

Abstract
Objective: To study the concurrent and construct validity and test-retest reliability in the practice setting of an outcome measure for myasthenia gravis (MG).
Methods: Eleven centers participated in the validation study of the Myasthenia Gravis Composite (MGC) scale. Patients with MG were evaluated at 2 consecutive visits. Concurrent and construct validities of the MGC were assessed by evaluating MGC scores in the context of other MG-specific outcome measures. We used numerous potential indicators of clinical improvement to assess the sensitivity and specificity of the MGC for detecting clinical improvement. Test-retest reliability was performed on patients at the University of Virginia.
Results: A total of 175 patients with MG were enrolled at 11 sites from July 1, 2008, to January 31, 2009. A total of 151 patients were seen in follow-up. Total MGC scores showed excellent concurrent validity with other MG-specific scales. Analyses of sensitivities and specificities of the MGC revealed that a 3-point improvement in total MGC score was optimal for signifying clinical improvement. A 3-point improvement in the MGC also appears to represent a meaningful improvement to most patients, as indicated by improved 15-item myasthenia gravis quality of life scale (MG-QOL15) scores. The psychometric properties were no better for an individualized subscore made up of the 2 functional domains that the patient identified as most important to treat. The test-retest reliability coefficient of the MGC was 98%, with a lower 95% confidence interval of 97%, indicating excellent test-retest reliability.
Conclusions: The Myasthenia Gravis Composite is a reliable and valid instrument for measuring clinical status of patients with myasthenia gravis in the practice setting and in clinical trials.
Outcome measures for myasthenia gravis (MG) differ in ease of use, time to completion, what is being measured, how it is being measured, and who reports symptoms and impairments (i.e., patient-reported vs physician-reported).1–15 Most of these scales are unweighted but a few have weighted test item response options.3,7,9 The Myasthenia Gravis Composite (MGC) scale consists of test items that measure symptoms and signs of MG, with weighted response options (table 1).3 The individual test items of the MGC were selected from existing MG-specific scales based on their performance during 2 randomized, controlled clinical trials of mycophenolate mofetil for MG.3,16,17 The test items were selected so as to be meaningful to both the physician and the patient, frequently abnormal in patients with active disease, and responsive to clinical change.3
Table 1 The Myasthenia Gravis Composite scale
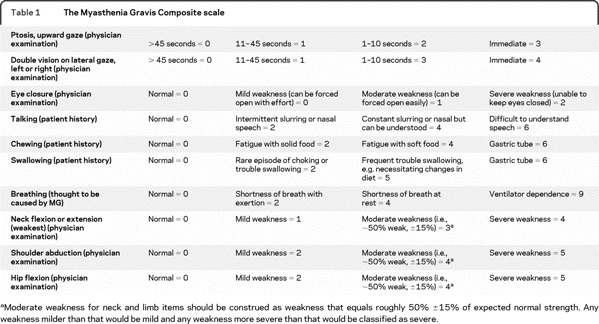
We present the results of validity and test-retest reliability analyses of the MGC in the patient care setting.
METHODS
Validity testing of the MGC was conducted at 11 neuromuscular centers (9 in the United States and 2 in Europe) during the routine care of subjects with MG, following approval by each center's ethical standards committee on human experimentation. The investigators at each center have special expertise in MG management. Patients over 17 years of age with a clinical diagnosis of MG were recruited for the study if the treating physician predicted that they would be seen back for follow-up within 6 months. Written informed consent was obtained for each patient enrolled. The diagnosis of MG was made by the MG specialists at each center, and was almost always confirmed by serologic and electrodiagnostic testing.
Prior to the study, the neck flexion test item of the original MGC was modified to include neck extension testing (worse score recorded). Also, physicians were asked to record the duration until development of ptosis or dysconjugate gaze to see if the time required for these 2 test items could be shortened. After learning that <1% of ocular score times were between 46 and 60 seconds, the ocular test items were modified so that 45 seconds was the maximum duration of testing.
At the initial visit, the following data were recorded: demographic information, disease characteristics (e.g., duration, antibody status, Myasthenia Gravis Foundation of America [MGFA] class), MGC, and the Choose 2 component of the MGC. The Choose 2 form was administered by the physician, who asked the patient which 2 of 10 listed MG symptoms were considered by the patient to be most important to treat. A Choose 2 subscore was derived from the corresponding MGC scores of the patient's 2 chosen items. Investigators at each site chose to also include either the MG–manual muscle test (MG-MMT) or MG–activities of daily living (MG-ADL) as part of their patient assessment at each visit.13,15 A gestalt impression of change was determined by the physician at the second visit, based on their overall assessment of the patient without considering change in any of these MG scale scores. The 15-item myasthenia gravis quality of life scale (MG-QOL15) was completed by the patient at each study visit.4 Patients were not instructed to omit pyridostigmine prior to their visits.
Test-retest reliability testing of the MGC was performed on 38 patients at 1 site (University of Virginia) from October 2008 to March 2009, by having the patient evaluated on the same day by one of the authors (T.M.B.) and also by a second physician, often a neurology resident or clinical neurophysiology fellow.
Concurrent validity was assessed by analyzing the correlation of the MGC scores with scores of other MG-specific outcome measures at visit 1 and visit 2. To assess longitudinal construct validity, we determined the sensitivity and specificity of the MGC for detecting clinical improvement. Because there is no gold standard for clinical improvement in MG and because every outcome measure has limitations, we compared the changes in MGC scores with several outcome measures, alone and in combination.
Test-retest reliability was estimated from the reliability coefficient and the 95% confidence intervals given in Fleis.18 Concurrent validity between scales at each visit was estimated from the Pearson and Spearman correlation coefficients. In all cases, these correlation estimates were nearly equal, and all the correlation coefficients presented in this article are Pearson correlations. For each definition of a gold standard for clinical improvement, sensitivity and specificity were estimated using standard formulas.19 Receiver operating characteristic (ROC) curves were used to assess the sensitivity and specificity corresponding to different cutoffs for the MGC.
RESULTS
Test-retest reliability.
A total of 38 patients with MG were evaluated independently on the same day by 2 neurologists (T.M.B. and another), who were blinded to the results of the other evaluation. The order of evaluators varied depending on the situation; often the other evaluator (e.g., clinical neurophysiology fellow) performed the first evaluation followed by T.M.B., but other times T.M.B. evaluated first followed by a resident or fellow. The mean MGC score at the first assessment was 4.9 (SD = 7.1; range = 0–35). The MGC score never differed by more than 4 points between evaluators; it was within 2 points in 92% of patients, and within 3 points in 95%. The test-retest reliability coefficient was 98%, with a lower 95% confidence interval [CI] of 97%, indicating excellent test-retest reliability. Score differences for individual test items were most frequent for ptosis (6/38) and double vision (6/38), followed by neck strength (3/38) testing.
Concurrent and longitudinal construct validity testing of the MGC.
A total of 175 patients with MG were enrolled at 11 sites from July 1, 2008, to January 31, 2009. A total of 151 patients were seen in follow-up (August 18, 2008, to August 1, 2009). The mean age at visit 1 was 58 years, and the mean disease duration was 7 years. A total of 53% were men and 47% were women. The mean time between visit 1 and visit 2 was 4.7 months (0.13–10.1 months). The 24 patients not seen at visit 2 did not differ from the other 151 patients in age (p = 0.85), MGC score at visit 1 (p = 0.73), MG-QOL15 score at visit 1 (p = 0.21), or gender (p = 0.65).
Breakdown by current MGFA Class at visit 1 is as follows: 22% Class I (ocular), 41% Class II (mild, generalized), 19% Class III (moderate, generalized), 3% Class IV (severe, generalized). A total of 15% of patients were in remission (Class 0) at visit 1. None were intubated and ventilated (Class V) at visit 1, but 14.3% had experienced at least 1 prior episode of myasthenic crisis. At visit 2, 24% were Class I, 42% Class II, 13% Class III, 0% Class IV, and Class V, and 20% were in remission. A total of 78% of patients were acetylcholine receptor antibody positive (AChR+) and 7% were muscle specific tyrosine kinase positive (MuSK+). A total of 10% of patients had negative serology for at least AChR (often both AChR and MuSK). Serologic status was unknown for 5%. Of the 175 subjects, 142 (82%) were follow-up outpatients, 23 (13%) were newly diagnosed, and 8 (5%) were hospitalized for MG at the time of the evaluation. A total of 110/175 (63%) patients were taking pyridostigmine at the time of their first visit.
The mean, median, SD, and range of MG scale scores for both visits are listed in table 2. The total MGC scores showed excellent concurrent validity with other MG-specific scales. At visit 1, the total MGC score had a correlation of 0.68 (95% CI 0.59–0.75) with the MG-QOL15 total score, 0.85 (95% CI 0.77–0.90) with the MG-ADL total score, and 0.80 (95% CI 0.72–0.86) with the MG-MMT total score. Nearly identical correlations were observed at visit 2.
Table 2 Myasthenia Gravis Composite scale scores at visits 1 and 2
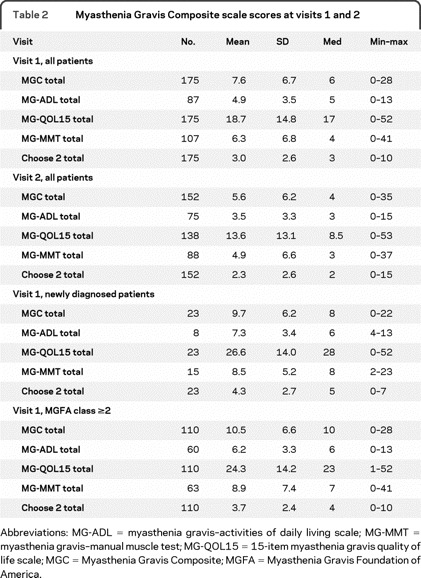
Responsiveness and longitudinal construct validity of the MGC were assessed by using different gold standards of clinical change (table 3). The figure 1 demonstrates the ROC curve for the MGC using the physician impression of improvement plus improvement in MG-QOL15 score (visit 2–visit 1) as the criterion for improvement. Analyses of sensitivities and specificities of the MGC revealed that a 3-point improvement in MGC score was optimal for signifying clinical improvement. Furthermore, a ≥3-point improvement in the MGC appears to be meaningful to most patients. For instance, of the 49 patients with a ≥3-point improvement in the MGC score (visit 2–visit 1), the mean improvement (i.e., decrease) in MG-QOL15 total score was 12.2 (SD = 11.4), compared to a mean MG-QOL15 improvement of 2.5 (SD = 8.7) among the 52 patients with an improvement in MGC of 0 to 2 points, and a mean MG-QOL15 worsening of 1.1 (SD = 8.8) points among the 37 patients whose MGC score worsened (visit 2–visit 1). In addition, 44 (90%) of the 49 patients who had a ≥3-point improvement in the MGC had an improved MG-QOL15 score at visit 2.
Table 3 Sensitivity and specificity of a ≥3-point improvement in MGC scale score to signify clinical improvement, using various criteria as the gold standard
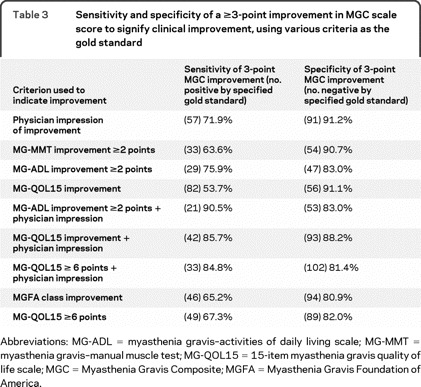
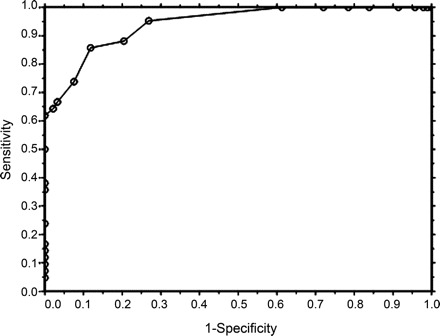
Figure Receiver operating characteristic (ROC) curve of the Myasthenia Gravis Composite
Receiver operating characteristic curve of the Myasthenia Gravis Composite, using physician impression of improvement plus improvement in 15-item myasthenia gravis quality of life scale (MG-QOL15) score at visit 2 as the criterion of improvement. The area under the ROC curve is 0.94, suggesting high accuracy.19
Using the physician impression of improvement plus improvement in the MG-QOL15 score as the gold standard for clinical improvement, the effect on sensitivity and specificity of different choices for cutoffs for the MGC, MG-ADL, and MG-MMT scores is shown in table 4. A 1- or 2-point drop in the MGC also provides high sensitivity with small sacrifices in specificity. The sensitivities and specificities for the MGC compare favorably with the MG-ADL and MG-MMT (table 4).
Table 4 Comparison of sensitivities and specificities of the MG-MMT, MG-ADL, and MGC at various cutpoints, using physician impression of improvement plus improvement in MG-QOL15 score at visit 2 as the criterion of improvement
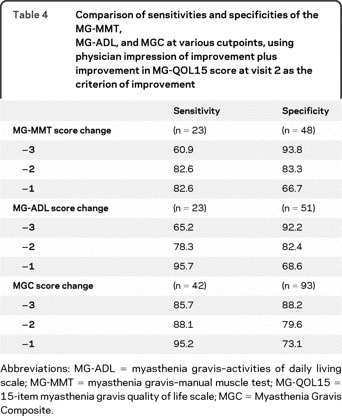
In the subgroup of patients with initial MGC scores of 5 or more, the sensitivity of a 3-point change in MGC was 94.6% (n = 42) and the specificity was 80.0% (n = 45). In the group with initial MGC score of 7 or more, the sensitivity was 100% and the specificity was 81%.
Analysis of the Choose 2 component of the MGC.
Eyelid droop (33%), double vision (36%), and trouble swallowing (36%) were the 3 most common domains selected by the patient as being most important during the Choose 2 component of the MGC. At each visit, the correlations for the MG-MMT, MG-ADL, and MG-QOL15 to the Choose 2 subscale score were lower than to the total MGC score. For all the gold standards of change listed in table 3, the Choose 2 component showed either lower sensitivity or lower specificity than the MGC total (data not shown). These results do not support the use of the Choose 2 component of the MGC over the total MGC score.
DISCUSSION
The notable attributes of the MGC include the following. 1) The test items were rigorously selected through a process that assessed item performance during 2 randomized, controlled trials involving >250 patients with MG.3 2) The MGC covers the 10 important functional domains most frequently affected by MG. 3) The proportion of bulbar and respiratory items to total number of items (4/10) is appropriate given the clinical importance of these domains. 4) The test items are appropriately weighted. For example, a maximum score for worst respiratory status is worth more points than the maximum score for worst eyelid strength. 5) The MGC is easy to administer, taking less than 5 minutes to complete, without the need for any equipment. 6) The MGC is easy to interpret, taking less than 10 seconds to calculate a total score. Also, the assessment of each of the 10 test items provides immediate insight into the status of that particular functional domain. 7) The MGC is reliable, as evidenced by the results of our test-retest assessment. 8) The MGC demonstrates concurrent and longitudinal construct validity in the MG practice care setting, based on the results of this prospective study conducted on >150 patients at 11 centers (table 3, table 4, figure).20 It should be noted that most of our patients were in good clinical condition at study entry, and, thus, changes in status between clinic visits were often small and sometimes even questionable. It is theoretically easier to validate an instrument that measures future clinical improvement of patients whose initial clinical status is poor (e.g., newly diagnosed patients and patients entering treatment trials), for which improvement following initiation of new treatment is likely and is often of high magnitude. In these situations even coarse instruments will identify improvement. The fact that the MGC demonstrated excellent longitudinal construct validity even in our practice-based setting provides further evidence of its sensitivity.
The sample enrolled in this study is likely representative of the overall population of patients with MG because it included consecutive patients seen for routine care at 11 centers. The 9 centers in the United States were located in 9 different states (4 East Coast, 4 Midwest, and 1 West Coast). The 2 centers in Europe were in Glasgow and Milan. Our sample population is similar in many characteristics to other cohorts of patients with MG and covered the spectrum of MG severity. For example, the serologic status, percentage of patients with previous crisis, and proportion of patients by MGFA Class were similar to reported cohorts said to be representative of MG.21–24 Also, the mean MG-ADL score at visit 1 in our cohort was similar to what it was for patients enrolled in the Aspreva MG trial.13 However, overall severity in our cohort was probably less than that of patients with MG entering most clinical trials because of exclusion from those trials of patients with milder disease (e.g., ocular MG [MGFA Class I] or patients in remission). For example, the mean MG-MMT (6.3) and MG-ADL (4.9) scores in our patients were lower than in the recent Muscle Study Group trial of mycophenolate mofetil for generalized MG.
The MGC can be used in everyday practice and in clinical trials. For clinical trials, we propose that a 3-point fall in MGC score be the cutpoint to indicate clinical improvement. The 3-point threshold demonstrated acceptable test-retest reliability and offered the best trade-off in terms of sensitivity and specificity when compared with the various criteria chosen to signify clinical improvement. A 3-point improvement in MGC also appears to be meaningful to most patients, as evidenced by a mean improvement in MG-QOL15 score of >12 points for those patients who had a ≥3-point improvement in the MGC.
The MGC differs from most scales in that it is a hybrid of physician-reported and patient-reported test items. This combination, however, is especially appropriate for an MG outcome measure for 3 major reasons. 1) MG manifests with fluctuating and fatigable symptoms and signs, which consequently limits the utility of measuring disease status solely based on a “snapshot” examination. 2) Manifestations of MG are evident not only to the physician but also to the patient (and others), and thus patient-reported measures of symptoms must be considered in assessing disease severity. 3) Dysfunction in chewing and swallowing are almost certainly more evident to the patient than to the physician. It is not surprising that certain patient-reported test items perform better than their physician-reported examination counterparts and thus justify inclusion in the MGC.
A limitation of our study is the lack of a single gold standard for clinical change. On the other hand, if there had been a gold standard then there would have been less reason to develop the MGC in the first place. To overcome this limitation of no gold standard, we looked at the performance of the MGC using several different criteria of change, each of which has its own limitations. For example, one problem with using MG-MMT or MG-ADL to assess MGC responsiveness is that the MGC contains 4 test items from each the MG-MMT and the MG-ADL. However, it was not reasonable to remove these shared items for the validity analysis because each item represents a functional domain that often acts independently of the other domains. Thus, assessing correlations with scales stripped of shared test items would offer little insight into the performance of the MGC. Limitations in using the physician impression of change as a comparator included physician uncertainty, limited physician memory of patient status at visit 1, and the fact that in some cases different physicians saw the patient at the 2 visits. We settled on the physician impression of improvement plus improvement in the patient-derived MG-QOL15 score as our gold standard for clinical improvement, and placed the most weight on this comparator to assess the longitudinal validity of the MGC and to determine the optimal cutpoint for signifying clinical improvement.
COINVESTIGATORS
MG Composite and MG-QOL15 Study Group: Principal Investigator: Ted M. Burns, MD (University of Virginia); Investigators: Mark Conaway, PhD, Guillermo Solorzano, MD (University of Virginia); Maria E. Farrugia, DPhil, MD (Institute of Neurological Sciences, Glasgow, UK); Donald B. Sanders, MD, Janice M. Massey, MD, Vern C. Juel, MD, Lisa D. Hobson-Webb, MD, Bernadette Tucker-Lipscomb, RN, MSN (Duke University); Carlo Antozzi, MD, Renato Mantegazza, MD (Neurological Institute Foundation “Carlo Besta,” Milan, Italy); David Lacomis, MD (University of Pittsburgh); Elliot Dimberg (Mayo Clinic, Florida); Srikanth Muppidi, MD, Gil I. Wolfe, MD (University of Texas, Southwestern); Mazen M. Dimachkie, MD, Richard J. Barohn, MD, Mamatha Pasnoor, MD, April L. McVey, MD, Laura Herbelin, BS (University of Kansas); Tahseen Mozaffar, MD, Vinh Q. Dang, MD, Sandhya Rao, MD (University of California, Irvine); Robert Pascuzzi, MD, Riley Snook, MD (Indiana University); Anthony A. Amato, MD (Brigham and Women's Hospital and Harvard Medical School).
DISCLOSURE
Dr. Burns serves as Section Editor of the Neurology® Podcast and receives research support from Alexion Pharmaceuticals, Inc., Knopp Neurosciences Inc., and the Myasthenia Gravis Foundation of America. Dr. Sanders has served/serves as a consultant for Accordant Health Service, Bayhill Therapeutics, BioMarin Pharmaceutical Inc., Anthera Pharmaceuticals, Jacobus Pharmaceutical Co. Inc., Baxter International Inc., and GlaxoSmithKline; and serves on speakers' bureaus for Athena Diagnostics, Inc. and Discovery Institute of Medical Education/Talecris Biotherapeutics. Dr. Conaway serves on the editorial board of Developmental Medicine and Child Neurology and has received research support from the Myasthenia Gravis Foundation of America. Dr. Farrugia reports no disclosures. Dr. Solorzano's wife has received speaker honoraria from Pfizer Inc. and she has received research support from Novo Nordisk, the NIH (2U54HD828934-16A1 revised [Co-I]), and the University of Virginia Children's Hospital. Dr. Massey serves on the editorial board of Seminars in Neurology; receives an educational grant from Allergan, Inc. for an annual workshop; receives research support from the NIH (ADD GRANT NUMBER [Site PI]); and serves on Board of Directors of the American Board of Psychiatry & Neurology (ABPN)–Neurology. Dr. Juel receives research support from Alexion Pharmaceuticals, Inc. Dr. Hobson-Webb has received honoraria for speaking or educational activities not funded by commercial entities and receives research support from Genzyme Corporation and the American Association of Neuromuscular and Electrodiagnostic Medicine. Dr. Tucker-Lipscomb has received research support from Alexion Pharmaceuticals, Inc. and the NIH/UAB. Dr. Antozzi serves on the editorial board of Neurological Sciences. Dr. Mantegazza serves on a scientific advisory board for GlaxoSmithKline; has received funding for travel from Sanofi-Aventis; has received a speaker honorarium from MEDA Pharmaceuticals Inc.; and receives research support from CARIPLO (Fondazione-Cassa di Risparmio delle Provincie Lombarde), AVASM (Associazione Volontari Aiuti Sclerosi Multipla), and AIM (Associazione Italiana per la lotta alla Miastenia). Dr. Pascuzzi reports no disclosures. Dr. Lacomis serves on the editorial board of the Journal of Clinical Neuromuscular Disease; has received publishing royalties for his contributions to UptoDate (2007–present); and receives research support from Knopp Neurosciences Inc. Dr. Dimberg has received honoraria from ePocrates.com and Harris Interactive. Dr. Muppidi reports no disclosures. Dr. Wolfe serves on scientific advisory boards and speakers' bureaus for Eli Lilly and Company and Talecris Biotherapeutics; serves as an Associate Editor of Muscle and Nerve; and has received research support from the NIH for the thymectomy trial in myasthenia gravis for which he serves as clinical chair (R01 NS0442685-01A2). He also received research support from Alexion Pharmaceuticals, Inc. Dr. Dimachkie has received funding for travel from Baxter International Inc.; serves on speakers' bureaus for Pfizer Inc. and Merck Serono; and receives research support from the FDA, Orphan Products Division (1R01FD003538 [Co-I]). Dr. Barohn serves on a speakers' bureau for Talecris Biotherapeutics; and receives research support from the FDA and the NIH, as principal investigator for 2 studies: 1R01FD003454 Phase II therapeutic trial of mexiletine in patients with nondystrophic myotonia and 1R01FD003538 Phase II trial of methotrexate in myasthenia gravis. Dr. McVey and Dr. Pasnoor report no disclosures. L. Herbelin has serves as a consultant for Alexion Pharmaceuticals, Inc. and has received research support from support from the FDA, Orphan Products Division (1R01FD003538 [Project manager]). Dr. Mozaffar serves on the Medical and Scientific Advisory Board for the Myositis Association; has received funding for travel from and serves on speakers' bureaus for Genzyme Corporation and Talecris Biotherapeutics; has served as a consultant for Avanir Pharmaceuticals and Baxter International Inc.; serves as Medical Director for Crescent Health Care; and receives research support from the NIH (RO15R01NS049203 [Co-I]). Dr. Dang, Dr. Rao, and Dr. Snook report no disclosures. Dr. Amato has served on the Medical Advisory Board for MedImmune; serves as an Associate Editor for Muscle & Nerve and on the editorial board of Neurology®; receives royalties from the publication of Neuromuscular Disease (McGraw-Hill, 2008); and receives research support from MedImmune and the NIH (RO1 NS049639-01A2 [PI] and U54 RR019482 [Co-I]).
Supplementary Material
Glossary
- AChR+
acetylcholine receptor antibody positive
- CI
confidence interval
- MG
myasthenia gravis
- MG-ADL
myasthenia gravis–activities of daily living scale
- MG-MMT
myasthenia gravis–manual muscle test
- MG-QOL15
15-item myasthenia gravis quality of life scale
- MGC
Myasthenia Gravis Composite
- MGFA
Myasthenia Gravis Foundation of America
- MuSK+
muscle specific tyrosine kinase positive
- ROC
receiver operating characteristic.
Footnotes
Study funding: Supported in part by the Myasthenia Gravis Foundation of America.
Disclosure: Author disclosures are provided at the end of the article.
Received November 17, 2009. Accepted in final form February 3, 2010.
REFERENCES
- 1.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann NY Acad Sci 1998;841:769–772. [DOI] [PubMed] [Google Scholar]
- 2.Barohn RJ. Standards of measurement in myasthenia gravis. Ann NY Acad Sci 2003;998:432–439. [DOI] [PubMed] [Google Scholar]
- 3.Burns TM, Conaway MR, Cutter GR, Sanders DB. The construction of an efficient evaluative instrument for myasthenia gravis: the MG Composite. Muscle Nerve 2008;38:1553–1562. [DOI] [PubMed] [Google Scholar]
- 4.Burns TM, Conaway M, Cutter G, Sanders DB, The Muscle Study Group. Less is more, or almost as much: the development of a fifteen-item quality-of-life instrument for patients with myasthenia gravis. Muscle Nerve 2008;38:957–963. [DOI] [PubMed] [Google Scholar]
- 5.Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med 1982;307:769–775. [DOI] [PubMed] [Google Scholar]
- 6.Farrugia ME, Robson MD, Clover L, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain 2006;129:1481–1492. [DOI] [PubMed] [Google Scholar]
- 7.Gajdos P, Sharshar T, Chevret S. Standards of measurements in myasthenia gravis. Ann NY Acad Sci 2003;998:445–452. [DOI] [PubMed] [Google Scholar]
- 8.Jaretzki A, Barohn RJ, Ernstoff RM, et al, for the Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Myasthenia gravis: recommendations for clinical research standards. Neurology 2000;55:16–23. [DOI] [PubMed] [Google Scholar]
- 9.Mantegazza R, Antozzi C, Pelucchetti D, Sghirlanzoni A, Cornelio F. Azathioprine as a single drug or in combination with steroids in the treatment of myasthenia gravis. J Neurol 1988;235:449–453. [DOI] [PubMed] [Google Scholar]
- 10.Mullins LL, Carpentier MY, Paul RH, Sanders DB, Muscle Study Group. A disease-specific measure of quality of life for myasthenia gravis. Muscle Nerve 2008;38:947–956. [DOI] [PubMed] [Google Scholar]
- 11.Palace J, Newsom-Davis J, Lecky B, Myasthenia Gravis Study Group. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 1998;50:1778–1783. [DOI] [PubMed] [Google Scholar]
- 12.Padua L, Evoli A, Aprile I, et al. Myasthenia gravis outcome measure: development and validation of a disease-specific self-administered questionnaire. Neurol Sci 2002;23:59–68. [DOI] [PubMed] [Google Scholar]
- 13.Sanders DB, Tucker-Lipscomb B, Massey JM. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann NY Acad Sci 2003;998:440–444. [DOI] [PubMed] [Google Scholar]
- 14.Sharshar T, Chevret S, Mazighi M, et al. Validity and reliability of two muscle strength scores commonly used as endpoints in assessing treatment of myasthenia gravis. J Neurol 2000;247:286–290. [DOI] [PubMed] [Google Scholar]
- 15.Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology 1999;52:1487–1489. [DOI] [PubMed] [Google Scholar]
- 16.Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008;71:394–399. [DOI] [PubMed] [Google Scholar]
- 17.Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008;71:400–406. [DOI] [PubMed] [Google Scholar]
- 18.Fleis J. The Design and Analysis of Clinical Experiments. New York: John Wiley and Sons; 1986
- 19.Rosner B. Fundamentals of Biostatistics, 6th edition. Belmont, CA: Thomson Brooks/Cole; 2006
- 20.Swets JA. Measuring the accuracy of diagnostic systems. Science 1988;240:1285–1293. [DOI] [PubMed] [Google Scholar]
- 21.Mantegazza R, Beghi E, Pareyson D, et al. A multicentre follow-up study of 1152 patients with myasthenia gravis in Italy. J Neurol 1990;237:339–344. [DOI] [PubMed] [Google Scholar]
- 22.Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve 2008;37:141–149. [DOI] [PubMed] [Google Scholar]
- 23.Oosterhuis HJGH. Studies in myasthenia gravis: part 1: a clinical study of 180 patients. J Neurol Sci 1964;1:512–546. [DOI] [PubMed] [Google Scholar]
- 24.Keesey JC. Clinical evaluation and management of myasthenia gravis. Muscle Nerve 2004;29:484–505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.