

Abstract
Background: The identification of mutations in the TARDBP and more recently the identification of mutations in the FUS gene as the cause of amyotrophic lateral sclerosis (ALS) is providing the field with new insight about the mechanisms involved in this severe neurodegenerative disease.
Methods: To extend these recent genetic reports, we screened the entire gene in a cohort of 200 patients with ALS. An additional 285 patients with sporadic ALS were screened for variants in exon 15 for which mutations were previously reported.
Results: In total, 3 different mutations were identified in 4 different patients, including 1 3-bp deletion in exon 3 of a patient with sporadic ALS and 2 missense mutations in exon 15 of 1 patient with familial ALS and 2 patients with sporadic ALS.
Conclusions: Our study identified sporadic patients with mutations in the FUS gene. The accumulation and description of different genes and mutations helps to develop a more comprehensive picture of the genetic events underlying amyotrophic lateral sclerosis.
The profile of genes mutated in amyotrophic lateral sclerosis (ALS) has expanded considerably since 2006. The primary causative gene remains the zinc copper superoxide dismutase gene (SOD1) as mutations in SOD1 explain ∼15%–20% of familial ALS (FALS) cases, which altogether represents ∼2% of the combined sporadic (SALS) and FALS cases.1 However, several mutations were recently reported in the TAR-DNA binding protein (TARDBP) gene encoding the TDP-43 protein at the ALS10 locus.2 The TARDBP mutation search was initiated following the discovery that TDP-43 is a major constituent of aggregates found in patients with ALS or frontotemporal dementia (FTD).3 Mutations are almost exclusively situated at the glycine-rich C-terminal portion of TARDBP in its sixth and last exon. The identification of mutations in TARDBP helped convince researchers studying the ALS6 locus on chromosome 16 that the FUS gene was a good candidate for harboring mutations in patients with ALS.
The presence of a homozygous mutation in a recessive family with ALS from Cape Verde combined with heterozygous changes in dominant ALS pedigrees that helped map the ALS6 locus led to the conclusion that FUS was the causative gene in that region.4,5 The product encoded by the FUS gene (FUS) has a function similar to that of the TDP-43 protein. It is an RNA-binding protein with hnRNP properties and it has a prior history of involvement in the nervous system: mouse glutamate receptors help regulate the localization of FUS to dendritic spines.6 Moreover, FUS helps in actin organization of dendritic spines via mRNA transport of the actin-stabilizing protein Nd1-L.7 Initially, FUS/TLS (fusion/translated in liposarcomas) was identified as a t(12;16) translocation product which combines its N-terminal portion with the C-terminal portion of the CHOP gene leading to round cell liposarcomas.8 FUS knockout mice have been generated and no neurologic defects were reported.9,10
We sought to validate the results recently obtained on chromosome 16 by sequencing the FUS gene in a panel of FALS and SALS cases. This led to the identification of 2 mutations that were previously reported4 and 1 novel mutation.
METHODS
Standard protocol approvals, registrations, and patient consents.
Protocols were approved by the ethics committee on human experimentation of the Centre Hospitalier de l’Université de Montréal and the Comite d’Ethique de la Salpêtrière. All patients gave written informed consent, after which patient information and blood were collected.
Subjects.
Patients were collected from the province of Quebec, Canada (n = 100), and from France (n = 100) between 2004 and 2009. DNA was extracted from peripheral blood using standard protocols. A total of 80 patients with FALS and 120 patients with SALS were screened for the 15 coding exons of the FUS gene as well as 190 ethnically matched controls and 285 patients with schizophrenia or autism as part of an unrelated project. An additional cohort of 285 patients with SALS were screened for variants in exon 15, considering that most of the mutations already identified are located in the c-terminus of the protein.4
Gene screening.
Primers were designed using the ExonPrimer software from the UCSC human genome browser Web site (www.genome.ucsc.edu). Twelve sets of primers were sufficient to cover the 15 exons in FUS (NM_004960.2). Primer sequences and amplification conditions are listed in table e-1 on the Neurology® Web site at www.neurology.org. PCR products were sequenced at the Genome Quebec Innovation Center. Variants were tested in patients and controls using the same procedure of direct sequencing.
Protein sequence alignment.
Cluster analysis was performed using the Clustal W method. The closest homologue in several species was retrieved by use of NCBI’s BLAST program (figure e-1).
Phosphorylation sites prediction.
The phosphorylation site prediction scores corresponding to the deletion p.S57 were obtained using the NetPhos neural network-based method (table e-2).
RESULTS
The complete sequencing of the FUS gene in 200 patients with ALS and the sequencing of exon 15 for an additional 285 SALS cases led to the identification of 2 missense mutations and 1 3-bp deletion in 3 patients with SALS and 1 patient with FALS (table). These mutations were not found in 285 patients with schizophrenia or autism used as a non-ALS disease cohort, nor in 190 controls matched for age and ethnicity. A 3-bp heterozygous deletion (c.169_171delTCT, p.S57del) was identified in a patient with sporadic ALS. This TCT deletion results in the loss of the serine-57 residue and an overall decreased phosphorylation score (table e-2). Two mutations were present at amino acid 521: an arginine to cysteine (c.1561 C>T, p.R521C) in 1 patient with SALS, and an arginine to a histidine (c. 1562 G>A, p.R521H) in 1 patient with SALS and 1 patient with FALS (figure). Notably, these 2 missense mutations are the same as reported by Kwiatkowski et al.4 We were unable to test for segregation in the patient with FALS as no additional family members were available. All the patients with mutations had a typical ALS profile. No documented history of FTD or cognitive impairment was present in these patients.
Table Clinical and genetic profile of patients with ALS with mutations in the FUS gene
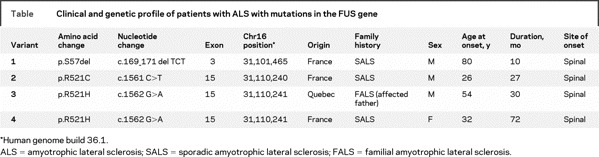
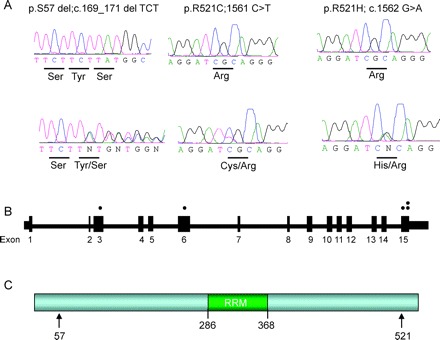
Figure Sequence traces and position of mutations in FUS
(A) Sequence trace for the wild-type allele is presented over top of the sequence of the mutated allele. The amino acid that is changed is listed below. (B) Schematic (not to scale) of the FUS gene. Dots represent the exons in which mutations were identified. In the lower panel, the amino acid position of the mutations is indicated by the arrows. The position of the RNA-recognition motif (RRM) is also highlighted in green.
A rare missense variant (c.188 A>G, p.N63S) was detected in a SALS case; however, it was also present in 8 of 190 controls, suggesting that it is a benign polymorphism. This particular base pair change is not well conserved across species and the serine residue is present in the rhesus monkey. Additionally, a 9-bp deletion (c.676_684delGGCGGCGGC) was detected in exon 6, which results in the loss of 3 glycine residues (p.G226_G228del). This variant was detected in a patient with ALS as well as 1 in 190 controls (table e-3).
The entire FUS gene was sequenced in 285 patients with schizophrenia and autism, and only 1 coding variant was detected, a GGC insertion in exon 6, resulting in the introduction of a glycine residue (c.684_685insGGC, p.G228_G229insG). This is at the same location where a 9-bp deletion was detected in a patient with ALS and a control individual, suggesting that this particular glycine stretch is prone to expansion/contractions. Thus, the frequency of FUS variants is not particularly high in a population of patients without ALS (table e-3).
DISCUSSION
Our study identified 3 SALS cases with mutations in the FUS gene. These patients were labeled sporadic considering that the cases were isolated and that the family history was negative.
Two mutations that were previously described by Kwiatkowski et al.4 were also identified in this study, the p.R521H and p.R521C mutations. One new deletion was also identified in a patient with SALS. The overall percentage of mutations identified in this study was 1 of 80 FALS or 1.25%, and 3 of 405 SALS or 0.74%. This is less than the original reports. Also, our study only detected heterozygous changes, while the other reports described homozygous and heterozygous changes.4,5
Future identification of more sporadic cases with missense and deletion mutations in FUS will provide a more comprehensive picture of the proportion of mutations involved in ALS pathology. The accumulation and description of different mutations in ALS cases and future detection of mutations in more patients with SALS will help us understand the genetic mechanisms involved in this neurodegenerative disease. The identification of new genes represents a highly informative event for the selection of candidate genes to be investigated in the future, considering that the genetic factors underlying a substantial proportion of ALS cases remains unknown. Further investigation of the function of those genes will progressively stimulate the development of drug treatment and therapy for the disease.
ACKNOWLEDGMENT
The authors thank the patients involved in this study and Mélanie Benard, Isabelle Thibault, and Pierre Provencher for sample collection and organization.
DISCLOSURE
V.V. Belzil, Dr. Valdmanis, Dr. Dion, Dr. Daoud, Dr. Kabashi, A. Noreau, Dr. Gauthier, P. Hince, and A. Desjarlais report no disclosures. Dr. Bouchard served on a scientific advisory board for Serono Canada; held a corporate appointment with Teva Neuroscience; receives research support from Sanofi-Aventis and Biogen Idec; and has given expert written testimony in a medical court proceeding. Dr. Lacomblez serves on scientific advisory boards and receives unrestricted research support from GlaxoSmithKline, Novartis, Eisai, Teva, and Trophos. Dr. Salachas and Dr. Pradat report no disclosures. Dr. Camu receives honoraria as consultant to Sanofi-Aventis and Serono. Dr. Meininger serves on the editorial board of Amyotrophic Lateral Sclerosis. Dr. Dupre serves on an NIH grant review panel; has received honoraria and funding for travel from Genzyme and Teva; and serves as a consultant to Vithcom. Dr. Rouleau reports no disclosures.
Supplementary Material
Glossary
- ALS
amyotrophic lateral sclerosis
- FALS
familial amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- SALS
sporadic amyotrophic lateral sclerosis.
Footnotes
Editorial, page 1172
See also page 1180
Supplemental data at www.neurology.org
e-Pub ahead of print on September 9, 2009, at www.neurology.org.
*These authors contributed equally.
Supported by the Association pour la Recherche sur la Sclérose Latérale Amyotrophique (ARS), the Association Française contre les Myopathies (AFM), the French Group on MND, and Genethon for DNA extraction and cell lines. N.D., V.V.B., and G.A.R. are supported by the Canadian Institutes of Health Research, E.K. by ALS Canada, and P.N.V. by the Fonds de Recherche en Santé Québec (FRSQ).
Disclosure: Author disclosures are provided at the end of the article.
Received March 17, 2009. Accepted in final form July 17, 2009.
REFERENCES
- 1.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 2.Liscic RM, Grinberg LT, Zidar J, Gitcho MA, Cairns NJ. ALS and FTLD: two faces of TDP-43 proteinopathy. Eur J Neurol 2008;15:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 4.Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 5.Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii R, Okabe S, Urushido T, et al. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 2005;15:587–593. [DOI] [PubMed] [Google Scholar]
- 7.Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. J Cell Sci 2005;118:5755–5765. [DOI] [PubMed] [Google Scholar]
- 8.Rabbitts TH, Forster A, Larson R, Nathan P. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 1993;4:175–180. [DOI] [PubMed] [Google Scholar]
- 9.Hicks GG, Singh N, Nashabi A, et al. Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 2000;24:175–179. [DOI] [PubMed] [Google Scholar]
- 10.Kuroda M, Sok J, Webb L, et al. Male sterility and enhanced radiation sensitivity in TLS(−/−) mice. EMBO J 2000;19:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.