

Abstract
Knowledge about the different mechanisms underlying the aging process has increased exponentially in the last decades. The fact that the basic mechanisms involved in the aging process are believed to be universal allows the use of different model systems, from the simplest eukaryotic cells such as fungi to the most complex organisms such as mice or human. As our knowledge on the aging mechanisms in those model systems increases, our understanding of human aging and the potential interventions that we could approach rise significantly. Among the different mechanisms that have been implicated in the aging process, DNA repair is one of the processes which have been suggested to play an important role. Here, we review the latest investigations supporting the role of these mechanisms in the aging process, stressing how beneficial the use of different model systems is. We discuss how human genetic studies as well as several investigations on mammalian models and simpler eukaryotic organisms have contributed to a better understanding of the involvement of DNA repair mechanisms in aging.
1. Introduction
Our cells are constantly exposed to endogenous and exogenous agents that induce damage to the cellular macromolecules such as DNA, RNA, proteins, and lipids. The sources of this damage include a broad range of agents such as industrial chemicals and combustion products present in our environment, UV radiation from the sun, and endogenous metabolic byproducts. In contrast to damaged lipids and proteins, damaged DNA, which carries the inherited genetic information of the cell, cannot be replaced. Therefore, formation of DNA lesions can have profound consequences for genomic stability due to the effect that these lesions can have on polymerase fidelity and processivity. Spontaneous reactions, such as hydrolysis, are the major sources of DNA damage. They can lead to deaminated bases and abasic sites in the DNA. Another very prominent type of endogenous DNA damage, oxidative DNA damage, is caused by reactive oxygen species (ROS), which are formed continuously as a consequence of normal aerobic metabolism during mitochondrial respiration but also by inflammatory responses. ROS can also be formed by a number of external factors including UV- and ionizing radiation and chemical mutagens. DNA repair mechanisms have evolved to remove the majority of all DNA lesions, but if these mechanisms are not sufficiently efficient, it will lead to DNA damage accumulation, which is likely to result in mutations and cellular dysfunction. Due to close proximity of the mitochondrial DNA to the inner mitochondrial membrane, the mitochondrial genome is more heavily exposed to ROS than the nuclear DNA and therefore also more likely to experience DNA damage. Thus, the mitochondrial free radical theory of aging [1] postulates that organisms age due to the accumulation of DNA damage and mutations in the mitochondrial DNA, leading to mitochondrial and eventually cellular dysfunction. This paper explores some of the recent research, which has been performed in order to uncover the relationship between DNA damage, DNA repair mechanisms, and the aging process, and emphasis is given to the use of eukaryotic model systems for this area of research.
2. DNA Damage and Aging
Among the wide variety of known DNA lesions, 8-oxo-deoxyguanosine (8oxoG) has received a lot of attention due to its mutagenicity and because of the possible correlation between its accumulation and pathological processes like cancer, degenerative diseases, and aging. However, an increasing number of studies also include other types of DNA lesions. Numerous studies report measurement of DNA damage in nuclear and mitochondrial DNA from tissues of young and old organisms, with variable outcomes. Although still controversial, several careful studies do show that 8oxoG accumulates with age. Thus, in a recent study Gan and coworkers used a sensitive LC-MS/MS method to demonstrate that 8oxoG increases with age in DNA in a number of different mouse tissues, with the largest age-dependent increase in brain [2]. Likewise, it was shown in another recent study that the oxidative DNA lesion 8,5′-cyclopurine-2′-deoxynucleoside accumulates with age in a tissue specific manner in mouse [3]. Using a high-performance liquid chromatography—electrochemical—detector Lee and coworkers showed a positive correlation between the level of 8oxoG in DNA and age in human gastric tissue [4]. Importantly, Hudson and coworkers have shown that 8oxoG increases three-fold with age in mitochondrial DNA of rat heart [5] and a number of other studies have reported similar results for mitochondria of other tissues including postmitotic tissues (reviewed in [6]).
Beside being exposed to endogenous ROS, the DNA of for example skin cells may also be heavily exposed to environmental factors such as UV irradiation due to sun light exposure. Photodamage leads to thymine dimers, 6–4 photoproducts, and ROS that damage genomic DNA and give rise to mutations in coding or regulatory DNA sequences of critical genes. The protective repeated DNA sequences at the end of our chromosomes, the telomeres, are disproportionately damaged by both UV and ROS, due to their greater proportion of target TT and G bases compared with the chromosomes overall. Such damage is postulated to disrupt the telomeric loop, expose the TTAGGG overhang, and promote aging (reviewed in [7]). UV radiation produced ROS may also activate cell surface receptors, which eventually leads to reduced dermal matrix formation. In dermal fibroblasts, UV irradiation also induces mitochondrial DNA deletions, leading to compromised synthesis of mitochondrial proteins, further increase of reactive oxygen species (ROS), and decreased ability of the cell to generate energy.
Epidemiological studies suggest that smoking is another important environmental factor in skin aging. Exposure to tobacco smoke causes DNA single-strand breaks, aromatic adducts, and oxidative damage to DNA, chromosome aberrations, and micronuclei (reviewed in [8]). ROS in tobacco smoke also increases the expression of matrix metalloproteinases, and the elevated enzyme levels are suggested to lead to the degradation of collagen and elastic fibers in the skin [9]. Finally, DNA double strand breaks pose severe problems for cells and this type of DNA damage also seems to increase with age in various tissues [10, 11].
3. Nuclear and Mitochondrial DNA Repair Pathways
Due to the serious consequences that DNA damage accumulation may have on cellular function and survival, different pathways of DNA repair have evolved in order to prevent it. DNA repair pathways have mainly been investigated in the nucleus; however, some of the known pathways have also been described in mitochondria (Figure 1).
Figure 1.
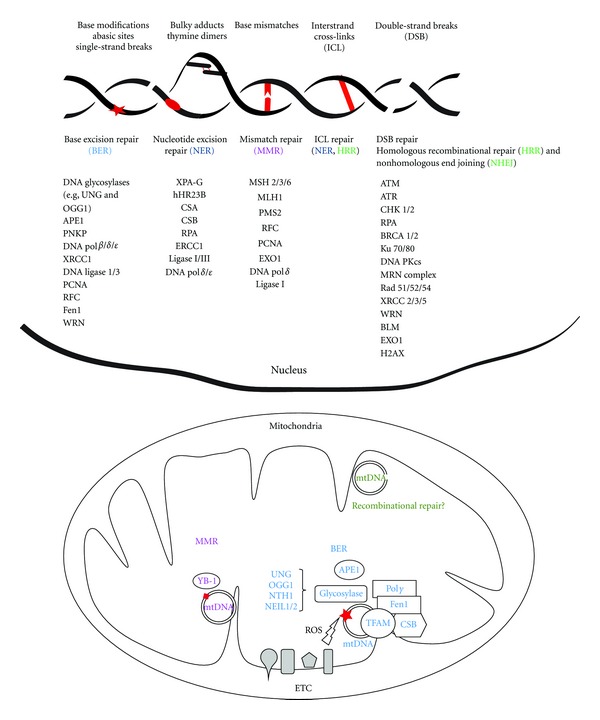
Nuclear and mitochondrial DNA repair mechanisms. DNA repair lesions are represented in red and DNA repair mechanisms are coded with specific colors. Only the central proteins involved in each pathway are represented. Enzymes involved in mitochondrial DNA repair are in some cases splice variants of the corresponding nuclear DNA repair enzymes. More detailed information about the different pathways is included in the text. BER: base excision repair; NER: nucleotide excision repair; MMR: mismatch repair; HRR: homologous recombinational repair; NHEJ: nonhomologous end joining; ETC: electron transport chain; ROS: reactive oxygen species.
3.1. Nucleotide Excision Repair (NER)
In nuclear DNA, the NER mechanism can remove numerous types of helix distorting and bulky lesions. Additionally, NER is central for the repair of DNA cross-links. Briefly, the NER pathway includes damage recognition, opening of the DNA helix, incision of the nucleotides surrounding the lesion, gap filling replication, and ligation. NER has two subpathways with different ways of detecting lesions: the global genome (GG) NER and transcription coupled (TC) NER.
Damage recognition of the NER is accomplished through sequential actions of multiple proteins. For GG-NER destabilization of the base pairing is detected by xeroderma pigmentosum complementation group C (XPC) protein in complex with the human homologue Rad23B (hHR23B) protein, which is suggested by many studies to be the first protein factor to arrive at the lesion, and it ensures a broad spectrum of substrate specificity. Additionally, UV-damaged DNA-binding protein (UV-DDB) recognizes particular types of lesions, such as UV-induced photoproducts, thereby recruiting XPC and extending the substrate specificity [12]. For TC-NER, damage recognition is believed to be caused by the blockage of the transcribing RNA polymerase II on the damaged DNA template. TC-NER is then initiated by the Cockayne Syndrome complementation group B (CSB) protein, later followed by the CSA gene product. In both GG-NER and TC-NER, the lesion recognition step is followed by recruitment of TFIIH. The XPB and XPD helicases from the 10-subunit TFIIH complex unwind the DNA around the lesion. The initial open complex is stabilized by XPG and XPA, verifying the lesion, and by RPA, which binds the opposite intact single-stranded DNA. The structure-specific endonucleases XPG and ERCC1/XPF cleave 3′ and 5′of the lesion, respectively. The resulting 24–32 nucleotide fragment, containing the lesion, is removed and the remaining single-strand gap filled by the replication machinery and the resulting nick is sealed by ligase I or ligase III [13, 14]. NER does not seem to take place in mitochondria (reviewed in [15]).
3.2. Base Excision Repair (BER)
The BER pathway is responsible for the repair of a broad spectrum of DNA base adducts. BER takes place both in nuclei and mitochondria and is therefore the main guardian against endogenously derived DNA lesions in the nucleus as well as the mitochondria [16, 17]. Thus, the BER pathway is very critical for the maintenance of genomic stability [18]. As described in the introduction, one of the most prominent theories of aging, the mitochondrial free radical theory of aging, states that free radicals generated in mitochondria are involved in the intrinsic aging process, mainly due to the accumulation of oxidative damage and derived mutations in mtDNA [19, 20]. The relevance of BER mechanisms have been highlighted by studies in yeast and animal models reporting that defects in BER enzymes shorten chronological life span and are associated with aging or age-related diseases [21, 22].
Briefly, the BER pathway is initiated by removal of the modified base by a lesion-specific mono- or bifunctional DNA glycosylase, which leaves an apurinic/apyrimidinic site (AP site). AP sites are incised by AP endonuclease 1 (APE1) generating single-strand breaks (SSBs). End processing of SSBs is necessary, since they contain obstructive 3′ or 5′ termini, and this is performed by polymerase β (Polβ), APE1, or polynucleotide kinase 3′-phosphatase (PNKP) depending on the specific terminus. Completion of the BER pathway is performed by either of two subpathways: short-patch BER or long-patch BER [23]. In the nucleus, the final steps of the short-patch BER pathway include filling of the single nucleotide gap by Polβ assisted by the scaffold protein XRCC1, and subsequently ligation by ligase 3α. In the long-patch BER pathway, one or two polymerases (Polβ and Polδ/ε) fill in the 2–13 nucleotides gap assisted by additional proteins (PCNA, RFC, and Fen1) followed by ligation by ligase I. Recently, both subpathways have been described to take place in mitochondria as well [24, 25]. In mitochondria, the only polymerase in charge of the gap filling is the polymerase γ (polγ) (reviewed in [18]).
3.3. Mismatch Repair (MMR)
Mismatched nucleotides cause genomic instability and are generated when errors of DNA polymerase escape their proofreading activity or, for example, polymerase slippage occurs at repetitive sequences. The MMR pathway was previously considered exclusively to take place in the nucleus, but several reports have indicated some form of mitochondrial MMR activity (reviewed in [15]). Briefly, the eukaryotic nuclear MMR pathway can be divided into four consecutive steps. (i) Recognition and binding of a mismatch by either a MSH2-MSH6 or MSH2-MSH3 heterodimeric ATPase complex. MSH2-MSH6 preferentially recognizes base-base mismatches and insertion-deletion loops of 1-2 nucleotides while MSH2-MSH3 has preference for larger insertion-deletion loops. The mismatch-bound MSH2-MSH6 (or MSH2-MSH3) complex recruits the MLH1-PMS2 complex in order to form a ternary complex. A proliferating cell nuclear antigen (PCNA) clamp recruits MMR proteins to the replication fork while the clamp loader replication factor C (RFC) loads PCNA. A strand-specific nick or gap, which may reside either 5′ or 3′ to the mismatch, is sufficient to direct repair in 5′- and 3′-directed MMR, respectively. (ii) Excision is performed by the exonuclease EXO1 in both 3′- and 5′-directed MMR. RPA binds to protect the single-stranded DNA during the excision and to facilitate the following DNA repair synthesis. (iii) Repair synthesis is accurately performed by Polδ. (iv) Ligation of the remaining nicks after DNA synthesis is performed by ligase I (reviewed in [26]). MMR proteins have been identified in yeast and coral mitochondria but MMR complexes, as we know them from the nucleus, have not yet been detected in human mitochondria. Still, human mitochondria do seem to have mismatch-repair activity, which involves proteins distinct from nuclear MMR. One of these proteins is the repair factor YB-1 [27].
3.4. Recombinational Repair
DNA double-strand breaks (DSBs) are some of the most hazardous DNA lesions, since they can lead to genome rearrangements. The DSB repair pathway is regulated by several phosphorylation events, starting immediately after DSB formation, where large numbers of the histone protein H2AX are phosphorylated (γH2AX) and accumulate in the chromatin around the break [28, 29]. Several DSB damage response proteins accumulate in foci around DSBs and send signals via signal transducers to a set of downstream effectors, which affect events like DNA repair, cell cycle checkpoints, telomere maintenance, and transcription. Two major mechanisms exist to repair DSBs: homologous recombinational repair (HRR) and nonhomologous end joining (NHEJ). The main difference between the two major DSB repair pathways is the error-prone nature of NHEJ in contrast to the error-free HRR. The balance between the DSB repair pathways differs among species, cell types, and during the phases of the cell cycle [30].
The NHEJ pathway takes place throughout the cell cycle, whereas HRR repairs DSBs during the S and G2 phase of the cell cycle [31]. Recognition of DSBs in NHEJ is performed by the Ku70/80 heterodimer, which also recruits DNA PKcs forming a DNA PK complex on both sides of the DSB [32, 33]. The DNA ends are processed followed by autophosphorylation of DNA PKcs that regulates the final ligation [34]. HRR uses homologous sequences in sister chromatids to repair DSBs, especially those formed at collapsed replication forks. Phosphorylation of BRCA1 (by ATM, ATR, and CHK2) regulates the MRN end processes. Then the 3′ssDNA ends are bound to RPA, and following several phosphorylation steps and the action of different enzymes (Rad52, BRCA2, and CDKs), the 3′ssDNA ends are bound to Rad51 leading to the formation of a nucleoprotein filament that invades a homologous sequence. The invading and complimentary strands extend and once all intermediates are repaired, the HRR pathway is terminated. DSBs are believed to contribute to mtDNA rearrangements observed during aging [35]. Although some reports have suggested mitochondrial recombinational repair in model organisms (yeast and Drosophila melanogaster) [36–38], our knowledge of mammalian mtDNA recombination is still limited (reviewed in [39]).
4. Human Aging and Single-Nucleotide Polymorphisms (SNPs) in Genes Related to DNA Repair Mechanisms
The involvement of genetic factors on longevity has been investigated by heritability studies using twin cohorts. The concept of heritability is valuable for identifying to which extend individual genetic differences contribute to observed individual differences for a specific trait in a study population [40]. Several of those heritability studies have suggested that additive genetic factors account for a quarter to one-third of the variation in human lifespan [41–43]. Thus, epidemiological geneticists have been searching for genes involved in longevity, and recent population studies have investigated correlations of numerous single-nucleotide polymorphisms (SNPs) with longevity in order to identify longevity genes. One gene that has been described to strongly relate to longevity is the forkhead box O3A (FOXO3) [44, 45], which is a transcription factor involved in apoptosis and protection from oxidative stress [46]. Additionally, FOXO3 regulates the stress resistance of cells by facilitating the repair of damaged DNA [47].
Similar studies with other genes have led to the conclusion that it may be possible to identify genetic variants that affect lifespan, inspiring human geneticists to investigate different candidate genes that may be involved in human longevity. Genes related to the maintenance of DNA stability are among those candidate genes.
Initial findings were obtained by analyzing single or a few SNPs in selected candidate genes. The human exonuclease 1 (EXO1) gene was identified as a potential longevity candidate gene by investigating SNPs in approximately 400 German and French centenarians [48]. EXO1 is a 5′-3′ exonuclease participating in MMR and homologous recombination [49]. Interestingly, EXO1 also interacts with the WRN protein. As will be described in more detail later, WRN protein is crucial for the maintenance of genomic stability [50]. Additionally, an association study on SNPs in the WRN gene suggested associations between WRN polymorphisms, longevity, and age-associated diseases in Mexican and Finnish populations [51]. Hence, WRN is suggested to be a longevity candidate gene. The antioxidant enzymes superoxide dismutases (SOD1 and SOD2) are responsible for converting superoxide radicals, which are harmful to macromolecules and to oxygen and hydrogen peroxide. Variations in SOD1 and SOD2 were shown to influence human longevity, and thus also suggested as candidate genes for longevity [52].
Despite that some studies suggest central DNA repair and antioxidant genes as candidate longevity genes, it has been difficult to replicate these findings in independent study populations. Thus, identification of universal longevity polymorphisms is a difficult task, due to the fact that genetics of longevity seems to be extremely complicated.
It is now widely accepted that complex traits, such as longevity, are determined by numerous genes with small effects, and results from single SNP analysis provide limited biological insight. Instead of analyzing single SNPs, recent studies have investigated combined effect of a group of SNPs located in genes involved in the same biological pathway. Successive to the HapMap project [53], the tagging SNP approach is considered a stronger approach than investigating few gene variations, since the most common gene variations in the entire gene is covered. The most comprehensive study of large collections of longevity candidate variations to date has identified novel SNPs to be associated with human longevity [54]. By a case control design, including 1089 oldest-old and 736 middle-aged, Danes, Soerensen and coworkers investigated several rare allele variants in genes involved in DNA metabolism, such as BER genes (NTHL1, Polβ, and WRN) and DSB repair genes (RAD52, H2AFX, XRCC5, and WRN). By a longitudinal study approach, SNPs located in genes coding for DNA repair and antioxidant gene products were identified to associate with longevity [54]. Rare allele variants of MLH1, involved in MMR, and XRCC5, involved in DSB repair, as well as rare allele variants of XDH and TXNRD1, pro- and antioxidant enzymes, were advantageous, whereas the presence of rare allele variants in H2AFX, related to nucleosome formation and DNA damage signaling, appeared disadvantageous for longevity. Together, the studies suggest new candidate variations for human longevity located in pro-/antioxidant, DNA damage signaling and DNA repair genes, and highlight the importance of functional studies of the molecular effects of these newly presented candidate variants [54].
Increased telomere length has been associated with longevity in human leukocytes [55], and decreased telomere length in leukocytes has been associated with increased mortality [56]. At the telomere ends, T-loops are formed, which stabilize the telomeric region and prevent the ends from being recognized as DNA breaks by DNA repair proteins. The shelterin complex holds the T-loop together, protecting the telomeres [57]. Absence of T-loops will allow NHEJ at the telomeric ends, resulting in chromosomal fusion. The telomere length is regulated by telomerases and associated factors [58]. Recent studies have investigated the associations between telomere length and SNPs in telomerase and shelterin genes [55, 59–62]. The most promising results have been reported in relation to TERC and TERT genes; interestingly two of these studies have also described associations between the TERT and TERC genes and human longevity [55, 61].
5. Age Regulation of DNA Repair in Human Population Studies
Previous studies investigating SSBR in peripheral blood mononuclear cells (PBMCs) from differently aged donors suggest minor or no effect of age [63–66]; however, the endogenous level of SSBs was found to be significantly higher at old age [67–69]. An age-related decline of NHEJ functions in brain tissue has been suggested from animal studies [70, 71]. In line with these results, PBMCs from elderly humans showed reduced nuclear localization and DNA binding of the NHEJ specific Ku70/80 complex compared to PBMCs from young humans [72]. In another study, PMBCs showed an age-related decline of Ku70 levels [73], and a different study reported declining levels of Ku80 with age [74]. Using a host cell reactivation assay, Wei and coworkers found that the human NER capacity in PMBCs declines with approximately 0.6% per year [75]. Together, these studies suggest biological changes in DNA repair with age, while powerful functional studies are missing.
6. Animal Models as a Useful Approach to Human Aging
As previously described, DNA repair pathways have evolved as important systems for maintenance of DNA stability and cell survival. Proteins in the main DNA repair pathways are highly conserved [76, 77], and such high degree of conservation indicates that DNA repair pathways are fundamental mechanisms in cell survival. As observed in other DNA repair pathways, many of the genes involved in the BER pathway are highly conserved from bacteria to humans [78–80].
Since aging mechanisms have been proposed to be universal [81], animal models have become an excellent tool for investigation of the molecular mechanisms involved in the human aging process, including DNA repair pathways. One of the main advantages of animal models is that most of them have relatively short life span and that genetic modification is possible. Along with mammalian models, aging research studies and DNA metabolism include unicellular organisms and multicellular eukaryotes such as D. melanogaster, Caenorhabditis elegans, [82–85], or filamentous fungi like Podospora anserina [80, 86]. Investigations in these models have contributed notably to the understanding of the role of DNA repair mechanisms in the aging process in humans, particularly those taking place in mitochondria.
One of the first studies using mammalian models for investigating the relation between aging and DNA repair mechanisms was the one performed by Hart and Setlow [87]. They investigated the repair of UV-light induced DNA damage in fibroblasts of different mammalian species, reporting that DNA repair activity was inversely correlated to the maximum life span potential (MLSP) [87]. In a similar recent investigation on C. elegans strains showing different longevities, long-lived strains showed higher rates of UV-induced DNA damage repair than the wildtype nematodes [83]. Comparative studies using different mammalian species have also been used in order to investigate the correlation between BER mechanisms and MLSP. Brown and Stuart reported no correlation, when nuclear BER activities were analyzed in mammalian dermal fibroblasts [88]. Up to now, no investigation has been performed analyzing the correlation between mitochondrial BER and MLSP.
Compiling evidence supports that during aging alterations in BER capacity occur, age-related deficiencies in those mechanisms play an important role in cell function and survival, particularly in postmitotic tissues. As mentioned above, the mtBER pathway is highly conserved among mammalian species, and mice are widely used for investigating the role of mtBER in the aging process. Mitochondrial BER capacity has been described to be organ-specific, with the brain being one of the tissues with the lowest capacity [89]. Various studies have investigated the role of changes in mitochondrial BER in age-related functional decline, showing tissue specific age-related changes. Although some studies have observed a reduction in mtBER with aging in rat liver mitochondria [90], most of the investigations in murine mitochondria from hepatic and cardiac tissues have reported increases in APE1 activity and increases or no changes in DNA glycosylase activities during aging [91–93]. On the other hand, a general decline in DNA glycosylase activity has been observed in brain cortical mitochondria in rats [94] and mice [95, 96]. These results support the idea that mtBER plays a critical role in the maintenance of the central nervous system during aging [97]. We have reported significant differences in mtBER among various mouse brain regions during aging. Thus, we found that in cortical mitochondria, DNA glycosylase activities peaked at middle age followed by a significant drop at old age. However, only minor changes were observed in hippocampal mitochondria during the whole lifespan of the animals. Mitochondrial AP endonuclease activity increased in old animals in both brain regions [96]. The cortical region and the cerebellum have been described to accumulate less mtDNA lesions with aging and to be more resistant to oxidative stress conditions [98–100]. Interestingly, those regions showed higher BER capacity than hippocampus, which has been described to be a much more vulnerable region in the brain [98, 99]. A recent investigation also supports the relevance of mtBER in brain aging and stresses the importance of the central nervous system heterogeneity in these processes, since the age-related decline in brain mtBER seems to occur specifically at the synapses [101]. Although investigations on mtBER in humans are scarce, results obtained on mitochondrial, nuclear, or total BER from central nervous system cell cultures or even postmortem neuronal human tissue suggest that the results obtained from mouse studies can be transferred into humans. Thus, increasing Ogg1 activity in mitochondria from oligodendrocytes after targeting hOGG1 into mitochondria increases cell survival and protects against induced-oxidative stress [102]. Moreover, Weissman and coworkers reported that a significant decline in total BER takes place in the cortical area of Alzheimer's patients [103]. Postmitotic tissues are specially affected during aging, and an age-related decline has also been reported in mtBER in skeletal muscle [104], which has been suggested to contribute to age-related sarcopenia. Studies in the aging model P. anserina have also shown that aging is associated with a decrease in mitochondrial BER [80], likely contributing to the observed mtDNA instability in the aged fungi [105].
An important approach in the study of the role of DNA repair in the aging process is the development of several knockout (KO) mice. Among them, those models being deficient in essential enzymes in the BER pathway have resulted in embryonic lethality, such as APE1 or Polβ or ligase III [106–108], revealing the importance of these enzymes in cellular survival. Since DNA glycosylases have a certain grade of overlapping activity, KO mice for these enzymes generally result in elevated levels of DNA lesions but no apparent aging phenotype, although some KO mice show higher incidence of cancer [109–111].
Mice mutated in Ku70 or Ku80, proteins involved in repair of DSBs, display an early aging phenotype [112], and are therefore an interesting model for studying the role of NHEJ in aging. Also, Ercc1 and Xpf mutant mice, with a defect in NER and cross-link repair, display reduced lifespan and a broad range of aging-associated changes at an early age, and thus provides another valuable model for studying the role of DNA repair mechanisms in aging [113]. Mice carrying a specific XPD point mutation found in several patients suffering from trichothiodystrophy (TTD) recapitulate the human disorder to a great extent, but in addition they display pathology consistent with accelerated aging (reviewed in [114]).
A mouse model developed in the last decade, the mitochondrial mutator mouse, appears as an interesting model in which to investigate the causative link between mtDNA mutation accumulation and aging, and the role that mtBER may play in the process [115, 116]. Moreover, the generation of this knock-in mouse expressing proof-reading deficient Polγ, but conserved replicative function, is considered by some authors as an important support of accelerated mtDNA mutation rate resulting in increased aging rate [117, 118]. In various tissues of this mouse model, mtDNA point mutations as well as mtDNA deletions accumulate at a much higher rate than in the wildtype mice [115, 116]. However, whether it is the accumulation of mtDNA deletions or point mutations that drive the premature aging phenotype and the precise mechanisms are still discussed [119, 120].
Some animal models have also been used in order to investigate the role of DNA repair mechanisms on age-related diseases, mainly age-related neurodegenerative diseases. Thus, different mouse models have been created in attempts to elucidate the underlying molecular mechanisms for disorders such as Alzheimer's (AD), Parkinson's (PD), or Huntington's disease (HD), and some of them have been used to investigate the potential role of DNA repair in those disorders [14, 121]. Investigations of age-related neurodegenerative diseases on humans are normally limited to postmortem tissue samples; consequently, animal models of those diseases are very valuable. However, the results observed in animal models do not always match those found in humans. For instance, although a general decline in BER has been described in postmortem cortical tissue in AD patients [103], no changes have been observed in BER mechanisms when the investigations have been performed in the APP and 3xTgAD mouse models [101, 122]. Interestingly, these particular models, unlike humans, do not display neurodegeneration despite the fact that both amyloid-β plaques and neurofibrillary tangles accumulate. Two well-established mouse models for HD have related DNA repair mechanisms with the progression of the disease. Whereas investigations on the HdhQ111 knock-in mice have supported the role of mismatch repair proteins in HD [123, 124], studies on R6/1 mice have revealed the contribution of BER enzymes, particularly OGG1, to CAG expansion in somatic cells that takes place in HD [121]. A recent report suggests that CSB may also be involved in promoting CAG repeat expansion in HD [125, 126].
7. Premature Aging Syndromes
Finally, another important approach to human aging is the study of genetically inherited premature aging syndromes. Such syndromes have been widely used as valuable tools in understanding the normal aging process [127]. Similarly to age-related neurodegenerative diseases, the investigation of human tissues in these syndromes is limited due to scarcely available tissue samples. Thus, research on mouse models and established human cell lines has been critical for understanding some of the mechanisms involved in premature aging syndromes and furthermore increased the understanding of the normal aging process in humans. The DNA repair defective disease Cockayne syndrome (CS), which is a segmental premature aging syndrome, is associated with severe developmental deficiencies and neurodegeneration. Although transcription coupled NER (TC-NER) is a prominent DNA repair pathway affected in this syndrome, an increasing number of investigations indicate that BER is affected as well [128]. In the last years, the investigations that have been carried out on different models of CS, particularly on the models of CSB, have dramatically improved our knowledge about this syndrome and the several roles that the CSB protein plays in genomic stability and cellular survival. Along with the alterations in TC-NER, deficiency in the repair of 8oxoG was initially reported both in whole cell extracts [129] and in mitochondria from mammalian CSB-deficient cells [130]. Moreover, CSB seems to play a role in general mitochondrial maintenance [131]. In accordance, the CSB protein, which was thought to be present exclusively in the nucleus, has recently been shown to localize to mitochondria [132]. This investigation suggests that CSB protein plays a direct role in mtBER by interacting and stabilizing BER proteins in the protein-DNA complexes associated with the inner mitochondrial membrane when mtDNA repair takes place. In addition, it has recently been reported that CSB-deficient cells show higher free radical production and accumulation of damaged mitochondria together with altered mitochondrial autophagy [133]. Thus, the authors suggest that CSB may act as an mtDNA damage sensor, inducing mitochondrial autophagy in response to stress.
Werner syndrome (WS) is another genetically inherited premature aging syndrome, which has been studied intensely with regard to the molecular role of the affected gene product, WRN. The syndrome is characterized by premature graying and thinning of hair, cataracts, diabetes mellitus, osteoporosis, and a number of other typical age-associated deficiencies. The affected WRN protein is a member of the so-called RecQ helicases and orthologs exist in a wide variety of organisms such as Saccharomyces cerevisiae, Schizosaccharomyces pombe, C. elegans, and Xenopus laevis (reviewed in [134]). Based on cellular and biochemical studies of WRN-deficient cell lines, the WRN protein has been suggested to have multiple roles in DNA repair. Hence, the protein interacts with several DNA repair proteins, which are involved in BER, NHEJ, or HHR. This is in agreement with the fact that WS cells show phenotypes such as nonhomologous chromosome exchanges and large chromosomal deletions, caused by deficiency of DSBR [135], and accumulation of, for example, Fapy lesions, which are induced by ROS [136].
In conclusion increasing evidence suggests that DNA repair mechanisms are involved in the aging process. Additional studies using various model systems will help us gain an even better understanding on the functional relationship between these associations.
Authors' Contributions
All authors contributed equally to this paper.
References
- 1.Harman D. Role of free radicals in aging and disease. Annals of the New York Academy of Sciences. 1992;673:126–141. doi: 10.1111/j.1749-6632.1992.tb27444.x. [DOI] [PubMed] [Google Scholar]
- 2.Gan W, Nie B, Shi F, et al. Age-dependent increases in the oxidative damage of DNA, RNA, and their metabolites in normal and senescence-accelerated mice analyzed by LC-MS/MS: urinary 8-oxoguanosine as a novel biomarker of aging. Free Radical Biology and Medicine. 2012;52(9):1700–1707. doi: 10.1016/j.freeradbiomed.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Clauson CL, Robbins PD, et al. The oxidative DNA lesions 8, 5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell. 2012;11(4):714–716. doi: 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee BM, Kwack SJ, Kim HS. Age-related changes in oxidative DNA damage and benzo(a)pyrene diolepoxide-I (BPDE-I)-DNA adduct levels in human stomach. Journal of Toxicology and Environmental Health A. 2005;68(19):1599–1610. doi: 10.1080/15287390500182818. [DOI] [PubMed] [Google Scholar]
- 5.Hudson EK, Hogue BA, Souza-Pinto NC, et al. Age-associated change in mitochondrial DNA damage. Free Radical Research. 1998;29(6):573–579. doi: 10.1080/10715769800300611. [DOI] [PubMed] [Google Scholar]
- 6.Lee HC, Wei YH. Mitochondria and aging. Advances in Experimental Medicine and Biology. 2012;942:311–327. doi: 10.1007/978-94-007-2869-1_14. [DOI] [PubMed] [Google Scholar]
- 7.Yaar M, Gilchrest BA. Photoageing: mechanism, prevention and therapy. British Journal of Dermatology. 2007;157(5):874–887. doi: 10.1111/j.1365-2133.2007.08108.x. [DOI] [PubMed] [Google Scholar]
- 8.Husgafvel-Pursiainen K. Genotoxicity of environmental tobacco smoke: a review. Mutation Research. 2004;567(2-3):427–445. doi: 10.1016/j.mrrev.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Morita A. Tobacco smoke causes premature skin aging. Journal of Dermatological Science. 2007;48(3):169–175. doi: 10.1016/j.jdermsci.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Singh NP, Ogburn CE, Wolf NS, Van Belle G, Martin GM. DNA double-strand breaks in mouse kidney cells with age. Biogerontology. 2001;2(4):261–270. doi: 10.1023/a:1013262327193. [DOI] [PubMed] [Google Scholar]
- 11.Swain U, Subba Rao K. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mechanisms of Ageing and Development. 2011;132( 8–9):374–381. doi: 10.1016/j.mad.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 12.Sugasawa K. Multiple DNA damage recognition factors involved in mammalian nucleotide excision repair. Biochemistry. 2011;76(1):16–23. doi: 10.1134/s0006297911010044. [DOI] [PubMed] [Google Scholar]
- 13.de Boer J, Hoeijmakers JHJ. Nucleotide excision repair and human syndromes. Carcinogenesis. 2000;21(3):453–460. doi: 10.1093/carcin/21.3.453. [DOI] [PubMed] [Google Scholar]
- 14.Jeppesen DK, Bohr VA, Stevnsner T. DNA repair deficiency in neurodegeneration. Progress in Neurobiology. 2011;94(2):166–200. doi: 10.1016/j.pneurobio.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gredilla R, Bohr VA, Stevnsner T. Mitochondrial DNA repair and association with aging—an update. Experimental Gerontology. 2010;45(7-8):478–488. doi: 10.1016/j.exger.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Letters. 2000;476(1-2):73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 17.Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 18.Gredilla R. DNA damage and base excision repair in mitochondria and their role in aging. Journal of Aging Research. 2010;2011 doi: 10.4061/2011/257093.257093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harman D. Free radical theory of aging. Mutation Research. 1992;275(3–6):257–266. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 20.Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. American Journal of Physiology. 2007;292(1):R18–R36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- 21.Maclean MJ, Aamodt R, Harris N, et al. Base excision repair activities required for yeast to attain a full chronological life span. Aging Cell. 2003;2(2):93–104. doi: 10.1046/j.1474-9728.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 22.Marusyk A, DeGregori J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochimica et Biophysica Acta. 2008;1785(1):1–11. doi: 10.1016/j.bbcan.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson DM, III, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6(4):544–559. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Zheng L, Zhou M, Guo Z, et al. Human DNA2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Molecular Cell. 2008;32(3):325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Copeland WC, Longley MJ. DNA2 resolves expanding flap in mitochondrial base excision repair. Molecular Cell. 2008;32(4):457–458. doi: 10.1016/j.molcel.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiricny J. The multifaceted mismatch-repair system. Nature Reviews Molecular Cell Biology. 2006;7(5):335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 27.de Souza-Pinto NC, Mason PA, Hashiguchi K, et al. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair. 2009;8(6):704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muslimovic A, Ismail IH, Gao Y, Hammarsten O. An optimized method for measurement of gamma-H2AX in blood mononuclear and cultured cells. Nature Protocols. 2008;3(7):1187–1193. doi: 10.1038/nprot.2008.93. [DOI] [PubMed] [Google Scholar]
- 29.Bonner WM, Redon CE, Dickey JS, et al. GammaH2AX and cancer. Nature Reviews Cancer. 2008;8(12):957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shrivastav M, Miller CA, De Haro LP, et al. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair. 2009;8(8):920–929. doi: 10.1016/j.dnarep.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Research. 2008;18(1):134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 32.Calsou P, Delteil C, Frit P, Drouet J, Salles B. Coordinated assembly of Ku and p460 subunits of the DNA-dependent protein kinase on DNA ends is necessary for XRCC4-ligase IV recruitment. Journal of Molecular Biology. 2003;326(1):93–103. doi: 10.1016/s0022-2836(02)01328-1. [DOI] [PubMed] [Google Scholar]
- 33.Yoo S, Dynan WS. Geometry of a complex formed by double strand break repair proteins at a single DNA end: recruitment of DNA-PKcs induces inward translocation of Ku protein. Nucleic Acids Research. 1999;27(24):4679–4686. doi: 10.1093/nar/27.24.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Block WD, Yu Y, Merkle D, et al. Autophosphorylation-dependent remodeling of the DNA-dependent protein kinase catalytic subunit regulates ligation of DNA ends. Nucleic Acids Research. 2004;32(14):4351–4357. doi: 10.1093/nar/gkh761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukui H, Moraes CT. Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Human Molecular Genetics. 2009;18(6):1028–1036. doi: 10.1093/hmg/ddn437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graziewicz MA, Longley MJ, Copeland WC. DNA polymerase γ in mitochondrial DNA replication and repair. Chemical Reviews. 2006;106(2):383–405. doi: 10.1021/cr040463d. [DOI] [PubMed] [Google Scholar]
- 37.Morel F, Renoux M, Lachaume P, Alziari S. Bleomycin-induced double-strand breaks in mitochondrial DNA of Drosophila cells are repaired. Mutation Research. 2008;637(1-2):111–117. doi: 10.1016/j.mrfmmm.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 38.Sickmann A, Reinders J, Wagner Y, et al. The proteome of Saccharomyces cerevisiae mitochondria. Proceedings of the National Academy of Sciences United States of America. 2003;100(23):13207–13212. doi: 10.1073/pnas.2135385100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu P, Demple B. DNA repair in mammalian mitochondria: much more than we thought? Environmental and Molecular Mutagenesis. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 40.van Dongen J, Slagboom PE, Draisma HHM, et al. The continuing value of twin studies in the omics era. Nature Reviews Genetics. 2012;13(9):640–653. doi: 10.1038/nrg3243. [DOI] [PubMed] [Google Scholar]
- 41.McGue M, Vaupel JW, Holm N, Harvald B. Longevity is moderately heritable in a sample of Danish twins born 1870–1880. Journals of Gerontology. 1993;48(6):B237–B244. doi: 10.1093/geronj/48.6.b237. [DOI] [PubMed] [Google Scholar]
- 42.Herskind AM, McGue M, Holm NV, Sørensen TIA, Harvald B, Vaupel JW. The heritability of human longevity: a population-based study of 2872 Danish twin pairs born 1870–1900. Human Genetics. 1996;97(3):319–323. doi: 10.1007/BF02185763. [DOI] [PubMed] [Google Scholar]
- 43.Ljungquist B, Berg S, Lanke J, McClearn GE, Pedersen NL. The effect of genetic factors for longevity: a comparison of identical and fraternal twins in the Swedish Twin Registry. Journals of Gerontology A. 1998;53(6):M441–M446. doi: 10.1093/gerona/53a.6.m441. [DOI] [PubMed] [Google Scholar]
- 44.Willcox BJ, Donlon TA, He Q, et al. FOXO3A genotype is strongly associated with human longevity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(37):13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soerensen M, Dato S, Christensen K, et al. Replication of an association of variation in the FOXO3A gene with human longevity using both case-control and longitudinal data. Aging Cell. 2010;9(6):1010–1017. doi: 10.1111/j.1474-9726.2010.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Experimental Diabetes Research. 2012;2012:7 pages. doi: 10.1155/2012/939751.939751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran H, Brunet A, Grenier JM, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3A through the Gadd45 protein. Science. 2002;296(5567):530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 48.Nebel A, Flachsbart F, Till A, et al. A functional EXO1 promoter variant is associated with prolonged life expectancy in centenarians. Mechanisms of Ageing and Development. 2009;130(10):691–699. doi: 10.1016/j.mad.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Liberti SE, Rasmussen LJ. Is hEXO1 a cancer predisposing gene? Molecular Cancer Research. 2004;2(8):427–432. [PubMed] [Google Scholar]
- 50.Sharma S, Sommers JA, Driscoll HC, Uzdilla L, Wilson TM, Brosh RM. The exonucleolytic and endonucleolytic cleavage activities of human exonuclease 1 are stimulated by an interaction with the carboxyl-terminal region of the Werner syndrome protein. Journal of Biological Chemistry. 2003;278(26):23487–23496. doi: 10.1074/jbc.M212798200. [DOI] [PubMed] [Google Scholar]
- 51.Castro E, Edland SD, Lee L, et al. Polymorphisms at the Werner locus: II., 1074Leu/Phe, 1367Cys/Arg, longevity, and atherosclerosis. American Journal of Medical Genetics A. 2000;95(4):374–380. [PubMed] [Google Scholar]
- 52.Yashin AI, De Benedictis G, Vaupel JW, et al. Genes and longevity: lessons from studies of centenarians. Journal of Gerontology. 2000;55(7):B319–B328. doi: 10.1093/gerona/55.7.b319. [DOI] [PubMed] [Google Scholar]
- 53.International HapMap Consortium. The International HapMap Project. Nature. 2003;426(6968):789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 54.Soerensen M, Dato S, Tan Q, et al. Human longevity and variation in GH/IGF-1/insulin signaling, DNA damage signaling and repair and pro/antioxidant pathway genes: cross sectional and longitudinal studies. Experimental Gerontology. 2012;47(5):379–387. doi: 10.1016/j.exger.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atzmon G, Cho M, Cawthon RM, et al. Evolution in health and medicine Sackler colloquium: genetic variation in human telomerase is associated with telomere length in Ashkenazi centenarians. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(supplement 1):1710–1717. doi: 10.1073/pnas.0906191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimura M, Hjelmborg JVB, Gardner JP, et al. Telomere length and mortality: a study of leukocytes in elderly danish twins. American Journal of Epidemiology. 2008;167(7):799–806. doi: 10.1093/aje/kwm380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes and Development. 2005;19(18):2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 58.Collins K, Mitchell JR. Telomerase in the human organism. Oncogene. 2002;21(4):564–579. doi: 10.1038/sj.onc.1205083. [DOI] [PubMed] [Google Scholar]
- 59.Levy D, Neuhausen SL, Hunt SC, et al. Genome-wide association identifies OBFC1 as a locus involved in human leukocyte telomere biology. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(20):9293–9298. doi: 10.1073/pnas.0911494107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Codd V, Mangino M, Van Der Harst P, et al. Common variants near TERC are associated with mean telomere length. Nature Genetics. 2010;42(3):197–199. doi: 10.1038/ng.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soerensen M, Thinggaard M, Nygaard M, et al. Genetic variation in TERT and TERC and human leukocyte telomere length and longevity: a cross-sectional and longitudinal analysis. Aging Cell. 2012;11(2):223–227. doi: 10.1111/j.1474-9726.2011.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deelen J, Uh HW, Monajemi R, et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenancepathways. doi: 10.1007/s11357-011-9340-3. Age. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Müller WU, Bauch T, Stüben G, Sack H, Streffer C. Radiation sensitivity of lymphocytes from healthy individuals and cancer patients as measured by the comet assay. Radiation and Environmental Biophysics. 2001;40(1):83–89. doi: 10.1007/s004110000087. [DOI] [PubMed] [Google Scholar]
- 64.Zana M, Szécsényi A, Czibula A, et al. Age-dependent oxidative stress-induced DNA damage in Down's lymphocytes. Biochemical and Biophysical Research Communications. 2006;345(2):726–733. doi: 10.1016/j.bbrc.2006.04.167. [DOI] [PubMed] [Google Scholar]
- 65.Trzeciak AR, Barnes J, Ejiogu N, et al. Age, sex, and race influence single-strand break repair capacity in a human population. Free Radical Biology and Medicine. 2008;45(12):1631–1641. doi: 10.1016/j.freeradbiomed.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trzeciak AR, Mohanty JG, Jacob KD, et al. Oxidative damage to DNA and single strand break repair capacity: relationship to other measures of oxidative stress in a population cohort. Mutation Research. 2012;736(1-2):93–103. doi: 10.1016/j.mrfmmm.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mutlu-Türkoğlu Ü, Ilhan E, Öztezcan S, Kuru A, Aykaç-Toker G, Uysal M. Age-related increases in plasma malondialdehyde and protein carbonyl levels and lymphocyte DNA damage in elderly subjects. Clinical Biochemistry. 2003;36(5):397–400. doi: 10.1016/s0009-9120(03)00035-3. [DOI] [PubMed] [Google Scholar]
- 68.Humphreys V, Martin RM, Ratcliffe B, et al. Age-related increases in DNA repair and antioxidant protection: a comparison of the Boyd Orr Cohort of elderly subjects with a younger population sample. Age and Ageing. 2007;36(5):521–526. doi: 10.1093/ageing/afm107. [DOI] [PubMed] [Google Scholar]
- 69.Møller P. Assessment of reference values for DNA damage detected by the comet assay in human blood cell DNA. Mutation Research. 2006;612(2):84–104. doi: 10.1016/j.mrrev.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 70.Ren K, Peña De Ortiz S. Non-homologous DNA end joining in the mature rat brain. Journal of Neurochemistry. 2002;80(6):949–959. doi: 10.1046/j.0022-3042.2002.00776.x. [DOI] [PubMed] [Google Scholar]
- 71.Vyjayanti VN, Rao KS. DNA double strand break repair in brain: reduced NHEJ activity in aging rat neurons. Neuroscience Letters. 2006;393(1):18–22. doi: 10.1016/j.neulet.2005.09.053. [DOI] [PubMed] [Google Scholar]
- 72.Frasca D, Barattini P, Tirindelli D, et al. Effect of age on DNA binding of the ku protein in irradiated human peripheral blood mononuclear cells (PBMC) Experimental Gerontology. 1999;34(5):645–658. doi: 10.1016/s0531-5565(99)00026-1. [DOI] [PubMed] [Google Scholar]
- 73.Ju YJ, Lee KH, Park JE, et al. Decreased expression of DNA repair proteins Ku70 and Mre11 is associated with aging and may contribute to the cellular senescence. Experimental and Molecular Medicine. 2006;38(6):686–693. doi: 10.1038/emm.2006.81. [DOI] [PubMed] [Google Scholar]
- 74.Scarpaci S, Frasca D, Barattini P, Guidi L, Doria G. DNA damage recognition and repair capacities in human naïve and memory T cells from peripheral blood of young and elderly subjects. Mechanisms of Ageing and Development. 2003;124(4):517–524. doi: 10.1016/s0047-6374(03)00030-7. [DOI] [PubMed] [Google Scholar]
- 75.Wei Q, Matanoski GM, Farmer ER, et al. DNA repair and aging in basal cell carcinoma: a molecular epidemiology study. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(4):1614–1618. doi: 10.1073/pnas.90.4.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aravind L, Walker DR, Koonin EV. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Research. 1999;27(5):1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gros L, Saparbaev MK, Laval J. Enzymology of the repair of free radicals-induced DNA damage. Oncogene. 2002;21(58):8905–8925. doi: 10.1038/sj.onc.1206005. [DOI] [PubMed] [Google Scholar]
- 78.Olsen LC, Aasland R, Wittwer CU, Krokan HE, Helland DE. Molecular cloning of human uracil-DNA glycosylase, a highly conserved DNA repair enzyme. EMBO Journal. 1989;8(10):3121–3125. doi: 10.1002/j.1460-2075.1989.tb08464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krokan HE, Standal R, Slupphaug G. DNA glycosylases in the base excision repair of DNA. Biochemical Journal. 1997;325(1):1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soerensen M, Gredilla R, Müller-Ohldach M, et al. A potential impact of DNA repair on ageing and lifespan in the ageing model organism Podospora anserina: decrease in mitochondrial DNA repair activity during ageing. Mechanisms of Ageing and Development. 2009;130(8):487–496. doi: 10.1016/j.mad.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Strehler BL. Time, Cells and Aging. New York, NY, USA: Academic Press; 1977. [Google Scholar]
- 82.Oliveira BF, Nogueira-Machado JA, Chaves MM. The role of oxidative stress in the aging process. The Scientific World Journal. 2010;10:1121–1128. doi: 10.1100/tsw.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hyun M, Lee J, Lee K, May A, Bohr VA, Ahn B. Longevity and resistance to stress correlate with DNA repair capacity in Caenorhabditis elegans. Nucleic Acids Research. 2008;36(4):1380–1389. doi: 10.1093/nar/gkm1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meyer JN, Boyd WA, Azzam GA, Haugen AC, Freedman JH, Van Houten B. Decline of nucleotide excision repair capacity in aging Caenorhabditis elegans. Genome Biology. 2007;8(5, article R70) doi: 10.1186/gb-2007-8-5-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morinaga H, Yonekura SI, Nakamura N, Sugiyama H, Yonei S, Zhang-Akiyama QM. Purification and characterization of Caenorhabditis elegans NTH, a homolog of human endonuclease III: essential role of N-terminal region. DNA Repair. 2009;8(7):844–851. doi: 10.1016/j.dnarep.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 86.Scheckhuber CQ, Osiewacz HD. Podospora anserina: a model organism to study mechanisms of healthy ageing. Molecular Genetics and Genomics. 2008;280(5):365–374. doi: 10.1007/s00438-008-0378-6. [DOI] [PubMed] [Google Scholar]
- 87.Hart RW, Setlow RB. Correlation between deoxyribonucleic acid excision repair and life span in a number of mammalian species. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(6):2169–2173. doi: 10.1073/pnas.71.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brown MF, Stuart JA. Correlation of mitochondrial superoxide dismutase and DNA polymerase β in mammalian dermal fibroblasts with species maximal lifespan. Mechanisms of Ageing and Development. 2007;128(11-12):696–705. doi: 10.1016/j.mad.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 89.Karahalil B, Hogue BA, De Souza-Pinto NC, Bohr VA. Base excision repair capacity in mitochondria and nuclei: tissue-specific variations. FASEB Journal. 2002;16(14):1895–1902. doi: 10.1096/fj.02-0463com. [DOI] [PubMed] [Google Scholar]
- 90.Leclère R, Torregrossa R, Garcia C, Vara E, Treguerres JAF, Gredilla R. Aging, estrogens and mitochondrial DNA repair in rat liver. In: Delgado MD, editor. Advances in Comparative Endocrinology, Volume 6. 2012. In press. [Google Scholar]
- 91.Mitra S, Izumi T, Boldogh I, Bhakat KK, Chattopadhyay R, Szczesny B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair. 2007;6(4):461–469. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 92.Souza-Pinto NC, Croteau DL, Hudson EK, Hansford RG, Bohr VA. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Research. 1999;27(8):1935–1942. doi: 10.1093/nar/27.8.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Souza-Pinto NC, Hogue BA, Bohr VA. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radical Biology and Medicine. 2001;30(8):916–923. doi: 10.1016/s0891-5849(01)00483-x. [DOI] [PubMed] [Google Scholar]
- 94.Chen D, Cao G, Hastings T, et al. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. Journal of Neurochemistry. 2002;81(6):1273–1284. doi: 10.1046/j.1471-4159.2002.00916.x. [DOI] [PubMed] [Google Scholar]
- 95.Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiology of Aging. 2006;27(8):1129–1136. doi: 10.1016/j.neurobiolaging.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 96.Gredilla R, Garm C, Holm R, Bohr VA, Stevnsner T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiology of Aging. 2010;31(6):993–1002. doi: 10.1016/j.neurobiolaging.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007;145(4):1318–1329. doi: 10.1016/j.neuroscience.2006.08.061. [DOI] [PubMed] [Google Scholar]
- 98.Brown MR, Sullivan PG, Dorenbos KA, Modafferi EA, Geddes JW, Steward O. Nitrogen disruption of synaptoneurosomes: an alternative method to isolate brain mitochondria. Journal of Neuroscience Methods. 2004;137(2):299–303. doi: 10.1016/j.jneumeth.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 99.Filburn CR, Edris W, Tamatani M, Hogue B, Kudryashova I, Hansford RG. Mitochondrial electron transport chain activities and DNA deletions in regions of the rat brain. Mechanisms of Ageing and Development. 1996;87(1):35–46. doi: 10.1016/0047-6374(96)01696-x. [DOI] [PubMed] [Google Scholar]
- 100.Hota SK, Barhwal K, Singh SB, Ilavazhagan G. Differential temporal response of hippocampus, cortex and cerebellum to hypobaric hypoxia: a biochemical approach. Neurochemistry International. 2007;51(6-7):384–390. doi: 10.1016/j.neuint.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 101.Gredilla R, Weissman L, Yang JL, Bohr VA, Stevnsner T. Mitochondrial base excision repair in mouse synaptosomes during normal aging and in a model of Alzheimer’s disease. Neurobiology of Aging. 2010;33(4):694–707. doi: 10.1016/j.neurobiolaging.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Druzhyna NM, Hollensworth SB, Kelley MR, Wilson GL, LeDoux SP. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. GLIA. 2003;42(4):370–378. doi: 10.1002/glia.10230. [DOI] [PubMed] [Google Scholar]
- 103.Weissman L, Jo DG, Sørensen MM, et al. Defective DNA base excision repair in brain from individuals with Alzheimer’s disease and amnestic mild cognitive impairment. Nucleic Acids Research. 2007;35(16):5545–5555. doi: 10.1093/nar/gkm605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Szczesny B, Tann AW, Mitra S. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: susceptibility of skeletal muscles to oxidative injury. Mechanisms of Ageing and Development. 2010;131(5):330–337. doi: 10.1016/j.mad.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Borghouts C, Kimpel E, Osiewacz HD. Mitochondrial DNA rearrangements of Podospora anserina are under the control of the nuclear gene grisea. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(20):10768–10773. doi: 10.1073/pnas.94.20.10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ludwig DL, MacInnes MA, Takiguchi Y, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutation Research. 1998;409(1):17–29. doi: 10.1016/s0921-8777(98)00039-1. [DOI] [PubMed] [Google Scholar]
- 107.Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Human Molecular Genetics. 2005;14(13):1775–1783. doi: 10.1093/hmg/ddi184. [DOI] [PubMed] [Google Scholar]
- 108.Puebla-Osorio N, Lacey DB, Alt FW, Zhu C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Molecular and Cellular Biology. 2006;26(10):3935–3941. doi: 10.1128/MCB.26.10.3935-3941.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klungland A, Rosewell I, Hollenbach S, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(23):13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ocampo MTA, Chaung W, Marenstein DR, et al. Targeted deletion of mNth1 reveals a novel DNA repair enzyme activity. Molecular and Cellular Biology. 2002;22(17):6111–6121. doi: 10.1128/MCB.22.17.6111-6121.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chan MK, Ocampo-Hafalla MT, Vartanian V, et al. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair. 2009;8(7):786–794. doi: 10.1016/j.dnarep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li H, Vogel H, Holcomb VB, Gu Y, Hasty P. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Molecular and Cellular Biology. 2007;27(23):8205–8214. doi: 10.1128/MCB.00785-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gregg SQ, Robinson AR, Niedernhofer LJ. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair. 2011;10(7):781–791. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Diderich K, Alanazi M, Hoeijmakers JHJ. Premature aging and cancer in nucleotide excision repair-disorders. DNA Repair. 2011;10(7):772–780. doi: 10.1016/j.dnarep.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 116.Kujoth GC, Hiona A, Pugh TD, et al. Medicine: mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 117.Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genetics. 2007;3(2, article e24) doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kukat A, Trifunovic A. Somatic mtDNA mutations and aging—facts and fancies. Experimental Gerontology. 2009;44(1-2):101–105. doi: 10.1016/j.exger.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 119.Edgar D, Trifunovic A. The mtDNA mutator mouse: dissecting mitochondrial involvement in aging. Aging. 2009;1(12):1028–1032. doi: 10.18632/aging.100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Vermulst M, Wanagat J, Loeb LA. On mitochondria, mutations, and methodology. Cell Metabolism. 2009;10(6, article 437) doi: 10.1016/j.cmet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 121.Yang JL, Sykora P, Wilson DM, III, Mattson MP, Bohr VA. The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency. Mechanisms of Ageing and Development. 2011;132(8–9):405–411. doi: 10.1016/j.mad.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Weissman L, de Souza-Pinto NC, Mattson MP, Bohr VA. DNA base excision repair activities in mouse models of Alzheimer’s disease. Neurobiology of Aging. 2009;30(12):2080–2081. doi: 10.1016/j.neurobiolaging.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wheeler VC, Lebel LA, Vrbanac V, Teed A, te Riele HT, MacDonald ME. Mismatch repair gene Msh2 modifies the timing of early disease in HdhQ111 striatum. Human Molecular Genetics. 2003;12(3):273–281. doi: 10.1093/hmg/ddg056. [DOI] [PubMed] [Google Scholar]
- 124.Dragileva E, Hendricks A, Teed A, et al. Intergenerational and striatal CAG repeat instability in Huntington’s disease knock-in mice involve different DNA repair genes. Neurobiology of Disease. 2009;33(1):37–47. doi: 10.1016/j.nbd.2008.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kovtun IV, Johnson KO, McMurray CT. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging. 2011;3(5):509–514. doi: 10.18632/aging.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goula AV, Berquist BR, Wilson DM, III, Wheeler VC, Trottier Y, Merienne K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genetics. 2009;5(12) doi: 10.1371/journal.pgen.1000749.e1000749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bohr VA, Sander M, Kraemer KH. Rare diseases provide rare insights into DNA repair pathways, TFIIH, aging and cancer. DNA Repair. 2005;4(2):293–302. doi: 10.1016/j.dnarep.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 128.Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mechanisms of Ageing and Development. 2008;129(7-8):441–448. doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dianov G, Bischoff C, Sunesen M, Bohr VA. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Research. 1999;27(5):1365–1368. doi: 10.1093/nar/27.5.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stevnsner T, Nyaga S, De Souza-Pinto NC, et al. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002;21(57):8675–8682. doi: 10.1038/sj.onc.1205994. [DOI] [PubMed] [Google Scholar]
- 131.Osenbroch PO, Auk-Emblem P, Halsne R, et al. Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS Journal. 2009;276(10):2811–2821. doi: 10.1111/j.1742-4658.2009.07004.x. [DOI] [PubMed] [Google Scholar]
- 132.Aamann MD, Sorensen MM, Hvitby C, et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB Journal. 2010;24(7):2334–2346. doi: 10.1096/fj.09-147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Scheibye-Knudsen M, Ramamoorthy M, Sykora P, et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. Journal of Experimental Medicine. 2012;209(4):855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair. 2010;9(3):331–344. doi: 10.1016/j.dnarep.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Singh DK, Ahn B, Bohr VA. Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging. Biogerontology. 2009;10(3):235–252. doi: 10.1007/s10522-008-9205-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Das A, Boldogh I, Jae WL, et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. Journal of Biological Chemistry. 2007;282(36):26591–26602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]