

Abstract
Purpose.
To determine connexin 43 (Cx43) localization in mitochondria and investigate the effects of high glucose (HG) on mitochondrial Cx43 (mtCx43) expression and whether altered mtCx43 channel activity is involved in promoting apoptosis in retinal endothelial cells.
Methods.
MtCx43 localization was determined using immunostaining, green fluorescent protein (GFP)-tagged Cx43 followed by confocal imaging, and Western blot analysis using protein isolated from mitochondria of rat retinal endothelial cells (RRECs). To assess HG effects on mtCx43 expression, RRECs were grown in normal (5 mM) or HG (30 mM) medium for 7 days, and mtCx43 protein level assessed by Western blot analysis. To determine if mtCx43 channel inhibition affected mitochondrial morphology, RRECs grown sparsely were left untreated or treated with β-glycerrhetinic acid (β-GA), an inhibitor of connexin channels, and imaged using confocal microscopy. Additionally, mitochondria isolated from RRECs were treated with β-GA, and cytochrome c release assessed by Western blot.
Results.
Cx43 localization on the mitochondria of RRECs was confirmed with immunofluorescence staining using Cx43 antibody and GFP-tagged Cx43 imaged in live cells. Western blot analysis indicated that Cx43 was located primarily on the inner mitochondrial membrane, and mtCx43 protein level was significantly reduced in RRECs grown in HG condition. Treatment of RRECs with β-GA significantly decreased mtCx43 phosphorylation, induced mitochondrial fragmentation, and isolated mitochondria treated with β-GA showed increased cytochrome c release.
Conclusions.
HG-induced downregulation of mtCx43 protein resulting in decreased channel activity may promote mitochondrial morphology changes and cytochrome c release, suggesting a novel mechanism for hyperglycemia-induced apoptosis in diabetic retinopathy.
The study reports high glucose (HG) downregulates mitochondrial connexin 43 (mtCx43) expression and contributes to retinal vascular cell death. Additionally, HG reduces mtCx43 channel activity, induces mitochondrial fragmentation, and cytochrome c release resulting in retinal endothelial cell death. This study investigated the effects of altered mtCx43 expression on cellular dysfunction associated with demise of vascular cells in early stages of diabetic retinopathy. In particular, the findings indicate that HG-induced downregulation of mtCx43 contributes to mitochondrial shape change and subsequent cytochrome c release, which in turn, promotes apoptosis in retinal endothelial cells.
Introduction
High glucose (HG)-induced mitochondrial dysfunction has been shown to play a significant role in the onset and progression of apoptosis, which is a characteristic hallmark of diabetic retinopathy.1–3 Mitochondria in most cell types exist in long, tubular networks that are carefully regulated by the rates of mitochondrial fusion and fission.4 Increased fragmentation of the mitochondrial networks has been shown to be a critical step in the progression of apoptosis.5 Such fragmentation of the mitochondrial network can lead to further mitochondrial dysfunction and increased cytochrome c release,5 and we have recently demonstrated that HG condition promotes mitochondrial fragmentation in retinal endothelial cells and pericytes concomitant with increased apoptosis.6,7 Recent studies in cardiac myocytes have shown mitochondrial connexin 43 (mtCx43) as an important regulator mitochondrial function and initiation of apoptosis8; yet little is known about the role of mtCx43 in retinal vascular cells. Thus, identifying the role of mtCx43 in maintenance of mitochondrial morphology and cytochrome c release in retinal endothelial cells could provide a better understanding on how apoptosis is triggered in diabetic retinopathy.
Specifically, it is unknown how HG affects Cx43 expression in the mitochondria of retinal endothelial cells. Cx43 is normally found in the plasma membrane where it performs its well-studied function of forming gap junction channels. Cx43 proteins of adjacent cells dock together to form the gap junction channel, and these channels act as the primary mechanism of intercellular communication.9 HG conditions have been shown to downregulate Cx43 expression and reduce gap junction intercellular communication (GJIC) in endothelial cells and pericytes,10–12 ultimately contributing to apoptosis.13,14 However, it is unknown where Cx43 localizes on the mitochondria of retinal endothelial cells, and whether its expression and possible channel activity play a role in mitochondrial functionality.
Only within the last decade has the presence of Cx43 in the mitochondria of various cell types been reported.8,15–17 However, the exact localization and functional role of Cx43 in the mitochondrial membranes remains unclear. Increased mtCx43 has been linked to ischaemic preconditioning and cardioprotection, possibly through regulation of mitochondrial ion homeostasis and swelling.17,18 In addition, studies have demonstrated the role of mtCx43 in controlling apoptosis initiation, where inhibition of mtCx43 channels promoted Ca2+ and cytochrome c release from myocyte mitochondria.8 A study showed inhibition of mtCx43 expression can influence mitochondrial metabolism, a vital function of the mitochondria and plays a role in initiating apoptosis.15 Overall, recent studies suggest the role of Cx43 in mitochondrial integrity and apoptosis initiation.19 However, more research is needed to understand the functional role of mtCx43 in other cell types, and how it is affected in different diseases.
Preventing accelerated apoptosis of retinal endothelial cells in diabetic retinopathy is an important therapeutic endpoint in treating this diabetic complication. Importantly, identifying novel molecular players in mitochondrial morphology and apoptosis regulation could provide new insight into understanding how HG induces apoptosis in the retinal vasculature. In this study, we have investigated specific localization of Cx43 in the mitochondria, outer/inner membrane, and matrix. In addition, we examined the effects of HG on Cx43 expression in the mitochondria of retinal endothelial cells, and whether channel inhibition of mtCx43 may play a role in mitochondrial fragmentation and subsequent apoptosis that is observed in HG conditions.
Materials and Methods
Cell Culture
Rat retinal endothelial cells (RRECs) were isolated from rat retinas as previously described,20 and third to fifth passage cells were used for this study. For experiments, RRECs were grown in Dulbecco's modified Eagle medium (DMEM) (Invitrogen, Grand Island, NY) containing 10% fetal bovine serum (FBS) and antibiotics. To assess HG effects on mtCx43 expression, cells were grown on 100-mm dishes (Western blot) or glass coverslips (immunofluorescence) for 7 days in normal (5 μM) or HG (30 mM) medium until confluent.
To assess the effects of inhibiting mitochondrial gap junction channels on mitochondrial morphology, RRECs were grown on poly-D-lysine-coated glass slide-bottom dishes (MatTek, Ashland, MA). Cells were plated sparsely and grown for 24 hours under normal (5 mM) medium. Four hours prior to imaging, one set of cells was treated with β-glycerrhetinic acid (β-GA), an inhibitor of gap junction channels (50 μM), and another set treated with glycyrrhizic acid (GZ; 50 μM), a chemical analog of β-GA that does not inhibit gap junction channels. To examine the effects of GA and GZ treatment on phosphorylation of mtCx43, cells were grown on 100-mm dishes in N medium. Four hours prior to protein harvesting, one set of cells was treated with β-GA (50 μM) and another set treated with GZ (50 μM).
Transfection
RRECs were grown on poly-D-lysine-coated glass slide-bottom dishes (MatTek) and transfected with 4 μg of Cx43–green-fluorescent protein (GFP) DNA using Lipofectamine 2000 protocol (Invitrogen). After 48 hours post transfection, cells were stained with MitoTracker Red (250 nM; Molecular Probes, Eugene, OR) and imaged live.
Gel Electrophoresis and Western Blotting
Cells were washed with PBS and lysed with a 0.1% Triton-X-100 buffer containing 10 mM Tris, pH 7.5, 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride (PMSF). To isolate mitochondrial protein and cytosolic protein separately, the cellular extract was centrifuged 700g for 5 minutes. The supernatant was extracted and centrifuged again at 21,000g for 15 minutes. The supernatant was extracted as the cytosolic protein fraction. The remaining cellular pellet was washed with the Trition buffer and centrifuged at 21,000g for 15 minutes. The supernatant was discarded and the cellular pellet was washed with radio-immunoprecipitation assay (RIPA) buffer containing 1 mM PMSF. The washed pellet solution was centrifuged at 21,000g for 15 minutes, and the supernatant was extracted as the mitochondrial protein fraction.
An equal volume of 2× sample buffer was added to the protein samples followed by denaturation at 95°C for 5 minutes. Then, the protein samples were electrophoresed at 120 V for 50 minutes. Kaleidoscope molecular weight standards were run in separate lanes in each gel. After completion of electrophoresis, the protein samples were transferred to nitrocellulose membranes using a semi-dry apparatus with Towbin buffer system according to the Towbin et al. procedure.21 The membranes were blocked with 5% nonfat dry milk for 1 hour and then exposed to either rabbit anti-mouse Cx43 antiserum or anti-cytochrome C in 0.2% nonfat milk overnight. The following day, the blots were washed with Tris-buffered saline containing 0.1% Tween-20 and then incubated with rabbit anti-mouse immunoglobulin G (IgG) secondary antibody for 1 hour. The membrane was again washed as above, and then exposed to Immun-Star Chemiluminescent Protein Detection System (BioRad, Richmond, CA) to detect the protein signals on x-ray film. Protein loading in the gels was confirmed by Ponceau-S staining. Densitometry was conducted and analyzed using NIH Image analysis program (NIH, Bethesda, MD).
Immunofluorescence Staining and Analysis of mtCx43
To study the relative amounts of mitochondrial Cx43, immunofluorescence staining for Cx43 was performed on RRECs. Briefly, cells grown to confluency were stained with MitoTracker Red (250 nM) for 45 minutes and then fixed in ice-cold methanol for 15 minutes, washed in PBS, and treated with 2% BSA for 15 minutes to block nonspecific antibody binding. The cells were then incubated overnight at 4°C in a moist chamber with a monoclonal mouse anti–rat Cx43 antibody (Chemicon, Temecula, CA) diluted 1:200 in PBS containing 2% BSA. After three PBS washes, the cells were incubated for 1 hour with fluorescein isothiocyanate–conjugated goat anti-mouse IgG (Sigma, St. Louis, MO) diluted 1:200 in PBS containing 2% BSA. After three PBS washes, coverslips were mounted in Slow-Fade (Molecular Probes). The cells were viewed and photographed using confocal microscopy to acquire z-stack images of individual mitochondria. Mitochondrial Cx43 signals were then identified by sequentially viewing z-stack slices and counting Cx43 signals (green) that colocalized with mitochondria (red).
Fluorescent Probes
To visualize mitochondria in living or fixed RRECs, cells were incubated at 37°C in a humidified chamber, with 5% CO2 and with 250 nM MitoTracker Red for 45 minutes.
Confocal Microscopy
Cells were imaged live under confocal microscopy using a Zeiss LSM 710 Meta microscope (Carl Zeiss, Oberkochen, Germany) with a 63× oil immersion objective. Cells were kept at 37°C in a humidified microscope stage chamber containing 5% CO2. MitoTracker Red was subjected to 543 nm helium/neon laser excitation and emission was recorded through a bandpass 650 to 710 nm filter (Zeiss, Thornwood, NY). To observe individual mitochondria z-stack images were acquired in series of six slices per cell ranging in thickness from 0.5 to 0.8 μm per slice.
Mitochondrial Morphology Analysis
Acquired images of mitochondria were analyzed using ImageJ (National Institutes of Health, Bethesda, MD) by first processing with a median filter to obtain isolated and equalized fluorescent pixels. After converting to “masks” individual mitochondria were subjected to particle analysis for acquiring fill factor (FF) values (4π*Area/perimeter2) and lengths of major and minor axes, called aspect ratio (AR) values. A minimal value of 1 indicates a perfect circle for both parameters, and as mitochondria elongate and become more branched FF increases; likewise, as mitochondria become more elliptical AR increases.
Mitochondrial Isolation and Cytochrome c Release Assay
Mitochondria were isolated from RRECs using Mitochondrial Isolation from Cultured Cells Kit (Thermo, Rockford, IL). Purity of protein samples from cytosolic and mitochondrial cell fractions was determined by probing for voltage-dependent anion channel (VDAC1) (abundant outer mitochondrial membrane protein) and Hsp90 (cytosolic protein).
Cytochrome c release from isolated mitochondria was assayed by resuspending isolated mitochondria in mannitol-sucrose buffer (MSB) buffer (400 mM mannitol; 50 mM Tris-HCl, pH 7.2; 5 mg/mL BSA; 10 mM KH2PO4) and treating with indicated drugs for 4 hours at 30°C. Following the incubation, the suspension was centrifuged at 4000g and the supernatant was collected as the “Release” fraction and the mitochondria pellet was lysed with 2% CHAPS buffer.
Statistics
All data were expressed as mean ± SD. Comparisons between groups were performed with Student's t-test. A value of P less than 0.05 was considered statistically significant.
Results
Endogenous Connexin 43 and Plasmid-Driven Connexin 43-GFP Localizes to Mitochondria in Retinal Endothelial Cells
To determine if Cx43 localizes to mitochondria in RRECs, immunofluorescence and Cx43-GFP were used to visually observe Cx43 localization within the cell. Immunofluorescence images of RRECs pretreated with MitoTracker Red and stained with anti-Cx43 antibody showed colocalization of Cx43 on mitochondria (Fig. 1A). Abundant Cx43 expression on the cell membrane is observed, indicating plasma membrane-associated gap junction plaques, as previously reported.10,11
Figure 1. .

Connexin 43 localizes to mitochondria in RRECs. (A) Immunofluorescence stains of RRECs pretreated with MitoTracker Red and stained with anti-Cx43 antibody showed colocalization of Cx43 on mitochondria. (B) RRECs transfected with Cx43-GFP plasmid show Cx43 localization on mitochondria (stained with MitoTracker Red) in live RRECs. Open arrowheads indicate Cx43 localization on mitochondria. Closed arrowheads indicate Cx43 localization in gap junction plaques between cells. (C) Western blot shows Cx43 localization in protein isolated from mitochondria of RRECs. Hsp90 is used as cytosolic marker, while VDAC1 is mitochondrial marker. (D) Cx43 is predominately present on the inner mitochondrial membrane. COX-2 is used as control for mitochondrial inner membrane fraction, and VDAC1 is used as control for outer mitochondrial membrane fraction. P = pellet representing mitochondrial inner membrane fraction, S = supernatant representing mitochondrial outer membrane fraction, Cyto represents cytosolic fraction.
RRECs transiently or stably transfected with Cx43-GFP plasmid show localization of Cx43-GFP to gap junction plaques in the plasma membrane. In addition, Cx43-GFP signal was observed to localize on mitochondria in live RRECs (Fig. 1B). Since Cx43-GFP signal is seen throughout the cytosol, it is possible that the Cx43-GFP signal that localizes to mitochondria is not on the mitochondria but adjacent in the cytosol. Finally, we performed mitochondrial isolation from RRECs, in order to identify if Cx43 localizes to the mitochondrial fraction. In mitochondrial samples, Cx43 expression was detected by Western blot (Fig. 1C). VDAC1 expression was strong in the mitochondrial fraction, indicating the purity of the sample.
Connexin 43 Localization Is Primarily on the Inner Mitochondrial Membrane
Western blot analysis was performed with a protein obtained from fractions containing inner and outer mitochondrial membrane, which indicated Cx43 to be predominantly localized in the inner mitochondria membrane. This was verified using cytochrome c oxidase subunit 2 (COX-2) expression as control for inner mitochondria membrane fraction (Fig. 1D).
Mitochondrial Connexin 43 Is Downregulated by High Glucose Condition in Retinal Endothelial Cells
Cx43 protein expression in whole cell lysate has been shown to be downregulated by HG condition in vitro10,11 and in diabetes in vivo.13 HG-induced downregulation of Cx43 protein leads to reduced gap junction intercellular communication and influences apoptosis of retinal microvascular cells.14,22 Thus, we sought to investigate whether HG condition may also have an effect on Cx43 protein expression in the mitochondria. Mitochondria were isolated from RRECs grown in normal or HG condition, and Cx43 protein level was assessed by Western blot. Cx43 protein level was significantly reduced in mitochondria of RRECs grown in HG media compared with those grown in normal media (Fig. 2A; 59 ± 19% of control, P = 0.027). Concomittant with mitochondrial Cx43 downregulation under HG condition, we observed mitochondrial fragmentation, similar to previous studies6 (Fig. 2B).
Figure 2. .
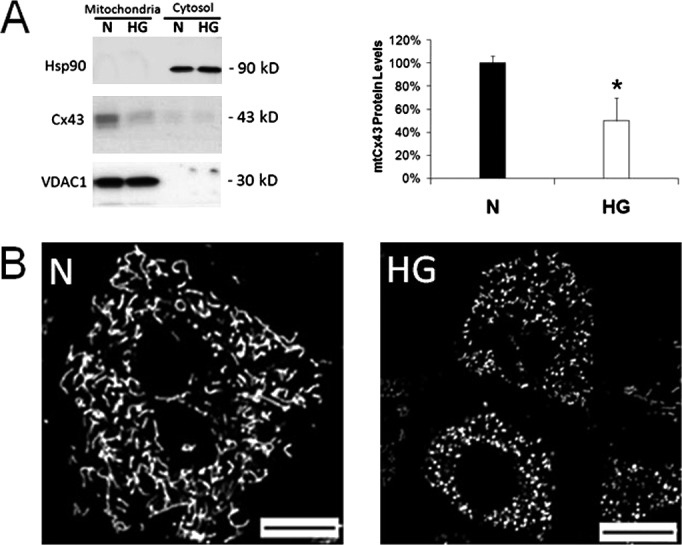
Mitochondrial connexin 43 is downregulated by high glucose in RRECs and is concomittant with mitochondrial fragmentation and cytochrome c release. (A) Mitochondria were isolated from RRECs grown in normal (N) or HG condition for 7 days, and Cx43 protein level was assessed by Western blot. Mitochondrial Cx43 protein level was significantly downregulated in RRECs grown in HG condition for 7 days. VDAC1, an abundant outer mitochondrial membrane protein, was used as control for mitochondrial protein, and Hsp90 for cytosolic fraction. *P < 0.05 (B) RRECs grown for 7 days in HG condition show mitochondrial fragmentation compared to cells grown in normal medium.
Connexin 43 Channel Activity Is Necessary for Maintenance of Mitochondrial Morphology
We have previously reported that mitochondria exist as long, tubular structures in RRECs, and that the mitochondrial network becomes fragmented when exposed to HG for six days.6 This mitochondrial fragmentation was associated with further mitochondrial dysfunction and commitment to apoptosis. To test whether downregulation of mitochondrial Cx43 may influence mitochondrial morphology change, RRECs plated sparsely without cell to cell contacts were treated with β-GA, an inhibitor of gap junction channels, or GZ, a chemical analog of β-GA that does not inhibit gap junction channels. Plating the cells without cell to cell contacts allowed us to investigate the effect of inhibiting Cx channels on mitochondria, rather than between cells. Treatment with β-GA significantly fragmented mitochondria toward a round, punctate morphology compared with untreated cells (Fig. 3A). GZ, a chemical analog of β-GA, that does not inhibit Cx channels, did not result in altered mitochondrial morphology (Fig. 3A). Quantifying the mitochondrial morphology change using FF and AR values verified that β-GA–treated cells display significantly lower FF and AR values compared with untreated and GZ-treated cells, indicating mitochondrial fragmentation (Fig. 3B; FF for β-GA–treated RRECs: 1.54 vs. 2.41 in untreated, P = 0.001, vs. 2.38 in GZ-treated, P = 0.001; AR: 1.86 vs. 2.11 in untreated, P = 0.010, vs. 2.02 in GZ-treated, P = 0.006).
Figure 3. .

Cx channel inhibitor disrupts mitochondrial morphology in retinal endothelial cells lacking cell-cell contacts. (A) Sparsely-plated RRECs exposed to β-GA, a Cx channel inhibitor, exhibited significant fragmentation of mitochondria into round, punctate morphology compared with those of untreated cells. Exposure to GZ, a chemical analog of β-GA that does not inhibit Cx channel activity, did not result in altered mitochondrial morphology. (B) Quantification of the mitochondrial morphology change using FF and AR values verified that β-GA–treated cells display significantly lower FF and AR values compared with untreated and GZ-treated cells, indicating mitochondrial fragmentation. *P < 0.05. (C) RRECs treated with β-GA showed decreased P2 and P1 forms of mtCx43, and increased P0 form indicating dephosphorylation of mtCx43. The P2 form is associated with active Cx43 channels, while P0 form is not.
In order to verify that β-GA treatment of RRECs lacking cell to cell contacts inhibits Cx43 channel activity on mitochondria, we assessed mitochondrial Cx43 phosphorylation patterns following β-GA or GZ treatment. RRECs treated with β-GA showed decreased phosphorylated 2 (P2) and phosphorylated 1 (P1) forms of mtCx43, and increased P0 form indicating dephosphorylation of mtCx43 (Fig. 3C; 36 ± 16% of control P2/P1, P = 0.007). The P2 form is associated with active gap junctions, while P0 form is not23; thus, β-GA treatment results in decreased active Cx43 channels on RREC mitochondria, in turn resulting in mitochondrial fragmentation similar to that induced by HG condition.
Treatment of Isolated Mitochondria with Cx Channel Inhibitor Causes Cytochrome c Release
To directly assess whether Cx43 channel inhibition by β-GA would compromise mitochondria leading to cytochrome c release, mitochondria were isolated from RRECs and treated with β-GA, GZ, or no treatment. During treatment mitochondria were suspended in MSB buffer. After treatment mitochondria were centrifuged and the supernatant was collected as the “Release” fraction. Western blot was then used to determine the extent of cytochrome c release from the mitochondria during treatment. Treatment of mitochondria for 4 hours with increasing concentrations of β-GA resulted in increasing release of cytochrome c from the mitochondria during the treatment, compared with GZ treatment (Fig. 4; 25 μM β-GA = 222 ± 66% of control, P < 0.05; 50 μM β-GA = 295 ± 83% of control, P < 0.05; 100 μM β-GA = 485 ± 111% of control, P < 0.01). In conclusion, decreased mtCx43 channel activity due to β-GA treatment or Cx43 downregulation under HG condition could promote cytochrome c release from mitochondria, leading to accelerated apoptosis of retinal endothelial cells.
Figure 4. .

Treatment of isolated mitochondria with Cx channel inhibitor causes cytochrome c release. (A) Mitochondria were isolated from RRECs, suspended in MSB buffer, and treated with β-GA, GZ, or no treatment. After treatment, mitochondria were centrifuged and the supernatant was collected as the “Mito-Release” fraction. Western blot analysis revealed that treatment of mitochondria for 4 hours with increasing concentrations of β-GA, Cx channel inhibitor, resulted in increasing release of cytochrome c from the mitochondria, compared with mitochondria treated with GZ, which does not inhibit Cx channel activity. Hsp90 was used as control for cytosolic fraction. VDAC1 was used as control for mitochondrial fraction. (B) Quantification of cytochrome c release from isolated mitochondria treated with β-GA. *P < 0.05. (C) RRECs exposed to β-GA for 24 hours also showed significant cytochrome c release compared to control.
Discussion
In this study we have identified Cx43 to be localized on mitochondria of RRECs, predominately in the inner mitochondrial membrane, and that it is downregulated by HG, which in turn, triggers cytochrome c release in the RRECs. Furthermore, we observed that mitochondrial Cx43 channel activity is necessary for maintenance of mitochondrial morphology, and importantly in isolated mitochondria inhibition of mitochondrial Cx43 channel activity–induced cytochrome c release. Results also indicate that HG-induced mitochondrial morphology changes in retinal vascular cells, which we previously reported,6 may be associated with dysfunction of mitochondrial Cx43 channel activity. While it is evident that HG-induced mitochondrial morphology changes and cytochrome c release leads to apoptosis,14 the underlying mechanisms of mitochondrial morphology disruption remain unclear. Our findings presented in this study suggest HG-induced mitochondrial Cx43 downregulation and reduced channel activity may contribute to cytochrome c release and promote apoptosis.
Several studies have shown that mitochondrial dysfunction plays a critical role in diabetes-induced apoptosis.1,3,24,25 Cx43 has been reported to be downregulated by HG and in diabetes where the canonical role of Cx43 as a gap junction channel is compromised, leading to reduced gap junction intercellular communication activity and triggering apoptosis.10–14 Here, we show that Cx43 located on the inner membrane is decreased by HG, which may result in overall decreased channel activity, provoking the release of cytochrome c. Presumably, these Cx43 on the mitochondrial inner membrane are functioning as hemi-channels (connexons); however, the limited studies on mitochondrial Cx43 have not ruled out other possible docked Cx43 channel activity in the mitochondrial membranes, or its functional role through interacting with key mitochondrial proteins.26 Cx43 hemi-channels on the plasma membrane of cells facilitate Na+ and Ca++ exchange between cells and the environment, H+ regulation and plasma membrane polarization, and other functions related to cell survival.27 Similarly, Cx43 hemi-channels in the mitochondrial inner membrane are involved in regulating mitochondrial ion exchange or mitochondrial volume and swelling.16 Other studies have shown mitochondrial Cx43 upregulation to promote ischaemic preconditioning, where mitochondrial ion regulation and swelling are established players in the preconditioning process.18 In addition, respiration defect was observed in isolated mitochondria from Cx43-deficient mice15; thus, findings from these studies and our current study point to the important role of Cx43 in normal mitochondrial functioning.
Mechanisms for Cx43 intracellular trafficking is not completely understood at this present time. Our studies using plasmid–Cx43-GFP showed a majority of the Cx43 plaques would localize on the cell plasma membrane, whereas a small percentage (<5%) would localize on the mitochondria. This difference in the distribution of Cx43 localization on mitochondria has been verified by more quantitative studies in rat myocardium.26,28 In retinal endothelial cells, overall Cx43 protein level both in the mitochondria and cell surface is significantly reduced under HG condition10; however, it is unclear whether HG alters this ratio. Presumably, an imbalance between the levels of Cx43 between the plasma membrane and the mitochondria may disrupt normal ion homeostasis. Cx43 is imported into the mitochondria by the transporter outer membrane (TOM) and transporter inner membrane (TIM) mitochondrial import machinery, and currently, it is believed that a certain percentage of the total Cx43 is targeted to the mitochondria.17 Further studies are needed to better understand Cx43 targeting to the mitochondria versus the cell membrane, and the consequences resulting from an imbalance between the two compartments.
Recent studies have stressed the importance of mitochondrial morphology changes and its association with disease processes. In diabetes, mitochondrial morphology changes have been shown to induce mitochondrial dysfunction through mitochondrial morphology changes in pancreatic islets,29 kidney podocytes,30 skeletal muscle,31 coronary endothelial cells,32 and retinal endothelial cells6 and pericytes.7 Maintenance of normal mitochondrial morphology has been shown to regulate several aspects of mitochondrial functioning, including mitochondrial metabolism, reactive oxygen species production, and induction of apoptosis.4 Importantly, our previous studies have shown how mitochondrial fragmentation under HG condition promotes mitochondrial oxygen consumption defects and increased apoptosis of both retinal endothelial cells6 and pericytes,7 two cell types that undergo cell death in the early stages of diabetic retinopathy.33 In a diabetic animal model we have observed Cx43 downregualtion in the retina and subsequent retinal pathology similar to those seen in human diabetic retinopathy.13 Here we present evidence that inhibition of normal mitochondrial Cx43 channel activity can lead to mitochondrial fragmentation, similar to the HG-induced mitochondrial morphology changes. This may point to an important role for HG-induced mitochondrial Cx43 inhibition in promoting mitochondrial fragmentation and subsequent apoptosis of retinal microvascular cells.
In this study we present data demonstrating the localization and important functional role of Cx43 on the mitochondria of retinal endothelial cells. As of yet, it is still unclear what is the functional role of Cx43 in the mitochondria, and whether it forms hemi-channels and what are the key molecules that are being exchanged through this channel. Only recently, Cx43 has been identified on the mitochondria of different cell types. It is clear that mitochondrial Cx43 plays an important role in mitochondrial function, specifically regulation of apoptosis.8,26 Our data demonstrates that HG is a critical player that drives mitochondrial Cx43-mediated cellular disturbances. Identifying means to improve proper functioning of Cx43 channel activity on mitochondria may hold therapeutic promise for treatment of diabetic retinopathy.
Footnotes
Supported by grants from the National Eye Institute (NIH EY018218 [SR]), Fight for Sight, Boston University Undergraduate Research Opportunities Program, and in part by a departmental grant from the Massachusetts Lions Organization.
Disclosure: K. Trudeau, None; T. Muto, None; S. Roy, None
References
- 1.Kowluru RA. Diabetic retinopathy: mitochondrial dysfunction and retinal capillary cell death. Antioxid Redox Signal. 2005;7:1581–1587 [DOI] [PubMed] [Google Scholar]
- 2.Nishikawa T, Araki E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9:343–353 [DOI] [PubMed] [Google Scholar]
- 3.Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and beta-cell failure in type 2 diabetes mellitus. Exp Diabetes Res. 2012;703538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scorrano L. Multiple functions of mitochondria-shaping proteins. Novartis Found Symp. 2007;287:47–55; discussion 55–49 [DOI] [PubMed] [Google Scholar]
- 5.Wasilewski M, Scorrano L. The changing shape of mitochondrial apoptosis. Trends Endocrinol Metab. 2009;20:287–294 [DOI] [PubMed] [Google Scholar]
- 6.Trudeau K, Molina AJ, Guo W, Roy S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: implications for diabetic retinopathy. Am J Pathol. 2010;177:447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trudeau K, Molina AJ, Roy S. High glucose induces mitochondrial morphology and metabolic changes in retinal pericytes. Invest Ophthalmol Vis Sci. 2011;52:8657–8664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goubaeva F, Mikami M, Giardina S, Ding B, Abe J, Yang J. Cardiac mitochondrial connexin 43 regulates apoptosis. Biochem Biophys Res Commun. 2007;352:97–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400 [DOI] [PubMed] [Google Scholar]
- 10.Sato T, Haimovici R, Kao R, Li AF, Roy S. Downregulation of connexin 43 expression by high glucose reduces gap junction activity in microvascular endothelial cells. Diabetes. 2002;51:1565–1571 [DOI] [PubMed] [Google Scholar]
- 11.Li AF, Sato T, Haimovici R, Okamoto T, Roy S. High glucose alters connexin 43 expression and gap junction intercellular communication activity in retinal pericytes. Invest Ophthalmol Vis Sci. 2003;44:5376–5382 [DOI] [PubMed] [Google Scholar]
- 12.Fernandes R, Girao H, Pereira P. High glucose down-regulates intercellular communication in retinal endothelial cells by enhancing degradation of connexin 43 by a proteasome-dependent mechanism. J Biol Chem. 2004;279:27219–27224 [DOI] [PubMed] [Google Scholar]
- 13.Bobbie MW, Roy S, Trudeau K, Munger SJ, Simon AM, Roy S. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Invest Ophthalmol Vis Sci. 2010;51:3758–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li AF, Roy S. High glucose-induced downregulation of connexin 43 expression promotes apoptosis in microvascular endothelial cells. Invest Ophthalmol Vis Sci. 2009;50:1400–1407 [DOI] [PubMed] [Google Scholar]
- 15.Boengler K, Ruiz-Meana M, Gent S, et al. Mitochondrial connexin 43 impacts on respiratory complex I activity and mitochondrial oxygen consumption. J Cell Mol Med. 2012;16:1649–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miro-Casas E, Ruiz-Meana M, Agullo E, et al. Connexin43 in cardiomyocyte mitochondria contributes to mitochondrial potassium uptake. Cardiovasc Res. 2009;83:747–756 [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Sinovas A, Boengler K, Cabestrero A, et al. Translocation of connexin 43 to the inner mitochondrial membrane of cardiomyocytes through the heat shock protein 90-dependent TOM pathway and its importance for cardioprotection. Circ Res. 2006;99:93–101 [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP. Mitochondria and preconditioning: a connexin connection? Circ Res. 2006;99:10–12 [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Sinovas A, Cabestrero A, Lopez D, et al. The modulatory effects of connexin 43 on cell death/survival beyond cell coupling. Prog Biophys Mol Biol. 2007;94:219–232 [DOI] [PubMed] [Google Scholar]
- 20.Chronopoulos A, Trudeau K, Roy S, Huang H, Vinores SA, Roy S. High glucose-induced altered basement membrane composition and structure increases trans-endothelial permeability: implications for diabetic retinopathy. Curr Eye Res. 2011;36:747–753 [DOI] [PubMed] [Google Scholar]
- 21.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology. 1992;24:145–149 [PubMed] [Google Scholar]
- 22.Bobbie M, Raithel R, Simon A, Roy S. Effect of Connexin 43 Downregulation on the Development of Retinal Vascular Lesions in Diabetic Retinopathy. Paper presented at: Association for Research in Vision and Ophthalmology Annual Meeting; May 3–7, 2008; Fort Lauderdale, FL [Google Scholar]
- 23.Guan X, Wilson S, Schlender KK, Ruch RJ. Gap-junction disassembly and connexin 43 dephosphorylation induced by 18 beta-glycyrrhetinic acid. Mol Carcinog. 1996;16:157–164 [DOI] [PubMed] [Google Scholar]
- 24.Wang Q, Frolova AI, Purcell S, et al. Mitochondrial dysfunction and apoptosis in cumulus cells of type I diabetic mice. PLoS One. 2010;5:e15901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabadkai G, Duchen MR. Mitochondria mediated cell death in diabetes. Apoptosis. 2009;14:1405–1423 [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Meana M, Rodriguez-Sinovas A, Cabestrero A, Boengler K, Heusch G, Garcia-Dorado D. Mitochondrial connexin43 as a new player in the pathophysiology of myocardial ischaemia-reperfusion injury. Cardiovasc Res. 2008;77:325–333 [DOI] [PubMed] [Google Scholar]
- 27.Wilkins AS. Gap junction-mediated intercellular signalling in health and disease. Bioessays. 1998;20:686–688 [DOI] [PubMed] [Google Scholar]
- 28.Boengler K, Dodoni G, Rodriguez-Sinovas A, et al. Connexin 43 in cardiomyocyte mitochondria and its increase by ischemic preconditioning. Cardiovasc Res. 2005;67:234–244 [DOI] [PubMed] [Google Scholar]
- 29.Molina AJ, Wikstrom JD, Stiles L, et al. Mitochondrial networking protects beta-cells from nutrient-induced apoptosis. Diabetes. 2009;58:2303–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Wang Y, Long J, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jheng HF, Tsai PJ, Guo SM, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32:309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shenouda SM, Widlansky ME, Chen K, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ejaz S, Chekarova I, Ejaz A, Sohail A, Lim CW. Importance of pericytes and mechanisms of pericyte loss during diabetes retinopathy. Diabetes Obes Metab. 2008;10:53–63 [DOI] [PubMed] [Google Scholar]