

Abstract

The optimization of a series of pterin amides for use as Ricin Toxin A (RTA) inhibitors is reported. On the basis of crystallographic data of a previous furan-linked pterin, various expanded furans were synthesized, linked to the pterin, and tested for inhibition. Concurrently, heteroanalogues of furan were explored, leading to the discovery of more potent triazole-linked pterins. Additionally, we discuss a dramatic improvement in the synthesis of these pterin amides via a dual role by diazabicycloundecene (DBU). This synthetic enhancement facilitates rapid diversification of the previously challenging pterin heterocycle, potentially aiding future medicinal research involving this structure.
Keywords: Pterin, Ricin inhibitors, DBU, RTA, heterocycles
Ricin is a cytotoxin that is easily isolated from castor beans and castor oil byproducts.1 It is categorized as a type II ribosome inactivating protein, which contains a catalytic A chain with N-glycosidase activity and a lectin B chain assisting cellular uptake.2 The A chain of ricin (RTA) has been well characterized, and its mechanism of action is well understood.3 RTA depurinates a specific adenosine on rRNA, thus inhibiting protein synthesis and resulting in cell death, with toxic doses as low as 0.1–1 μg/kg, depending on the mode of administration.4 The ease in which it is acquired, low toxic dose, and lack of an antidote make ricin a notable chemical weapons threat. A number of antiricin agents have been reported, though many are thought to act by disrupting cellular trafficking, and their binding target is not clear.5 The highly evolved specificity pocket of RTA makes the discovery of small molecule active-site inhibitors quite challenging. To date, there have been limited reports of RTA inhibitors showing activity below the micromolar range. Cyclic nucleotides acting as transition state analogues have shown nanomolar range Kd, but these were active only at low pH.6
Recently we reported on the utility of 7-substituted pterins, based on 7-carboxypterin (7CP), as RTA inhibitors, which showed IC50 values as low as 210 μM in a luciferase translation-based assay.7 This was seen as a significant improvement over previous pterin-based inhibitors, which had activity at or above 600 μM.8 Within this series, we reported a furan-linked pterin amide having an IC50 of 380 μM. While this was inferior to 7CP, X-ray analysis of the binding mode indicated the furan ring bridging the gap between the primary binding pocket and a nearby secondary pocket. In its natural state, the primary binding pocket binds the targeted adenosine while the second pocket binds an adjacent guanosine in the RNA sequence.6a We postulated that furans derivatized at position 5 would fill space moving toward this second binding pocket, which could improve inhibitor binding affinity and target specificity. Another preliminary observation reported for this furan-linked pterin was that while the furan ring appeared held in place via hydrogen bonding to the Tyr-80 residue of RTA, the 4 Å distance between the heteroatoms indicated only a weak interaction. It thus follows that, by optimizing the interaction between Tyr-80 and the heterocycle, we should achieve superior inhibition. To accomplish this task, we investigated heteroanalogues of furan: thiophene, imidazole, and triazole.
Pterins are notorious for their insolubility, severely hindering the ease of derivitization.9 For this reason, functionalization of the pterin was held to the last step. Our initial method for forming the amide required suspension of the pterin methyl ester (1) in methanol in the presence of the amine at elevated temperatures (Scheme 1). Coupling directly to the carboxylic acid, as is common with amide bond formation, was not a viable option, as the solubility restrictions of 7CP were incompatible with typical activating agents and coupling conditions. The thiophene and imidazole-linked pterins (2–4) were synthesized from the respective amines (Scheme 1).
Scheme 1. Synthesis of Heteroanalogues of Furanylpterin.

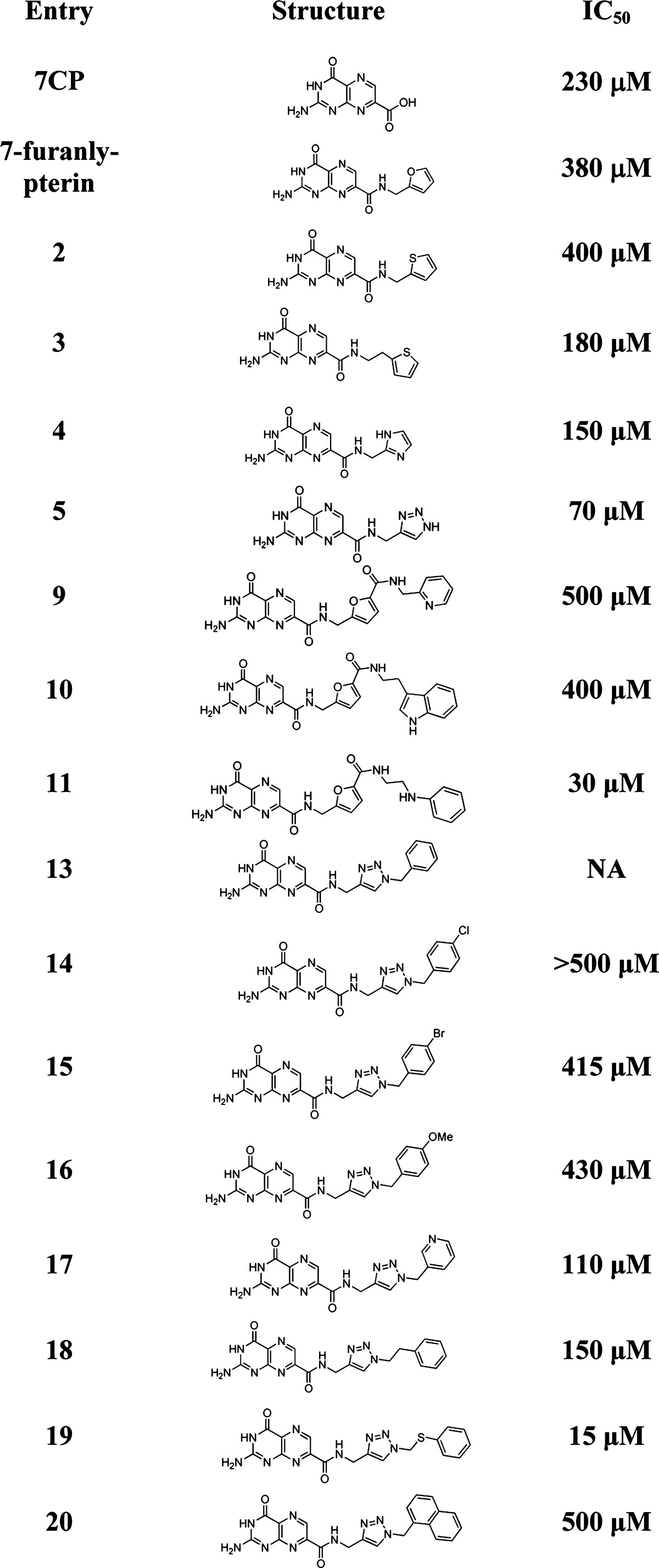
Full structure shown in Table 1.
When these structures were soaked into preformed RTA crystals, and analyzed crystallographically, the pterin was clearly observed bound into the specificity pocket, but there was limited electron density for the pendent. The IC50 values, obtained in the luciferase assay, were consistent with results seen for the parent furan compound, with only a slight improvement in activity. However, as crystallographic data only showed the pterin rigidly bound, the activity likely only stems from interactions with the pterin alone. Furthermore, as the 5-membered rings are not held in place, compounds 2–4 were not seen as prime candidates for rationally designed optimization.
Turning our attention to triazole, we synthesized 4-aminomethyltriazole and condensed it with the pterin ester 1. Compound 5 was tested for inhibition and was not only superior to the simple furan-linked pterin but also more potent than 7CP, with an IC50 of 70 μM.
The crystal structure of the RTA·5 complex showed the full density of the ligand and also revealed a firm hydrogen bond to Tyr-80, with a 2.8 Å distance between heteroatoms, as seen in Figure 1.
Figure 1.

X-ray structure of 5 bound into RTA. Hydrogen bonds to the protein are all between 2.6 and 2.9 Å. Density is from a 1.9 Å OMIT map contoured at 3σ. A water molecule is shown as a labeled sphere.
While this result shows great potential for triazoles, we still wished to optimize the lead furan-linked pterin via construction of expanded 2-aminomethylfurans substituted at the 5 position, hoping to bring the furan into the range of activity seen for the simple triazole 5.
We began with the synthesis of the desired expanded furans (6–8), starting from commercially available ethyl 5-chloromethyl-2-furancarboxylate, as shown in Scheme 2a.
Scheme 2. (a) Synthesis of Expanded Furan Derivatives. (b) Conjugation of Amines to 7-Methoxycarbonylpterin.

Our initial method of amidation gave acceptable yields with commercially available/simple amines, which can be used in large excess but became impractical with more complex synthetic amines, as seen by the low yield of compound 9 (Scheme 2b). Seeking to improve the conversion of ester to amide, we took note of the work by Vaidyanathan et al., who used DBU as a mild catalyst for amidation of esters.10 They proposed DBU attacking the ester, activating the carbonyl in situ, thus expediting aminolysis.
Upon adding DBU to the suspension of 1 in methanol, we quickly discovered an additional benefit of DBU, as the pterin fully dissolved. The solubility of unfunctionalized pterin in methanol is unprecedented. To investigate the nature of this solubility, we precipitated with dioxane, whereupon NMR of the yellow solid showed a 1:1 mixture of pterin to DBU, indicative of an organic salt. The pKa of the lactam NH of pterin has been reported as ∼8, within the range of DBU deprotonation.11 This solubility allowed reactions to be run at higher concentrations, decreasing the amount of amine required. With the DBU first acting to form an organic salt, 2 equiv of DBU were added to the pterin ester, in a minimal amount of methanol and 2 equiv of amine. Monitoring the reaction at room temperature revealed after 4 h the reaction had gone to completion. Isolation of the desired product was achieved by addition of aqueous hydrochloric acid, providing the product as a precipitate in acceptable yield. The proposed role of DBU in the amidation reaction is illustrated in Figure S1 (see the Supporting Information).
We screened other amine bases—triethylamine, Hunig’s base, and DABCO—and found none showed the remarkable improvement in solubility or rate acceleration.12 This supports the proposed nucleophilic character for DBU being crucial, as the tertiary amines could not proceed by such a mechanism.
Proceeding with the improved synthesis, we synthesized the expanded furan linked pterins 9–11 and tested them for RTA inhibition. Compounds 9 and 10 showed diminished activity relative to the lead furan compound. Additionally, the X-ray data for the RTA·inhibitor complexes only showed density for the pterin ring, indicating disordered and uncompensated binding of the pendent. Compound 11, however, showed a remarkable improvement in activity in the luciferase assay. At ∼30 μM, it was nearly an order of magnitude more potent than 7CP. This was a very encouraging result and justified our design of expanded furans. Surprisingly, the X-ray data of the bound inhibitor revealed a disordered binding of the pendent, but strong density displayed for the pterin ring alone.
Given the synthetic effort required for the expanded furans, compared with a simple “click chemistry”13 approach for triazoles, and the positive results for compound 5, focus was shifted from furan to expanded triazole inhibitors. Any number of azides can be reacted with propargyl amine to provide an expanded triazole to be further reacted with the pterin ester (1). However, to facilitate rapid diversification, we investigated an alternative synthetic route to the triazoles. Rather than fully synthesizing the heterocycle prior to condensation with the pterin, we first synthesized the propargyl-linked pterin amide (12), to react with various azides in parallel. In the absence of DBU, compound 12 failed to react. However, first isolating 12 as the highly soluble DBU salt allowed for synthesis of various triazoles via traditional “click” conditions. Compounds 13–20 were synthesized in this manner (Scheme 3).
Scheme 3. Synthesis of New Triazole-Linked Pterin.

Full structures are shown in Table 1.
These structures were chosen to probe the effect of the electron density of the aromatic side-chain, and the length of the linker (Table 1). Screening the newly synthesized triazole-linked pterins revealed many of them performed quite poorly. All compounds with benzyl substituents showed lackluster IC50’s in the 400–500 μM range. The pyridyl functionalized triazole gave better results, with an IC50 of roughly 110 μM. While less active than the parent triazole (5), this compound is still superior to 7CP and all other previously published pterin inhibitors. The only structure more potent than 5 was compound 19, with an IC50 of 15 μM. The X-ray data for the RTA·19 complex showed the binding of the ligand was still somewhat disordered, as there was strong density for the pterin ring but only limited density for the triazole ring and possible density observed for the heavy sulfur atom (see the Supporting Information). This indicates the triazole ring is relatively fixed in position, but the methylene spacer to the thiophenol adopts multiple geometries. Furthermore, the benzene ring of the thiophenol is freely rotating. It is possible that the free rotation has displaced trapped water molecules from the active-site, giving rise to favorable entropic effects, compensating the limited enthalpically favorable interactions. This hypothesis is partially supported by the disappearance of two water molecules previously observed in the space near the disordered thiophenol. We are currently investigating ITC experiments which may further elucidate the nature of the protein–ligand interaction. Compound 19 is currently one of the most potent RTA inhibitors to date.
Table 1. Summary of Results.
A representative dose–response curve from the luciferase assay for compound 19 is shown in Figure 2. RTA activity was determined through normalization of luciferase counts with and without inhibitor. Percent RTA activity is plotted as a function of inhibitor concentration (see Supporting Information for experimental details).
Figure 2.

Compound 19 versus RTA. RTA activity is plotted as a function of inhibitor concentration and fit to a hyperbolic decay function.
In summary, we have synthesized three expanded furans to optimize a previous furan-linked pterin. Toward this goal, we achieved success with compound 11. In the process, we improved our synthetic strategy through the use of DBU, which gave remarkable solubility to a notoriously insoluble heterocycle. We screened various heteroanalogues of furan, leading to improved interaction with RTA through a triazole-linked pterin (5). With the beneficial solubility, we rapidly constructed a library of new pterins via click chemistry. While the small triazole library only identified a single potent inhibitor (19), the ease of diversification highlights a significant advance in pterin functionalization. The use of the DBU salt of 12 for rapid diversification should represent an important contribution to medicinal chemistry, as this is a readily available, stable source of pterin that can be easily incorporated into a range of biologically and medicinally relevant compounds.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant AI 075509, by Robert A. Welch Foundation Grant F1225, and by the College of Natural Sciences support to the Center for Structural Biology. Assistance for this work was provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, the Institute for Cellular and Molecular Biology, the University of Texas at Austin.
Glossary
Abbreviations
- RTA
ricin toxin A
- PTA
pteroic acid
- 7CP
7-carboxy pterin
- DBU
1,8-diazabicyclounde-7-cene
Supporting Information Available
Experimental procedures and compound characterizations, proposed DBU mechanism, X-ray data for compound 19, additional dose response curves, and details for the in vitro assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Toho University Miyama, Funabashi, Japan.
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Franz D.Textbook of Military Medicine: Aspects of Chemical and Biological Warfare; 1997. [Google Scholar]
- a Lord J. M.; Roberts L. M.; Robertus J. D. Ricin: structure, mode of action, and some current applications. FASEB J. 1994, 82201–8. [PubMed] [Google Scholar]; b Robertus J. The structure and action of ricin, a cytotoxic N-glycosidase. Semin. Cell Biol. 1991, 2123–30. [PubMed] [Google Scholar]
- a Monzingo A. F.; Robertus J. D. X-ray analysis of substrate analogs in the ricin A-chain active site. J. Mol. Biol. 1992, 22741136–45. [DOI] [PubMed] [Google Scholar]; b Ready M. P.; Kim Y.; Robertus J. D. Site-directed mutagenesis of ricin A-chain and implications for the mechanism of action. Proteins 1991, 103270–8. [DOI] [PubMed] [Google Scholar]; c Montfort W.; Villafranca J. E.; Monzingo A. F.; Ernst S. R.; Katzin B.; Rutenber E.; Xuong N. H.; Hamlin R.; Robertus J. D. The 3-Dimensional Structure of Ricin at 2.8-A. J. Biol. Chem. 1987, 262115398–5403. [PubMed] [Google Scholar]; d Mlsna D.; Monzingo A. F.; Katzin B. J.; Ernst S.; Robertus J. D. Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993, 23429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. J.; Ravikumar K.; Shen H.; Suh J. K.; Kerwin S. M.; Robertus J. D. Structure-based design and characterization of novel platforms for ricin and shiga toxin inhibition. J. Med. Chem. 2002, 45190–8. [DOI] [PubMed] [Google Scholar]
- a Wahome P. G.; Bai Y.; Neal L. M.; Robertus J. D.; Mantis N. J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Stechmann B.; Bai S. K.; Gobbo E.; Lopez R.; Merer G.; Pinchard S.; Panigai L.; Tenza D.; Raposo G.; Beaumelle B.; Sauvaire D.; Gillet D.; Johannes L.; Barbier J. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 1412231–42. [DOI] [PubMed] [Google Scholar]
- a Ho M. C.; Sturm M. B.; Almo S. C.; Schramm V. L. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc. Natl. Acad. Sci. U. S. A. 2009, 1064820276–81. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chen X.-Y.; Berti P. J.; Schramm V. L. Transition-State Analysis for Depurination of DNA by Ricin A-Chain. J. Am. Chem. Soc. 2000, 122286527–6534. [Google Scholar]
- Pruet J. M.; Jasheway K. R.; Manzano L. A.; Bai Y.; Anslyn E. V.; Robertus J. D. 7-Substituted pterins provide a new direction for ricin A chain inhibitors. Eur. J. Med. Chem. 2011, 46, 3608–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X.; Hollis T.; Svinth M.; Day P.; Monzingo A. F.; Milne G. W. A.; Robertus J. D. Structure-based identification of a ricin inhibitor. J. Mol. Biol. 1997, 26651043–1049. [DOI] [PubMed] [Google Scholar]
- a Waring P. The Synthesis of 6-Aminomethyl-5,6,7,8-Tetrahydropterin. Aust. J. Chem. 1988, 415667–676. [Google Scholar]; b Taylor E. C.; Ray P. S. Pteridines. 52. Convenient Synthesis of 6-formylpterin. Synth. Commun. 1987, 17161865–1868. [Google Scholar]; c Pruet J. M.; Robertus J. D.; Anslyn E. V. Acyl radical insertion for the direct formation of new 7-substituted pterin analogs. Tetrahedron Lett. 2010, 51182539–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price K. E.; Larrivee-Aboussafy C.; Lillie B. M.; McLaughlin R. W.; Mustakis J.; Hettenbach K. W.; Hawkins J. M.; Vaidyanathan R. Mild and efficient DBU-catalyzed amidation of cyanoacetates. Org. Lett. 2009, 1192003–6. [DOI] [PubMed] [Google Scholar]
- Brown D. J.Pteridines; Wiley: New York, 1988; pp xxvii, 730. [Google Scholar]
- Of the amines tested, DBU is the most basic. There is enough difference in the basicity of DBU and DABCO that the latter may not deprotonate the pterin to a large enough extent to fully form the organic salt. However, the remaining bases are all similar in strength, with protonated DBU and Hunig’s base only differing by 0.5 pKa units. Thus, all are capable of forming the organic salt, yet only DBU should form a salt “greasy” enough to dissolve in methanol. Nevertheless, much of the utility of DBU stems from the in situ activation of the ester, which would not be possible with the three tertiary amines. pKa's for the four bases, among others, can be found at: www.cem.msu.edu/∼reusch/OrgPage/basicity.htm.
- Meldal M.; Tornoe C. W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 10882952–3015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.