm = 54 °C; GPC: Mw = 6.3 kDa; PDI = 1.09.
2.3.2. Synthesis of 2c
Procedure was same as that for 1b (2b part a) using 2,5-dihydroxyterephthalic acid (5.0 g, 25 mmol), zinc chloride (0.81 g, 5.9 mmol), and lauroyl chloride (87 mL, 378 mmol), yielding intermediate 1c as a yellow solid (5.1 g, 68 %). 1H NMR (CDCl3): δ = 0.80–0.93 (t, 6H, CH3), 1.20–1.45 (m, 32H, CH2), 1.70–1.81 (m, 4H, CH2), 2.51–2.65 (m, 4H, CH2), 7.70 (s, 2H, Ar-CH); 13C NMR (CDCl3): 14.32, 22.87, 29.34, 29.53, 29.67, 29.80, 29.82, 32.10, 34.37, 127.37, 128.74, 147.84, 165.27, 172.42; IR (NaCl cm−1): 2847, 1768, 1690 (C=O), 1268, 1177, 935, 897; Tm = 71 °C.
Procedure same as that for 2b (part b) using intermediate 1c (0.4 g, 0.7 mmol), mPEG (1.0 g, 0.2 mmol), DPTS (0.1 g, 0.3 mmol), and DCC (1 mL, 1.0 mmol), yielding 2c as a white solid (1.0 g, 91%). 1H NMR (CDCl3): δ 0.85 (t, 6H, CH3), 1.30 (m, 32H, CH2), 1.71 (m, 4H, CH2), 2.21 (m, 4H, CH2), 2.36 (m, 4H, CH2), 3.63 (m, ~0.4-kH, CH2), 7.65 (d, 1H, Ar-H), 8.10 (s, 1H, Ar-H); IR (NaCl cm−1): 3426, 2879, 1650 (C=O), 1108, 949, 842; Tm = 56 °C; GPC: Mw = 6.3 kDa; PDI = 1.07.
2.3.3. Synthesis of 2e
Procedure same as that for 1b (2b part a) using D-galacturonic acid (2.0 g, 9.2 mmol), zinc chloride (0.30 g, 2.2 mmol), and lauroyl chloride (10 mL, 46 mmol), yielding intermediate 1e as a white solid (1.2 g, 13 %). 1H NMR (CDCl3): δ = 0.80–0.86 (t, 12H, CH3), 1.21–1.38 (m, 64H, CH2), 1.50–1.65 (m, 8H,CH2), 2.20–2.28 (m, 8H, CH2), 4.61 (d, 1H, H), 5.01–5.08 (dxd, 1H, H), 5.20–5.29 (t, 1H, H), 5.60–5.69 (t, 1H, H), 6.35 (d, 1H, anomeric-H) 13C NMR (CDCl3): 14.33, 22.91, 22.91, 24.88, 24.92, 24.98, 25.14, 29.28, 29.32, 29.35, 29.50, 29.58, 29.71, 29.85, 32.15, 34.04, 34.16, 34.26, 34.30, 68.81, 68.92, 68.97, 70.15, 88.72, 171.58, 172.51, 172.84; IR (NaCl cm−1): 3492, 2920, 2851, 1740, 1715 (C=O), 1238, 1147; Tm = 51°C.
Procedure same as that for 2b (part b) using intermediate 1e (1.4 g, 1.5 mmol), mPEG (2.5 g, 0.5 mmol), DPTS (0.13 g, 0.4 mmol), and DCC (0.27 mL, 1.5 mmol) yielding 2e as a white solid (1.9g, 65%). 1H NMR (CDCl3): δ = 0.80–0.86 (t, 12H, CH3), 1.21–1.38 (m, 64H, CH2), 1.50–1.65 (m, 8H, CH2), 2.21–2.26 (m, 8H, CH2), 3.40–3.78 (m,~0.4-kH, CH2), 4.61 (d, 1H, H), 5.01–5.08 (dxd, 1H, H), 5.20–5.29 (t, 1H, H), 5.60–5.69 (t, 1H, H), 6.35 (d, 1H, anomeric-H); IR (NaCl cm−1): 2887 (C-H), 1742 (C=O), 1280, 1148, 1113 (C-O); Tm = 57 °C; GPC: Mw = 6.3 kDa; PDI = 1.07.
2.3.4. Synthesis of 2f
Procedure same as that for 1b (2b part a) using D-glucuronic acid (2.0 g, 10 mmol), zinc chloride (0.33 g, 2.4 mmol), and lauroyl chloride (12 mL, 51 mmol), yielding 1f as a white solid (4.7 g, 51%). 1H NMR (CDCl3): δ = 0.80–0.86 (t, 12H, CH3), 1.20–1.39 (m, 64H, CH2), 1.51–1.71 (m, 8H, CH2), 2.19–2.30 (m, 4H, CH2), 2.31–2.42 (m, 4H, CH2), 4.75 (s, 1H, H), 5.30–5.42 (qxd, 2H, H), 5.89 (s, 1H, H), 6.56 (d, 1H, anomeric-H) 13C NMR (CDCl3): 14.22, 22.90, 24.79, 24.98, 25.20, 29.26–29.88, 32.13, 34.09, 34.14, 34.16, 34.27, 65.98, 67.01, 68.28, 70.85, 89.37, 171.59, 172.56, 172.65, 172.84 (C=O): IR (NaCl cm−1): 3601, 2919, 2850, 1752, 1701 (C=O), 1299, 1172; Tm = 68 °C.
Procedure same as that for 2b (part a) using intermediate 1f (1.4 g, 1.5 mmol), mPEG (2.5g, 0.5 mmol), DPTS (0.13g, 0.4mmol), and DCC (0.27 mL, 1.5 mmol) yielding 2f as a white solid (1.5g, 51%). 1H NMR (CDCl3): δ = 0.88 (t, 12H, CH3), 1.28 (m, 64H, CH2),1.60 (m, 8H, CH2), 2.21 (m, 4H, CH2), 2.36 (m, 4H, CH2), 3.63 (m,~0.4-kH, CH2), 4.78 (s, 1H, CH), 5.40 (s, 2H, CH), 5.84 (s, 2H,CH), 6.53 (s, 1H, CH); IR (NaCl cm−1): 2883 (C-H), 1752 (C=O), 1280, 1148, 1114 (C-O); T = 55 °C; GPC: Mw = 6.3 kDa; PDI = 1.07.
2.4. Critical Micelle Concentration (CMC) Measurements
A solution of pyrene, the fluorescence probe molecule, was made up to a concentration of 5 × 10−6 M in acetone. Samples were prepared by adding 1 mL of pyrene solution to a series of vials and allowing the acetone to evaporate so that the final concentration of pyrene in all of the samples was 5 × 10−7 M. AMs were dissolved in HPLC grade water and diluted to a series of concentrations from 1 × 10−3 M to 1 × 10−10 M. AM-pyrene solutions (10 mL) were shaken overnight at 37 °C to allow partition of the pyrene into the micelles. Emission was performed from 300 to 360 nm, with 390 nm as the excitation wavelength. The maximum absorption of pyrene shifted from 332 to 334.5 nm on micelle formation.[29–31] The ratio of absorption of encapsulated pyrene (334.5 nm) to pyrene in water (332 nm) was plotted as the logarithm of polymer concentrations. The inflection point of the curve was taken as the CMC.
2.5. Cell Culture
Studies of polymer interactions were conducted using a tet-inducible cell line with controlled expression of scavenger receptor A (SR-A), human embryonic kidney (HEK) cells stably transfected with human SR-A (gift from Dr. Steven R. Post), which are referred to as HEK-SRA.[24,25] Cells were propagated in high glucose Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, 15 µg/mL Blasticidin and 100 µg/mL HygromycinB at 37 °C in 5% CO2. SR-A expression was induced with addition of 0.5 µg/mL tetracycline overnight and throughout the experiment. SR-A expression was confirmed via antibody binding assays (results not shown).
2.6. LDL Oxidation
Oxidized low density lipoprotein (oxLDL) was generated by incubating 50 µg/mL LDL purified from human plasma (Molecular Probes Eugene, OR) with 10 µM CuSO4 at 37 °C for 18 hr exposed to air.[32,33] Oxidation was terminated with 0.01% w/v EDTA (Sigma, St. Louis, MO).
2.7. OxLDL Accumulation in HEK-SRA Cells
The internalization of oxLDL by HEK-SRA cells was assayed by incubating boron-dipyrromethene (BODIPY)-labeled oxLDL (10 µg/mL) and 10−6 M polymers with cells for 24 hr at 37 °C and 5% CO2 in serum containing DMEM. Conditions included a control of medium alone without polymer intervention, and non-induced cells. Cells were washed with 1× PBS, fixed with 4% formaldehyde and imaged on a Nikon Eclipse TE2000-S fluorescent microscope to determine fluorescently tagged oxLDL accumulation. The images were analyzed with ImageJ 1.42q (NIH) and fluorescence data was normalized to cell count. The levels of oxLDL uptake were normalized to those obtained in the absence of polymers.
2.8. Statistical Analysis
Each in vitro experiment was performed at least twice and three replicate samples were investigated in each experiment. Five images per well were captured and analyzed. The results were then evaluated using analysis of variance (ANOVA). Significance criteria assumed a 95% confidence level (P<0.05). Standard error of the mean is reported in the form of error bars on the graphs of the final data.
2.9. Polymer and SR-A Modeling
The polymers were modeled according to their chemical structures illustrated in Figure 1 using the build module in molecular operating environment (MOE) (Chemical Computing Group, Inc., Montreal, Canada). The model polymer molecules were parameterized for Amber99[34] force field and energy minimized until convergence (grad = 0.001) was attained. The creation of the SR-A homology model was previously described.[15] Briefly, the 3D homology model of the SR-A collagen-like domain was generated using the program MODELLER35] with collagen type I chain A as template.
2.10. Docking and Scoring
Selected polymer models were docked to the collagen-like domain of SR-A using GOLD v3.2.[36] The GOLD program employs a genetic algorithm for docking flexible ligands into partially flexible receptor sites. The binding cavity was defined as residues Arg45 – Ser68 with an active site radius of 15 Å such that all major residues thought to be necessary for oxLDL binding were included. Dockings were performed with standard default settings; population size of 100, selection pressure of 1.1, number of operations at 100,000, number of islands at 5, and a niche size of 2. Twenty independent docking runs were performed for each polymer, which optimized the computational time required to dock and score non-redundant conformations. The docked pairs were ranked based on each GoldScore. The best ranking conformation of the polymer illustrated the most preferred conformation to interact with scavenger receptor and the binding energy was computed for the refined complexes using Equation 1.
| Equation 1 |
Where ΔEcomplex is the energy of the polymers docked to collagen-like domain of SR-A, ΔESR-A is the energy of the homology model of the scavenger receptor collagen-like domain, and ΔEPolymer is the energy of the polymer. Each structure (polymer model, homology model of the SR-A collagen-like domain, and the docked conformation of the pair) was parameterized using Amber99[34] force field and energy minimized until convergence (grad = 0.001) was attained. These minimized energies were used to estimate the binding energy from Equation 1.
3. RESULTS AND DISCUSSION
A series of AMs (Figure 1) were designed and synthesized to investigate the influence of altering the hydrophobic backbone on the resultant physicochemical properties. The hydrophobic domain was varied to investigate the influence of stereochemistry, rigidity and shape. The hypothesis was that specific stereochemical and conformational variations would alter polymer binding affinity to scavenger receptors and the corresponding capacity for inhibition of scavenger receptor-mediated oxLDL uptake.
Stereochemistry is widely recognized as an important design consideration for therapeutic molecules.[37–39] The hydroxyl stereochemistry of the polymers was altered by one carbon position to address the hypothesis that stereochemistry governs the spatial interactions of the carbohydrate with the receptor domains and serving to further enhance binding properties. Saccharic acid is a commercially available carbohydrate stereoisomer of mucic acid, differing in only one hydroxyl group position, and was chosen as the building block for the new AM, 2b, with altered hydroxyl stereochemistry (Figure 1). 2b was synthesized by modifying a previously published method for the preparation of 2a, which comprises mucic acid.[14] Briefly, the two-step procedure involves acylating the carbohydrate with lauroyl groups followed by coupling to PEG (Figure 2). In the initial acylation step, some modifications were required when saccharic acid was used in place of mucic acid. For example, to achieve an acceptable yield (56%) of 1b, the number of equivalents of acylating agent (lauroyl chloride) was significantly reduced (from 15 with mucic acid to 5 with saccharic acid) as isolation and purification proved problematic with a large excess of lauroyl chloride. Coupling of the PEG and 1b using DCC as the coupling agent and DPTS as the catalyst proceeded in excellent yields irrespective of hydroxyl stereochemistry (98% for 2a and 83% for 2b).
Because increased rigidity previously demonstrated efficient oxLDL inhibition,[15, 16] an aromatic component was introduced to further increase the rigidity. Thus, 2,5-dihydroxyterephthalic acid was chosen as the building block as it possesses both two carboxylic acids and two hydroxyl groups for functionalization. The synthesis of 2c was based on the same two-step procedure described above for 2b; no modifications were necessary and the overall yield was 62%. While 2c does not contain a sugar and is thus technically not a ‘carbohydrate-based’ polymer, it was included to ascertain the role of cyclic structures and flexibility in the five true, sugar-based AMs.
The effect of conformation on the resultant properties was further explored by preparing galacturonic and glucuronic acid-based AMs, 2e and 2f, respectively. Increasing the hydrophobic domain size was hypothesized to increase binding to the scavenger receptor pocket. The cyclic carbohydrates (galacturonic and glucuronic acid) were commercially available with carboxylic acid groups for PEG coupling and multiple hydroxyl groups for acylation. Following functionalization with aliphatic chains and PEG, both AMs based on these sugars (2e and 2f) are uncharged; unlike the other polymers (2a–2c), these two polymers lack a free carboxylic acid. As stereoisomers, the cyclic AMs 2e and 2f also provide insight into how hydroxyl stereochemistry influences bioactivity. Acylation of galacturonic and glucuronic acid proved more difficult than the linear carbohydrates due to the α and β anomeric forms. The acylation reaction was optimized (95 °C for 4–5 hours) to achieve the highest possible yield (13% for 1e and 51% for 1f). To prepare the AMs, the acylated cyclic saccharides, 1e and 1f, were coupled to PEG, resulting in 2e and 2f in 65% and 51% yield, respectively.
With this unique series of AMs, the influence of backbone architecture on the resultant physicochemical properties (hydrodynamic diameter, melting temperature (Tm) and critical micelle concentration (CMC)) was evaluated. The Tm values of all synthesized polymers were ~60 °C, demonstrating that the PEG domain dominates the polymer’s thermal behavior. Previous work indicated that the AM biological activity was dependent on their ability to form stable micelles.[15,16] The AMs with the lowest CMC values are extremely efficacious and the change in CMC may have a significant biological impact for the newly synthesized AMs. CMC values were measured using a previously reported fluorimetry technique using pyrene as the fluorescence probe.[29–31] Polymer 2b formed ~19 nm micelles based on dynamic light scattering (DLS) with a CMC in the same range (10−7 M) as the previously described 2a (Table 1). The similar hydrodynamic diameter and CMC values indicate that altering the hydroxyl stereochemistry by one position has little effect on polymer self-assembly into micelles. Incorporation of the rigid, aromatic 2, 5-dihydroxyterephthalic acid backbone of polymer 2c yielded micelles almost twice the size (~35 nm) of the linear analogs (2a and 2b), but with a comparable CMC value (10−7 M). The increase in size was anticipated as terephthalic acid is more sterically bulky than the linear carbohydrates. Similarly, the physicochemical properties of the cyclic carbohydrate-based polymers, 2e and 2f, differed from 2d, the uncharged linear analog of 2a. Compared to 2d, 2e and 2f have slightly larger hydrodynamic diameters (38 nm and 40 nm, respectively) and CMCs on the order of 10−5 M, an order of magnitude higher than that of 2d. Both results are likely due to the steric bulk of the cyclic carbohydrates as compared to their linear counterparts. This increased CMC suggests that micelles of the cyclic analogs (2e and 2f) are less stable under physiological conditions and may demonstrate decreased biological activity compared to micelles of 2d. These combined results suggest that although the AM structure is dominated by the mass of PEG, small changes to the hydrophobic domain have a significant impact on the micellar properties.
Table 1.
Physicochemical properties (hydrodynamic size, CMC and Tm) of AMs: as micelles(size and CMC) and in the solid state (Tm).
| Polymer | Sizea (nm) | CMC (M) | Tm (°C) |
|---|---|---|---|
| 2a | 20 | 1.20×10−7 | 56 |
| 2b | 19 | 1.71×10−7 | 59 |
| 2c | 35 | 1.58×10−7 | 56 |
| 2d | 27 | 1.67×10−6 | 59 |
| 2e | 38 | 1.00×10−5 | 55 |
| 2f | 40 | 1.30×10−5 | 57 |
Z-average size
The AMs were then evaluated for their ability to inhibit oxLDL internalization through the combination of molecular modeling and in vitro structure-activity relationship studies with HEK-SRA cells. For molecular modeling, the polymers were modeled according to their chemical structures and scaled to contain only 20 ethylene glycol repeat units instead of 110. We have seen previously that the PEG chain has little interaction with the collagen-like domain, and scaling allows for optimization of computational time. The aliphatic arms were modeled at full alkyl chain length to fully assess the role of the hydrophobic domain. The modeled polymers were docked to an SR-A collagen-like domain homology model using GOLD v.3.2[36] and ranked based on its GoldScore. Previous receptor blocking studies have indicated that the AMs bind specifically to HEK-expressed SR-A1.[26] As such, in vitro experiments were carried out via incubation of the HEK-SRA cells with 10−6 M polymers and fluorescently labeled oxLDL for 24 hr at 37 °C. To maintain a relatively constant concentration and to ensure we evaluated polymers in the same state (i.e., micellar form), these experiments were performed at or near the critical micelle concentration of the various polymers. The 24-hr time point allowed for saturation of the cells with oxLDL and showed maximal differences between the polymers. As controls, the basal uptake of oxLDL when SRA-expression was not induced and the basal uptake of oxLDL when no polymer was present were both evaluated. The previously synthesized 2a[14] and 2d[28], the uncharged analog of 2a, were compared to the newly synthesized anionic and neutral polymers. Altering the position of one hydroxyl group in the hydrophobic domain had a significant impact on the AMs ability to inhibit oxLDL uptake. 2b showed only ~10% oxLDL inhibition compared to the ~60% inhibition of 2a (Figure 3a). This data correlated well with the modeling studies which revealed that 2b has minimal hydrophobic interactions with SR-A, resulting in unfavorable binding (40 kcal/mol) which may be due to the significant degree of free space between the aliphatic arms of 2b and the protein binding pocket (see Figure S1). 2a, on the other hand, possesses a favorable binding energy (−29 kcal/mol) as the aliphatic arms remain in close contact with the SR-A model indicating a significant amount of hydrophobic interactions.
Figure 3.

Evaluation of AMs through a) in vitro inhibition of oxLDL uptake over 24 hr in HEK cells induced to express SR-A and b) energetics of AMs binding with the SR-A collagen-like domain via molecular modeling.
Incorporation of a rigid, aromatic hydrophobic component (2c) resulted in binding comparable to 2a. Although it has only half the number of aliphatic arms, 2c demonstrated comparable binding in both in vitro and molecular modeling experiments, inhibiting ~60% oxLDL uptake in HEK-SRA cells while showing a favorable binding energy (−26 kcal/mol) in modeling studies. This data correlates with previous findings that suggest increased rigidity positively affects oxLDL inhibition.[15,16] The incorporation of a cyclic aliphatic component showed slight improvements over 2d, with 2e and 2f inhibiting ~50% and ~40% oxLDL uptake, respectively. Given the lack of anionic groups in 2e and 2f, SR-A binding is primarily mediated through hydrophobic interactions as demonstrated by molecular modeling. Polymer 2f displays slightly better oxLDL inhibition and favorable binding energy, in comparison to 2e, suggesting that the aliphatic arms of 2f encircle the SR-A protein (Figure S1). While not as significant as the linear analogs, it is noteworthy that although 2e and 2f differ by only one stereocenter, they show different binding affinities to SR-A. This result once again illustrates how minute changes in the AM hydrophobic domains can significantly impact the polymer properties.
Notably, the ability to bind to HEK-SRA cells does not correlate to the solution stability of the AMs. The CMC value of 2b is comparable to that of 2a (10−7 M), yet was much less effective at inhibiting oxLDL uptake. 2e and 2f, however, formed less stable micelles when compared to their linear analog, 2d, but were much more efficacious in regards to oxLDL inhibition. This data suggests that the polymer behavior is dominated by the unimer, not the micelle, such that the individual polymer chains govern binding to scavenger receptors.
4. CONCLUSIONS
A series of AMs were designed to investigate the influence of hydrophobic domain stereochemistry and composition on aggregation and biological properties. While seemingly small with respect to the overall polymer compositions, minor alterations in backbone architecture resulted in significant differences in the physicochemical properties of the AMs, particularly in micelle size and solution stability. In vitro and molecular modeling experiments were implemented to examine AM binding to SR-A. These studies also demonstrated that minute changes in the polymer structure significantly affect SR-A binding affinities and consequently modulate the competitive inhibition of oxLDL uptake. Changing the stereochemistry of one hydroxyl group in the backbone (2b) significantly reduces oxLDL inhibition while incorporating an aromatic backbone (2c) results in oxLDL inhibition comparable to that of the previously published “gold standard”, 2a. Incorporation of a cyclic carbohydrate (2e and 2f) demonstrated increased oxLDL inhibition and more favorable binding energies than AMs based on the neutrally charged aliphatic analog (2d). These findings establish that the composition of the hydrophobic domain is a critical design factor influencing the biological and physicochemical properties of these polymers. Previous in vivo studies have demonstrated that the polymers, when loaded in a collagen matrix and implanted around the carotid artery, have been found to be effective at reducing plaque in rats fed a high fat diet.[23] These results, in conjunction with our ability to manipulate polymer carbohydrate compositions, suggest AMs as candidates for cardiovascular therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
Different components of this study were partially supported by NIH grant R21093753 to Prabhas V. Moghe, and NIH grant R01 HL107913 to Prabhas V. Moghe and Kathryn E. Uhrich. Nicole Plourde was partially supported by the Rutgers NSF DGE 0333196 IGERT Program on Biointerfaces. We acknowledge Professor William Welsh for access to computational facilities at UMDNJ-Informatics institute and for data interpretation. We also acknowledge the academic computing services of UMDNJ. We thank Dr. Steven R. Post from the Department of Pathology and Laboratory Medicine, University of Arkansas for Medical Sciences for providing Human embryonic kidney (HEK) cells stably transfected with human scavenger receptor A for these studies. Additionally, we would like to thank Dr. Bryan Langowski for his insight and critical review of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information Available
Molecular modeling was performed to demonstrate how the polymers interact with the SR-A homology model. This data is a qualitative representation of the AMs binding to SR-A. This material is available free of charge via the Internet at www.sciencedirect.com.
REFERENCES
- 1.Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235–1242. doi: 10.1038/nm1102-1235. [DOI] [PubMed] [Google Scholar]
- 2.Yoshimoto T, Takahashi Y, Kinoshita T, Sakashita T, Inoue H, Tanabe T. Growth stimulation and epidermal growth factor receptor induction in cyclooxygenase-overexpressing human colon carcinoma cells. Advances in experimental medicine and biology. 2002;507:403–407. doi: 10.1007/978-1-4615-0193-0_62. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein JL, Ho YK, Basu SK, Brown MS. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:333–337. doi: 10.1073/pnas.76.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Winther MP, van Dijk KW, Havekes LM, Hofker MH. Macrophage scavenger receptor class A: A multifunctional receptor in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:290–297. doi: 10.1161/01.atv.20.2.290. [DOI] [PubMed] [Google Scholar]
- 5.Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. The Journal of clinical investigation. 2000;105:1095–1108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annual review of biochemistry. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. The Journal of biological chemistry. 1997;272:20963–20966. doi: 10.1074/jbc.272.34.20963. [DOI] [PubMed] [Google Scholar]
- 8.Boullier A, Friedman P, Harkewicz R, Hartvigsen K, Green SR, Almazan F, et al. Phosphocholine as a pattern recognition ligand for CD36. J Lipid Res. 2005;46:969–976. doi: 10.1194/jlr.M400496-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Yoshiizumi K, Nakajima F, Dobashi R, Nishimura N, Ikeda S. 2,4-Bis(octadecanoylamino)benzenesulfonic acid sodium salt as a novel scavenger receptor inhibitor with low molecular weight. Bioorg Med Chem Lett. 2004;14:2791–2795. doi: 10.1016/j.bmcl.2004.03.082. [DOI] [PubMed] [Google Scholar]
- 10.Guaderrama-Diaz M, Solis CF, Velasco-Loyden G, Laclette J, Mas-Oliva J. Control of scavenger receptor-mediated endocytosis by novel ligands of different length. Mol Cell Biochem. 2005;271:123–132. doi: 10.1007/s11010-005-5756-3. [DOI] [PubMed] [Google Scholar]
- 11.Broz P, Benito SM, Saw C, Burger P, Heider H, Pfisterer M, et al. Cell targeting by a generic receptor-targeted polymer nanocontainer platform. J Control Release. 2005;102:475–488. doi: 10.1016/j.jconrel.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 12.Chnari E, Nikitczuk JS, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules can inhibit cellular uptake of differentially oxidized LDL. Biomacromolecules. 2006;7:597–603. doi: 10.1021/bm0506905. [DOI] [PubMed] [Google Scholar]
- 13.Chnari E, Lari HB, Tian L, Uhrich KE, Moghe PV. Nanoscale anionic macromolecules for selective retention of low-density lipoproteins. Biomaterials. 2005;26:3749–3758. doi: 10.1016/j.biomaterials.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 14.Tian L, Yam L, Zhou N, Tat H, Uhrich KE. Amphiphilic scorpion-like macromolecules: Design, synthesis, and characterization. Macromolecules. 2004;37:538–543. [Google Scholar]
- 15.Plourde NM, Kortagere S, Welsh W, Moghe PV. Structure-Activity Relations of Nanolipoblockers with the Atherogenic Domain of Human Macrophage Scavenger Receptor A. Biomacromolecules. 2009;10:1381–1391. doi: 10.1021/bm8014522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iverson NM, Sparks SM, Demirdirek B, Uhrich KE, Moghe PV. Controllable inhibition of cellular uptake of oxidized low-density lipoprotein: Structure-function relationships for nanoscale amphiphilic polymers. Acta Biomater. 2010;6:3081–3091. doi: 10.1016/j.actbio.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reeve MS, Mccarthy SP, Downey MJ, Gross RA. Polylactide Stereochemistry - Effect on Enzymatic Degradability. Macromolecules. 1994;27:825–831. [Google Scholar]
- 18.Sun T, Han D, Riehemann K, Chi L, Fuchs H. Stereospecific interaction between immune cells and chiral surfaces. Journal of the American Chemical Society. 2007;129:1496–1497. doi: 10.1021/ja0686155. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Gan H, Sun TL, Su BL, Fuchs H, Vestweber D, et al. Stereochemistry triggered differential cell behaviours on chiral polymer surfaces. Soft Matter. 2010;6:3851–3855. [Google Scholar]
- 20.Wang XG, H, Sun T. Chiral Design for Polymeric Biointerface: The Influence of Surface Chirality on Protein Adsorption. Advanced Functional Materials. 2011;21:6. [Google Scholar]
- 21.Kricheldorf HR, Nuyken O, Swift G. Handbook of Polymer Synthesis. 2 ed. Marcel-Dekker; 2005. [Google Scholar]
- 22.Chnari E, Nikitczuk JS, Wang J, Uhrich KE, Moghe PV. Engineered Polymeric Nanoparticles for Receptor-Targeted Blockage of Oxidized Low Density Lipoprotein Uptake and Atherogenesis in Macrophages. Biomacromolecules. 2006;7:1796–1805. doi: 10.1021/bm0600872. [DOI] [PubMed] [Google Scholar]
- 23.Iverson NM, Plourde NM, Sparks SM, Wang J, Patel EN, Shah PS, et al. Dual use of amphiphilic macromolecules as cholesterol efflux triggers and inhibitors of macrophage athero-inflammation. Biomaterials. 2011;32:8319–8327. doi: 10.1016/j.biomaterials.2011.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosswig N. Class A Scavenger Receptor-mediated Adhesion and Internalization Require Distinct Cytoplasmic Domains. The Journal of biological chemistry. 2003;278:34219–34225. doi: 10.1074/jbc.M303465200. [DOI] [PubMed] [Google Scholar]
- 25.Nikolic DM, Cholewa J, Gass C, Gong MC, Post SR. Class A scavenger receptor-mediated cell adhesion requires the sequential activation of Lyn and PI3-kinase. AJP: Cell Physiology. 2006;292:C1450–C1458. doi: 10.1152/ajpcell.00401.2006. [DOI] [PubMed] [Google Scholar]
- 26.York AW, Zablocki KR, Lewis DR, Gu L, Uhrich KE, Prud'homme RK, et al. Kinetically Assembled Nanoparticles of Bioactive Macromolecules Exhibit Enhanced Stability and Cell-Targeted Biological Efficacy. Advanced Materials. 2012;24:733–739. doi: 10.1002/adma.201103348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore JS, Stupp SI. Room-Temperature Polyesterification. Macromolecules. 1990;23:65–70. [Google Scholar]
- 28.Djordjevic J, Del Rosario LS, Wang JZ, Uhrich KE. Amphiphilic Scorpion-like Macromolecules as Micellar Nanocarriers. J Bioact Compat Pol. 2008;23:532–551. [Google Scholar]
- 29.Astafieva I, Zhong XF, Eisenberg A. Critical Micellization Phenomena in Block Polyelectrolyte Solutions. Macromolecules. 1993;26:7339–7352. [Google Scholar]
- 30.Meng FB, Zhang BY, Lian J, Wu YP, Li XZ, Yao DS. Mesomorphic Behavior and Optical Properties of Liquid-Crystalline Polysiloxanes Bearing Different Chiral Groups. J Appl Polym Sci. 2009;114:2195–2203. [Google Scholar]
- 31.Kalyanasundaram K, Thomas JK. Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems. Journal of the American Chemical Society. 1977;99:2039–2044. [Google Scholar]
- 32.Chang MY, Potter-Perigo S, Wight TN, Chait A. Oxidized LDL bind to nonproteoglycan components of smooth muscle extracellular matrices. J Lipid Res. 2001;42:824–833. [PubMed] [Google Scholar]
- 33.Oorni K, Pentikainen MO, Annila A, Kovanen PT. Oxidation of low density lipoprotein particles decreases their ability to bind to human aortic proteoglycans - Dependence on oxidative modification of the lysine residues. The Journal of biological chemistry. 1997;272:21303–21311. doi: 10.1074/jbc.272.34.21303. [DOI] [PubMed] [Google Scholar]
- 34.Wang JM, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 35.Sali A, Blundell TL. Comparative Protein Modeling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 36.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 37.Liu YM, Reineke TM. Hydroxyl stereochemistry and amine number within poly(glycoamidoamine)s affect intracellular DNA delivery. Journal of the American Chemical Society. 2005;127:3004–3015. doi: 10.1021/ja0436446. [DOI] [PubMed] [Google Scholar]
- 38.Krogsgaard-Larsen P, Liljefors T, Madson U. Textbook of Drug Design and Discovery. 2002. [Google Scholar]
- 39.Jeffrey GA, Wood RA. The crystal structure of galactaric acid (mucic acid) at −147°: An unusually dense, hydrogen-bonded structure. Carbohydrate Research. 1982;108:205–211. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.