

Abstract
Peptide macrocycles are found in many biologically active natural products. Their versatility, resistance to proteolysis and ability to traverse membranes has made them desirable molecules. Although technologies exist to synthesize such compounds, the full extent of diversity found among natural macrocycles has yet to be achieved synthetically. Cyanobactins are ribosomal peptide macrocycles encompassing an extraordinarily diverse range of ring sizes, amino acids and chemical modifications. We report the structure, biochemical characterization and initial engineering of the PatG macrocyclase domain of Prochloron sp. from the patellamide pathway that catalyzes the macrocyclization of linear peptides. The enzyme contains insertions in the subtilisin fold to allow it to recognize a three-residue signature, bind substrate in a preorganized and unusual conformation, shield an acyl-enzyme intermediate from water and catalyze peptide bond formation. The ability to macrocyclize a broad range of nonactivated substrates has wide biotechnology applications.
Marine natural products are a recognized source of diverse biologically active molecules1,2. These compounds have shown considerable promise as starting points in a wide range of therapeutic areas and have yielded a number of clinically approved therapeutics for cancer and pain3. Their exploitation, like that of natural products from the terrestrial sphere, is limited by low yields from natural sources, challenges in their organic synthesis and the inability to source large numbers of variants to optimize activity. For this reason, biotechnological or semisynthetic approaches beginning with natural starting materials have been used for several drugs (for example, ref. 4). Cyanobactins5 are ribosomal cyclic peptides (macrocycles) with an increasing list of interesting and important biological activities6. Understanding their biosynthesis should allow us to harness the relevant biosynthetic pathways and thus open up new avenues for the exploitation of these compounds.
Patellamides, members of the cyanobactin superfamily, are produced by Prochloron sp., obligate, uncultured symbionts of the sea squirt Lissoclinum patella7,8 (Fig. 1a). Patellamides display a variety of biological activities including cytotoxicity and the ability to reverse multiple-drug resistance in human leukemia cells9,10. Prochloron sp. produce cyclic peptides seven or eight amino acids in length that contain heterocyclized serine or threonine and cysteine residues, giving rise to oxazolines and thiazolines (CysThn), the latter of which are usually subsequently oxidized to thiazoles. (For a full description of modifications, see refs. 9–11.) The patellamide gene cluster consists of seven genes called patA–patG, five of which (patA, patD, patE, patF and patG) have been shown to be essential for patellamide biosynthesis11,12. The prepropeptide PatE consists of a 37-residue N-terminal leader sequence (containing a helix spanning residues 13–28 (ref. 13)) followed by two eight-residue cassettes7. Each cassette is flanked by a five-residue protease signature at the N terminus and a four-residue macrocyclization signature at the C terminus (Fig. 1a).
Figure 1.

Macrocyclization of patellamides. (a) The PatE prepropeptide consists of an N-terminal leader sequence followed by two eight-residue cassettes with the C-terminal macrocyclase recognition signal AYDG. X indicates any amino acid. The macrocyclization domain of PatG catalyzes the formation (dashed lines) of two cyclic peptides per prepropeptide. (b) PatGmac requires a heterocycle or proline (denoted Z) at the P1 position and the AYDG motif at the P1′–P4′ sites. An additional glutamate is often found at P5′ but is not required. (c) The test substrate used in this study can either give a linear peptide (curved line) with a mass of 716.375 Da or a macrocycle (octagon), which is 18 Da lighter.
Bioinformatic analysis of PatG identifies three domains: an N-terminal oxidase domain, a central subtilisin-like domain and a C-terminal domain of as-yet-unknown function. The subtilisin-like domain of PatG (PatGmac) has been identified as possessing the necessary macrocyclase activity for the patellamide biosynthetic pathway14,15. Subtilisin-like proteases typically recognize and cleave within a hexapeptide sequence (denoted P4, P3, P2, P1 before and P1′, P2′ after the cleavage site). The precise extent of recognition varies to include P5 and/or exclude P2′ (ref. 16). The corresponding binding sites on the protease are denoted S5–S2′. PatGmac has been reported to recognize the C-terminal macrocyclization signature (positions P1′–P4′) AYDG15 and to require a proline residue at the P1 site when unmodified peptides (Fig. 1b,c) are used15. Presumably, proline mimics the five-membered ring of the heterocycle in the natural product (Fig. 1b,c). The sequence requirements of the remainder of the cassette appear to be very relaxed, with no obvious preference or limitation.
Macrocyclization is common in both nonribosomal and ribosomal peptide biosynthetic pathways, presumably because of the significant advantages that macrocycles often have in stability, membrane permeability and activity over their linear counterparts. A number of synthetic and biotransformation strategies now exist for the generation of macrocyclic peptides for biology, but problems remain at a practical level, in terms of limitations on substrate diversity, yield and expense17. In nature, nonribosomal peptide macrocyclization is carried out by thioesterase (TE) domains, which contain the same Asp-His-Ser catalytic triad as serine proteases18,19. The substrate is first bound to a peptidyl carrier protein by a thioester linkage and then transferred (by transacylation) to the active site serine of a TE domain, forming a classical acyl-enzyme intermediate20. The N-terminal amine of the peptide then attacks the ester carbonyl, displacing the serine to form the macrocycle.
By analogy, macrocyclization of cyanobactins has been proposed to proceed via an acyl-enzyme intermediate14 that is attacked by the N terminus of the cassette to yield the macrocycle. However, TE domains and PatGmac work on quite distinct chemical entities; the activated thioester is a very different substrate from the peptide bond. PatGmac, with its ability to handle simple nonactivated peptide substrates, is potentially very valuable for organic synthesis and biotechnological applications. We set out to determine how the macrocyclase achieves this chemistry and the molecular basis of substrate recognition. We report the structure of a macrocyclase domain (PatGmac) in complex with a substrate mimic and characterize the enzyme biochemically. We have used these data to rationalize both PatGmac’s mechanism of macrocyclization and its recognition requirements. We have demonstrated this understanding by engineering altered specificity into the enzyme and enhancing its catalytic rate.
RESULTS
Overall structure of the PatG macrocyclase domain
The macrocyclase domain of PatG (PatGmac, residues 492–851) was overexpressed in Escherichia coli BL21 (DE3) cells and purified using established protocols21. The retention profile from gel filtration indicated that the domain was a monomer. The protein formed crystals belonging to the space group C2, with two biological monomers in the asymmetric unit. The structure was determined at 2.19-Å resolution by molecular replacement using the subtilisin Bacillus Ak.1 protease (AkP) (PDB: 1DBI) as a search model. Table 1 shows the data collection and refinement statistics. The refined model (PDB: 4AKS) includes residues 514–653, 659–685, 694–717, 728–745, 754–822 and 835–851 in chain A and residues 515–650, 660–688 and 692–850 in chain B. The missing residues are in loops and at the N terminus and are presumed to be disordered. PatGmac has a spherical shape with dimensions of approximately 53 Å × 42 Å × 48 Å. The protein contains a seven-stranded parallel β-sheet with two α-helices on each face, a fold common to all subtilisin-like proteases (Fig. 2a). However, the conserved metal ion of subtilisin-like proteases is not present in PatGmac, as the binding site is destroyed by sequence changes. PatGmac contains a catalytic triad consisting of Asp548 (located at the C terminus of the β-strand β1), His618 (in the middle of α4) and Ser783 (at the N terminus of α7). The carboxyl group of Asp548 is hydrogen-bonded to the side chain of His618 (2.9 Å), which is in turn hydrogen-bonded to the side chain of Ser783 (2.7 Å) (Fig. 2a).
Table 1.
Data collection and refinement statistics
| PatGmac | PatGmac + peptide | |
|---|---|---|
| Data collectiona,b | ||
| Space group | C2 | C2 |
| Cell dimensions | ||
| a, b, c (Å) | 132.08, 67.58, 97.34 | 135.63, 67.32, 137.87 |
| α, β, γ (°) | 90.0, 115.01, 90.0 | 90.0, 116.76, 90.0 |
| Resolution (Å) | 2.19 (2.24–2.19) | 2.63 (2.77–2.63) |
| Rmerge | 6.1 (49.8) | 10.7 (52.2) |
| I / σI | 13.7 (2.9) | 10.1 (2.3) |
| Completeness (%) | 99.5 (98.8) | 99.3 (96.4) |
| Redundancy | 3.6 (3.5) | 3.7 (3.1) |
| Refinement | ||
| Resolution (Å) | 33.79–2.19 | 21.42–2.63 |
| No. reflections | 38,196 | 31,502 |
| Rwork / Rfree | 0.203 / 0.224 | 0.191 / 0.218 |
| No. atoms | 4,877 | 5,108 |
| Protein | 4,653 | 4,897 |
| Ligand/ion | – | 69 |
| Water | 224 | 142 |
| B factors | 50.11 | 60.56 |
| Protein | 50.04 | 60.70 |
| Ligand/ion | – | 77.98 |
| Water | 51.5 | 47.19 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.009 | 0.009 |
| Bond angles (°) | 1.249 | 1.253 |
One crystal user per structure.
Values in parentheses are for highest-resolution shell.
Figure 2.
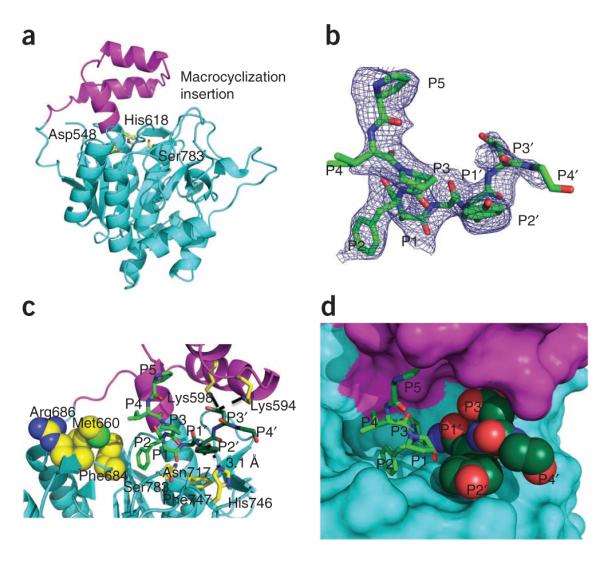
Structure of PatGmac. (a) PatGmac has a subtilisin-like core (cyan) and the conventional catalytic triad (yellow sticks). PatGmac has an insertion (magenta) that extends from β2 as a loop, then forms a helix-loop-helix motif and creates an N-terminal extension of α4, the helix that harbors His618. The insertion is found in other macrocyclases but is not conserved in length or sequence. (b) Difference electron density (Fo – Fc contoured at 3σ with phases calculated from a model which was refined with no peptide present) of the PIPFPAYDG substrate mimic bound to PatGmac H618A; three N-terminal residues (VPA) of the substrate mimic are disordered. (c) Interactions between the substrate mimic and PatGmac H618A. The proline (at position P1) of the substrate adopts a cis peptide conformation that results in the substrate pointing away from the protein. The side chains of Met660, Phe684 and Arg686 would prevent the binding of substrates that adopt an extended conformation. Lys598 and possibly Lys594 form salt bridges (dashed lines) with the P3′ residue (aspartate), whereas the P2′ tyrosine forms a hydrogen bond (dashed line) with His746 and interacts with Phe747 through π-stacking. (d) The active site where the acyl-enzyme intermediate would be formed is shielded from solvent by the macrocyclization insertion and the AYDG peptide (dark green).
Comparison of subtilisin-like protease AkP and PatGmac
The amino acid sequences of the subtilisin-like protease AkP and PatGmac are 28% identical, and pairwise superposition gives a Cα r.m.s. deviation of 1.23 Å over 145 structurally equivalent residues. The most striking difference is that PatGmac contains a helix-turn-helix insertion between β2 and α4 (residues Ala574–Lys610) that sits above the active site (Fig. 2a and Supplementary Fig. 1a); we denote this as the macrocyclization insertion. Eight of the residues in the insertion form a two-turn N-terminal extension of α4 when compared to the typical subtilisin structure. This results in the catalytic histidine being in the middle of this helix rather than at the end (Supplementary Fig. 1b). The other 29 residues form a helix-turn-helix motif. Four cysteines, which are highly conserved in PatG and its homologs (Supplementary Fig. 2), make two disulfide bonds: Cys685-Cys724 and Cys823-Cys834. The Cys685-Cys724 disulfide bond in PatGmac is different from that seen in subtilisins. Cys137 of AkP is equivalent to Cys685 of PatGmac, and it forms an intraloop disulfide bond with Cys139, making an 11-atom ring that is proposed to rigidify the active site. In contrast, PatGmac’s Cys685-Cys724 disulfide bond bridges two loops, one of which connects β4 to α6 adjacent to the active site (Supplementary Fig. 1c). As a result, Phe684 and Arg686 pack against the side chain of Met660, completely filling the substrate-binding pockets at positions S4 and S3. The Cys823-Cys834 disulfide bond links the ends of the loop that connects α8 to α9 at the C terminus of the domain and is distant from the active site.
PatGmac substrate complex
The VPAPIPFPAYDG peptide was chosen to match the residues equivalent to substrate positions P8–P4′, the eight-residue cassette and four-residue C-terminal macrocyclization signature. The proline residues were chosen to mimic the heterocycles of the natural substrate, and the peptide can in fact be macrocyclized by PatGmac, albeit slowly. The structure of the complex of PatGmacH618A (an inactive mutant) with peptide was determined at 2.63 Å by molecular replacement using PatGmac native as a search model (Table 1). The difference electron density for bound peptide in the active site of one protomer was unambiguous for PIPFPAYDG (P5–P4′) (Fig. 2b). The refined model (PDB 4AKT) contains residues 514–686, 694–719, 727–747, 754–823 and 833–851 in chain A and residues 515–651, 657–688 and 692–851 in chain B.
Substrate positions P5 (proline) and P4 (isoleucine) make no contact with the protein, whereas position P3 (proline) has weak van der Waals interactions with Tyr210. Position P2 (phenylalanine) also makes limited van der Waals contacts, and the side chain sits in a shallow pocket (Fig. 2c,d). The proline of position P1 adopts a cis peptide conformation, and the side chain makes van der Waals contacts with His618 (mutated alanine in complex) and Val622. The carbonyl of the P1–P1′ peptide is 4.3 Å from, and correctly oriented for nucleophilic attack by, the hydroxyl of Ser783. The side chain of Met784 sits on this face of the carbonyl, and the side chain of the absolutely conserved Asn717 points toward the opposite face in the correct position to stabilize the tetrahedral intermediate. The P1′ alanine Cα and side chain make only a few hydrophobic interactions, including contacts with Met784 and the protein backbone. It sits in a cavity that appears to be large enough for bulkier residues. The P2′ (tyrosine) residue makes extensive contacts with the protein: a π-stacking interaction with the highly conserved Phe747, a hydrogen bond to His746 (conserved as histidine or lysine in homologs) and a hydrogen bond between the tyrosine main chain oxygen and the nitrogen of Thr780 (Fig. 2c). The side chain of P3′ (aspartate) is oriented toward a large electropositive patch created by Arg589, Lys594 and Lys598. It makes a salt bridge with Lys598 and possibly Lys594, though the side chain of Lys594 is not well ordered (Fig. 2c). The P4′ glycine residue makes no contact with the protein, although the terminal carboxyl group is close to Lys594. The binding of the peptide is accompanied by changes in PatGmac at Phe684, as the main chain moves 2 Å at the Cα position to avoid a clash with the substrate.
Biochemical characterization of macrocyclization
The peptide VGAGIGFPAYDG was used as a substrate for PatGmac in biochemical assays (Fig. 1c). The ratio of macrocyclized to linear product using this substrate peptide was determined by ion counts obtained from liquid-chromatography electrospray-ionization mass spectrometry (LC-ESI MS). For native protein, only macrocyclized product (cycloVGAGIGFP) was detected (Table 2, Fig. 3a and Supplementary Fig. 3a).
Table 2.
Relative ion counts of linear cleaved and macrocyclized peptide substrate
| Unprocessed ion count (%) (M+H = 1,123) |
Linear ion count (%) (M+H = 717) |
Cyclic ion count (%) (M+H = 699) |
|
|---|---|---|---|
| PatGmac | 0 | 0 | 100 |
| PatGmacΔ1 | 8 | 92 | 0 |
| PatGmacΔ2 | <1 | >99 | 0 |
| PatGmac K598D | 0 | 100 | 0 |
| PatGmac K594D | 0 | 71 | 29 |
| PatGmac R589D | |||
| K594D K598D | 94 | 6 | 0 |
Figure 3.

Biochemical characterization of PatGmac and PatGmac mutants. (a–c) LC-ESI MS of macrocyclization reactions with PatGmac wild type (a), PatGmacΔ2 (b) and PatGmac K594D (c). Macrocyclized and linear products are indicated with octagons and curved lines, respectively. The error between observed and calculated mass is shown below the [M+H]+ and [M+Na]+ species. (d) Evidence for a stable acyl-enzyme intermediate between PatGmac and substrate.
PatGmac is a slow enzyme; turnover rates reported to date are ~1 per day14,15. Increasing the NaCl concentration from 150 mM to 500 mM improved rates by more than an order of magnitude. Increasing the pH from 8 to 9 further tripled the rate. Adding DMSO gave a small increase in rate but shifted the optimum pH; thus, a buffer containing 500 mM NaCl and 5% DMSO at pH 8 gave a reaction rate over 50 times greater (Supplementary Fig. 4). Under these conditions, about 7% linearized VGAGIGFP byproduct was observed, which could be separated from cycloVGAGIGFP by HPLC.
Mutants K594D and K598D as well as two deletion mutants, PatGmacΔ1 (lacking residues 578–608, the helix-loop-helix insertion motif) and PatGmacΔ2 (lacking residues 578–614, the helix-loop-helix insertion and the N-terminal extension of α4), consumed substrate at approximately the rate of native protein (Fig. 3 and Supplementary Fig. 3b). For K594D, approximately one-third of the product was macrocyclized and two-thirds were linear peptide. K598D and both deletions gave only linear VGAGIGFP (Fig. 3 and Supplementary Fig. 3b). The triple mutant R589D K594D K598D had a substantially slower catalytic rate and produced only linear substrate. All mutants purified normally and were folded, according to CD spectroscopy.
The substrate VGAGIGFPAYRG has a modified recognition sequence (aspartate to arginine); as expected, PatGmac wild type (and K594D and triple mutant R589D K594D K598D) reacted extremely slowly with this substrate, giving equal amounts of macrocyclized and linear products. PatGmac K598D produced cycloVGAGIGFP with only 8% linear product, at a rate over an order of magnitude faster than wild-type PatGmac with VGAGIGFPAYDG (Supplementary Fig. 5a,b). The precise nature of the N terminus of the substrate influenced the rate, as VGAGIGFPAYRG was processed an order of magnitude faster than GVAGIGFPAYRG.
Mutants S783A and H618A (both affecting the catalytic triad) gave no detectable reaction. MS clearly identified an acyl-enzyme intermediate (ITACThnITFCThn-PatGmac) during turnover (Fig. 3d).
DISCUSSION
The structure of PatGmac establishes that it belongs to the subtilisin class of proteases, possessing the characteristic catalytic Asp-His-Ser triad and core structure22. PatGmac cuts before an AYDG motif and macrocyclizes the preceding eight residues. Macrocyclization activity depends on Ser783 and His618; mutation of either residue inactivates the enzyme, pointing to an acyl-enzyme intermediate, which we have observed (Fig. 3d). The complex structure shows that the P1-P1′ bond of the substrate is in the correct position for nucleophilic attack by Ser783, with Asn717 stabilizing the tetrahedral intermediate that would subsequently collapse to the classical acyl-enzyme intermediate. The P1 (proline) residue adopts a cis peptide conformation, kinking the substrate peptide chain so that the side chain of P2 (phenylalanine) points into a nonspecific shallow pocket (Fig. 2c). As a result of the main chain kink, P3 and the preceding residues (P4 and P5) of the substrate point away from the protein surface. The nonspecific nature of the cavity that binds P2, and the lack of any additional contacts between PatGmac and the patellamide cassette, is consistent with the known lack of enzyme specificity for the cassette sequence14,15. Substrate recognition by PatGmac is focused on substrate residues C terminal to the cleavage site, in direct contrast to subtilisin-like proteases, where recognition is focused on substrate peptide residues N terminal to the cleavage site23. Further, substrates in proteases almost uniformly adopt an extended (β-strand–like) conformation. The binding of a substrate in this extended conformation is actively prevented in PatGmac, as the S3 and S4 recognition sites are, in fact, blocked by the main and side chains of a protein loop, which is itself stabilized by a disulfide bond conserved in PatGmac homologs (Fig. 2c). As a result, only a substrate whose P3 and P4 residues point away from the protein surface (thus making no recognition interaction) can bind to the protein. This bending away requires a cis peptide–like conformation between P2 and P1, which is energetically accessible only for proline (or for heterocyclized cysteine (thiazole or thiazoline) and serine or threonine (oxazoline) residues)14. For unmodified non-proline amino acids, the cis configuration would be too high in energy, and thus PatGmac cannot process such a substrate even though it possesses the AYDG macrocyclization signature.
Neither alanine nor glycine contributes notably to binding of the macrocyclization signature. The P1′ (alanine) residue of the signature sits in an open pocket, and indeed signatures with serine in that location are known15. The glycine at P4′ makes no interaction with the protein, although larger side chains could clash with Lys594, consistent with the preference, but not requirement, for glycine. It is the two other residues of the signature that make clearly favorable interactions with the enzyme. The aromatic ring of tyrosine (P2′) makes extensive interactions with the protein, consistent with the high degree of conservation of this residue (though some sequences have a phenylalanine) in macrocyclization signatures24. The aspartate (P3′), which binds to two conserved lysine residues (Lys594 and Lys598; Supplementary Fig. 2)), is also highly conserved (though in two substrates, glutamate is present in the signature)25.
To favor nucleophilic attack by the N terminus of the patellamide cassette over hydrolysis, it is essential for PatGmac to protect the acyl-enzyme intermediate from water. This protection must be efficient, given that no linear product is detected with our test substrate and native enzyme. PatGmac contains a large helix-loop-helix insertion adjacent to the active site, which results in the extension of the α4 helix and partial shielding of the active site and acyl-enzyme intermediate from water (Fig. 2d). This macrocyclization insertion is present in all PatG homologs and varies in length between 35 and 50 residues; secondary-structure predictions suggest that the helix-loop-helix motif is conserved (Supplementary Fig. 2). Removing the insertion by deletion mutation eliminates macrocyclization activity while retaining protease activity (hydrolysis), consistent with exposure of the acyl-enzyme intermediate to water.
The structure shows that the macrocyclization signature (AYDG) works together with the macrocyclization insert to complete the shielding of the acyl-enzyme intermediate from water (Fig. 2d). This model would require that AYDG remain bound at the active site after cleavage. Point mutations designed to disrupt the interaction do indeed reduce (K594D) or abolish (K598D) macrocyclase activity while retaining protease activity (Table 2). The more mobile K594D can rotate away from the S3′ site, minimizing the repulsive effect and thus retaining limited macrocyclization activity, whereas Lys594 is more conformationally restricted, and the effect of K598D mutation is consequently more profound (Fig. 3 and Table 2). This hypothesis predicts that a protein with reversed charge should process a macrocyclization signature with complementary charge swapped. The engineered PatGmac K598D mutant can indeed macrocyclize two peptides, containing an AYRG signature, that are not processed by wild-type enzyme, demonstrating the possibility of engineering in novel recognition elements.
A model of the acyl-enzyme intermediate with bound AYDG clearly shows that there is enough space around the alanine residue to allow it to move after cleavage and be positioned to attack the acyl-enzyme intermediate without clashing with PatGmac (Fig. 4a). As long as the AYDG peptide remains at the enzyme active site, the acyl-enzyme intermediate will exist in equilibrium with uncleaved substrate (Fig. 4b). This equilibrium exists for all proteases, but normally the rapid displacement of the cleaved portion by water drives the equilibrium to product. We propose that PatGmac retains the two product peptides (one anchored by the acyl-enzyme intermediate, the other anchored by salt bridge(s), hydrogen bonds and van der Waals interactions) at the active site, in equilibrium with uncleaved substrate, which is in turn in equilibrium with free substrate and enzyme. These equilibria would be disrupted only by the incoming N terminus of the cassette displacing the AYDG peptide, followed by the decomposition of the acyl-enzyme intermediate to give a macrocycle. In a chemical sense, the reaction is a transpeptidation. As already discussed, macrocyclization of polyketides by thioesterases proceeds via a thioester, rather than an ester, and operates on an activated substrate20. The sortase enzyme is a transpeptidase and is conceptually similar in that it attacks an unactivated peptide to form a thioester (in the first steps of the protease reaction). The covalent intermediate is decomposed by the peptide amino group to form a new peptide (or isopeptide) bond26. A third related enzyme class, the transglutaminase, forms a thioester intermediate, much as the protease does, but attacks a glutamine side chain rather than the peptide bond27. The intermediate is subsequently decomposed by an incoming amine nucleophile, forming a cross-link.
Figure 4.

Proposed mechanism for macrocyclization. (a) Model of the acyl-enzyme intermediate with AYDG remaining bound at the active site. (b) The acyl-enzyme intermediate is in equilibrium with the substrate. In PatGmac the N terminus of the substrate enters the active site, displacing AYDG and leading to macrocyclization. Mutations that disrupt binding of AYDG lead to linear product, as the substrate is hydrolyzed by water. The role of the histidine in deprotonating the incoming N terminus is speculative.
The PatG reaction is slow, but with optimization we have achieved rates that have been shown to be useful for other difficult reactions such as carbon-fluorine bond formation28. We note that previous work29 has shown that a cassette containing all l-amino acids and oxazolines and thiazolines is preorganized with the N and C termini close together, favoring macrocyclization. Such substrates may be processed more quickly than the test substrates to which we have access. Our mechanism is supported by several pieces of data. The acyl-enzyme intermediate is observed under reaction conditions, indicating its stability. Mutations aimed at disrupting binding of the macrocyclization signature do not prevent formation of the acyl-enzyme intermediate but instead lead to linear hydrolyzed product, consistent with a shift in the equilibrium between bound AYDG and water molecules (Fig. 4b). The nature of the side chain on the two N-terminal residues of the substrate does influence rate, as would be expected if the incoming N terminus docks to the protein. Raising the pH of the reaction increases the rate, consistent with increasing the amount of deprotonated incoming N terminus. It is possible, but remains untested, that His618 helps to deprotonate the incoming N terminus. The ability to accelerate the rate by almost two orders of magnitude by simple changes in buffer conditions greatly enhances the utility of the enzyme.
Cyanobactins are a large and diverse family of biologically active natural products synthesized and modified from ribosomally encoded peptides through a series of chemical transformations. PatGmac is the first subtilisin-like macrocyclase to be characterized, and the understanding of its mechanism and recognition requirements have allowed its rational re-engineering and improvement in rate. Opportunities for further engineering of the enzyme for use in biotransformation have been enabled by this work.
ONLINE METHODS
Protein cloning, expression and purification
PatGmac (PatG residues 492–851) was cloned from genomic DNA (Prochloron sp.) into the pHISTEV vector30 and expressed in Escherichia coli BL21 (DE3) grown on autoinduction medium31 for 48 h at 20 °C. Cells were harvested by centrifugation at 4,000g, 20 °C, for 15 min and resuspended in lysis buffer (500 mM NaCl, 20 mM Tris, pH 8.0, 20 mM imidazole and 3 mM β-mercaptoethanol (BME)) with the addition of complete EDTA-free protease inhibitor tablets (Roche) and 0.4 mg DNase (Sigma) g−1 wet cells. Cells were lysed by passage through a cell disruptor at 30 kPSI (Constant Systems), and the lysate was cleared by centrifugation at 40,000g, 4 °C for 45 min. Cleared lysate was applied to a Ni-NTA (Qiagen) column prewashed with lysis buffer, and protein was eluted with 250 mM imidazole. The protein was then passed over a desalting column (Desalt 16/10, GE Healthcare) in 100 mM NaCl, 20 mM Tris, pH 8.0, 20 mM imidazole, 3 mM BME. Tobacco etch virus (TEV) protease was added to the protein at a mass-to-mass ratio of 1:10, and the protein was digested for 1 h at 20 °C to remove the histidine tag. Digested protein was passed over a second Ni-NTA column, and the flow-through was loaded onto a MonoQ column (GE Healthcare) equilibrated in 100 mM NaCl, 20 mM Tris, pH 8.0, 3 mM BME. Protein was eluted from the MonoQ column through a linear NaCl gradient, eluting at 350 mM NaCl. Finally, the protein was subjected to size-exclusion chromatography (Superdex 75, GE Healthcare) in 150 mM NaCl, 20 mM Tris, pH 8.0, 3 mM BME and concentrated to 60 mg mL−1. All PatGmac point mutants were produced using the Phusion site-directed mutagenesis kit (Finnzymes) following the manufacturer’s protocol, and the lid deletion mutants were made with fusion PCR. All mutant proteins were expressed and purified as above.
Macrocyclization reactions
For macrocyclization reactions comparing final product ratios after substrate depletion, 100 μM peptide (VGAGIGFPAYDG) was incubated with 50 μM enzyme in 150 mM NaCl, 10 mM HEPES, pH 8, 1 mM TCEP for 120 h at 37 °C. Samples were analyzed by ESI or MALDI MS (LCT, Micromass or 4800 MALDI TOF/TOF Analyser, ABSciex).
For all other macrocyclization reactions, 100 μM peptide (VGAGIGFPAYDG, VGAGIGFPAYRG or GVAGIGFPAYRG) was incubated with 20 μM enzyme in a range of buffers for 24 h at 37 °C (Supplementary Figs. 4 and 5).
LC-MS analysis of products
LC-MS was performed using a Phenomenex Sunfire C18 column (4.6 mm × 150 mm). Solvent A was H2O containing 0.1% formic acid, and solvent B was methanol containing 0.1% formic acid. Gradient: 0–2 min, 10% B; 2–22 min, 10% B–100% B; 22–27 min, 100% B; 27–30 min 100% B–10% B. High-resolution mass-spectral data were obtained from a Thermo Instruments MS system (LTQ XL/LTQ Orbitrap Discovery) coupled to a Thermo Instruments HPLC system (Accela PDA detector, Accela PDA autosampler and Accela Pump). The following conditions were used: capillary voltage 45 V, capillary temperature 320 °C, auxiliary gas flow rate 10–20 arbitrary units, sheath gas flow rate 40–50 arbitrary units, spray voltage 4.5 kV, mass range 100–2,000 AMU (maximum resolution 30,000).
Crystallization, data collection and crystallographic analysis
Crystals of PatGmac were obtained in 19% PEG6000, 0.07 M calcium acetate, 0.1 M Tris, pH 9.0. The crystals were cryoprotected in 30% glycerol and flash cooled in liquid nitrogen. These crystals belonged to space group C2 with cell dimensions a = 132.1 Å, b = 67.6 Å, c = 97.3 Å, β = 115.0°. Crystals of PatGmac with peptide were obtained from a mixture of PatGmac with peptide (VPAPIPFPAYDG, 1:4 molar ratio) in 1.2 M sodium citrate, 0.1 M sodium cacodylate, pH 7.0. There was electron density for a peptide at one active site, but the quality of the map was poor. We reasoned this was due to low occupancy of the peptide and therefore soaked the complex crystals overnight in 7.5 mM peptide before data collection. These crystals belonged to space group C2 with a = 135.6 Å, b = 67.3 Å, c = 137.9 Å, β = 116.8°. Diffraction data of both structures were collected in house, each on a single crystal at 100 K on a Rigaku 007HFM rotating anode X-ray generator with a Saturn 944 CCD detector, and processed with xia2 (ref. 32). The structure of PatGmac was solved by molecular replacement with PHASER33,34 using the structure of AkP (PDB: 1DBI) as the search model, followed by automatic rebuilding with Phenix35. The structure of PatGmac with peptide was solved by molecular replacement using the PatGmac structure as the search model. Manual rebuilding was performed with COOT36, and refinement was performed using REFMAC5 (ref. 37) implemented in the CCP4 program suite38. The statistics of data collection and refinement are summarized in Table 1. All molecular graphics figures were generated with PyMOL (DeLano Scientific, LLC).
Synthesis of the peptide substrates
Fmoc amino acid derivatives, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and Fmoc-Gly-NovaSyn TGT resin were purchased from Novabiochem, Merck Biosciences, UK. Trifluoroacetic acid (TFA), N,N-diisopropylethylamine (DIEA), N,N-dimethylformamide (DMF) and piperidine were obtained from Sigma-Aldrich, UK, and used without further purification.
The peptides VGAGIGFPAYDG, VPAPIPFPAYDG and GVAGIGFPAYRG were synthesized manually using the standard Fmoc-based strategy39. Amino acids were sequentially coupled after removal of the Fmoc blocking group at each cycle. Fmoc deprotection steps were carried out with 20% piperidine in DMF (v/v) for 12 min ; coupling reactions were performed in DMF using a molar ratio of amino acid:HBTU:DIEA:resin of 5:5:10:1. Reactions were monitored using the Kaiser test.
The peptides were cleaved from the support and deprotected by treatment with a mixture consisting of 95% TFA, 2.5% triisopropylsilane (TIPS) 2.5% H2O (20 mL of mixture g−1 of peptide resin, 3 h at room temperature). The resin was then filtered and washed with TFA. The combined filtrates were concentrated under reduced pressure. The peptide was precipitated with cold diethyl ether and recovered by centrifugation. The peptide sequence was verified by MSMS analysis.
The peptide VGAGIGFPAYRG was purchased from Peptide Protein Research Ltd.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank J. Reeks for help with the manuscript. W.E.H. is supported as the recipient of a SULSA postdoctoral fellowship. J.K. is supported as the recipient of a DFG postdoctoral fellowship. M.C.M.S. acknowledges funding from the BBSRC project grant BB/F003439/1. Mass spectrometry is supported by the Wellcome Trust. The project is funded by the Leverhulm Trust Grant RPG-2012-504 (J.H.N. and M.J.).
Footnotes
Accession codes. Coordinates have been deposited in the Protein Data Bank, with accession codes 4AKS (PatGmac) and 4AKT (PatGmac with peptide).
Note: Supplementary information is available in the online version of the paper.
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat. Prod. Rep. 2012;29:144–222. doi: 10.1039/c2np00090c. [DOI] [PubMed] [Google Scholar]
- 2.Mayer AM, Rodriguez AD, Berlinck RG, Fusetani N. Marine pharmacology in 2007–8: marine compounds with antibacterial, anticoagulant, antifungal, anti-inflammatory, antimalarial, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous system, and other miscellaneous mechanisms of action. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2011;153:191–222. doi: 10.1016/j.cbpc.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery–an underexploited structural class. Nat. Rev. Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 4.Cuevas C, Francesch A. Development of Yondelis (trabectedin, ET-743). A semisynthetic process solves the supply problem. Nat. Prod. Rep. 2009;26:322–337. doi: 10.1039/b808331m. [DOI] [PubMed] [Google Scholar]
- 5.McIntosh JA, Donia MS, Schmidt EW. Ribosomal peptide natural products: bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009;26:537–559. doi: 10.1039/b714132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivonen K, Leikoski N, Fewer DP, Jokela J. Cyanobactins-ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 2010;86:1213–1225. doi: 10.1007/s00253-010-2482-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt EW, et al. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. USA. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long PF, Dunlap WC, Battershill CN, Jaspars M. Shotgun cloning and heterologous expression of the patellamide gene cluster as a strategy to achieving sustained metabolite production. ChemBioChem. 2005;6:1760–1765. doi: 10.1002/cbic.200500210. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt EW. The hidden diversity of ribosomal peptide natural products. BMC Biol. 2010;8:83. doi: 10.1186/1741-7007-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houssen WE, Jaspars M. Azole-based cyclic peptides from the sea squirt Lissoclinum patella: old scaffolds, new avenues. ChemBioChem. 2010;11:1803–1815. doi: 10.1002/cbic.201000230. [DOI] [PubMed] [Google Scholar]
- 11.Donia MS, et al. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat. Chem. Biol. 2006;2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- 12.Donia MS, Ravel J, Schmidt EW. A global assembly line for cyanobactins. Nat. Chem. Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houssen WE, et al. Solution structure of the leader sequence of the patellamide precursor peptide, PatE1–34. ChemBioChem. 2010;11:1867–1873. doi: 10.1002/cbic.201000305. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, McIntosh J, Hathaway BJ, Schmidt EW. Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J. Am. Chem. Soc. 2009;131:2122–2124. doi: 10.1021/ja8092168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McIntosh JA, et al. Circular logic: nonribosomal peptide-like macrocyclization with a ribosomal peptide catalyst. J. Am. Chem. Soc. 2010;132:15499–15501. doi: 10.1021/ja1067806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 17.Katoh T, Goto Y, Reza MS, Suga H. Ribosomal synthesis of backbone macrocyclic peptides. Chem. Commun. (Camb.) 2011;47:9946–9958. doi: 10.1039/c1cc12647d. [DOI] [PubMed] [Google Scholar]
- 18.Trauger JW, Kohli RM, Mootz HD, Marahiel MA, Walsh CT. Peptide cyclization catalysed by the thioesterase domain of tyrocidine synthetase. Nature. 2000;407:215–218. doi: 10.1038/35025116. [DOI] [PubMed] [Google Scholar]
- 19.Schneider A, Marahiel MA. Genetic evidence for a role of thioesterase domains, integrated in or associated with peptide synthetases, in non-ribosomal peptide biosynthesis in Bacillus subtilis. Arch. Microbiol. 1998;169:404–410. doi: 10.1007/s002030050590. [DOI] [PubMed] [Google Scholar]
- 20.Cane DE, Walsh CT. The parallel and convergent universes of polyketide synthases and nonribosomal peptide synthetases. Chem. Biol. 1999;6:R319–R325. doi: 10.1016/s1074-5521(00)80001-0. [DOI] [PubMed] [Google Scholar]
- 21.Liu H, Naismith JH. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr. Purif. 2009;63:102–111. doi: 10.1016/j.pep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dodson G, Wlodawer A. Catalytic triads and their relatives. Trends Biochem. Sci. 1998;23:347–352. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 23.Perona JJ, Craik CS. Structural basis of substrate specificity in the serine proteases. Protein Sci. 1995;4:337–360. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ziemert N, et al. Microcyclamide biosynthesis in two strains of Microcystis aeruginosa: from structure to genes and vice versa. Appl. Environ. Microbiol. 2008;74:1791–1797. doi: 10.1128/AEM.02392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donia MS, Schmidt EW. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem. Biol. 2011;18:508–519. doi: 10.1016/j.chembiol.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Popp MW, Ploegh HL. Making and breaking peptide bonds: protein engineering using sortase. Angew. Chem. Int. Edn Engl. 2011;50:5024–5032. doi: 10.1002/anie.201008267. [DOI] [PubMed] [Google Scholar]
- 27.Ahvazi B, Steinert PM. A model for the reaction mechanism of the transglutaminase 3 enzyme. Exp. Mol. Med. 2003;35:228–242. doi: 10.1038/emm.2003.31. [DOI] [PubMed] [Google Scholar]
- 28.Zhu X, Robinson DA, McEwan AR, O’Hagan D, Naismith JH. Mechanism of enzymatic fluorination in Streptomyces cattleya. J. Am. Chem. Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milne BF, Long PF, Starcevic A, Hranueli D, Jaspars M. Spontaneity in the patellamide biosynthetic pathway. Org. Biomol. Chem. 2006;4:631–638. doi: 10.1039/b515938e. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Naismith JH. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr. Purif. 2009;63:102–111. doi: 10.1016/j.pep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2009;43:186–190. [Google Scholar]
- 33.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D. Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr. D. Biol. Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 38.CCP4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D. Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 39.Cammish LE, Kates SA. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford Univ. Press; 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.