

Abstract
Calcium (Ca2+) is an ion vital in regulating cellular function through a variety of mechanisms. Much of Ca2+ signaling is mediated through the calcium-binding protein known as calmodulin (CaM)1,2. CaM is involved at multiple levels in almost all cellular processes, including apoptosis, metabolism, smooth muscle contraction, synaptic plasticity, nerve growth, inflammation and the immune response. A number of proteins help regulate these pathways through their interaction with CaM. Many of these interactions depend on the conformation of CaM, which is distinctly different when bound to Ca2+ (Ca2+-CaM) as opposed to its Ca2+-free state (ApoCaM)3.
While most target proteins bind Ca2+-CaM, certain proteins only bind to ApoCaM. Some bind CaM through their IQ-domain, including neuromodulin4, neurogranin (Ng)5, and certain myosins6. These proteins have been shown to play important roles in presynaptic function7, postsynaptic function8, and muscle contraction9, respectively. Their ability to bind and release CaM in the absence or presence of Ca2+ is pivotal in their function. In contrast, many proteins only bind Ca2+-CaM and require this binding for their activation. Examples include myosin light chain kinase10, Ca2+/CaM-dependent kinases (CaMKs)11 and phosphatases (e.g. calcineurin)12, and spectrin kinase13, which have a variety of direct and downstream effects14.
The effects of these proteins on cellular function are often dependent on their ability to bind to CaM in a Ca2+-dependent manner. For example, we tested the relevance of Ng-CaM binding in synaptic function and how different mutations affect this binding. We generated a GFP-tagged Ng construct with specific mutations in the IQ-domain that would change the ability of Ng to bind CaM in a Ca2+-dependent manner. The study of these different mutations gave us great insight into important processes involved in synaptic function8,15. However, in such studies, it is essential to demonstrate that the mutated proteins have the expected altered binding to CaM.
Here, we present a method for testing the ability of proteins to bind to CaM in the presence or absence of Ca2+, using CaMKII and Ng as examples. This method is a form of affinity chromatography referred to as a CaM pull-down assay. It uses CaM-Sepharose beads to test proteins that bind to CaM and the influence of Ca2+ on this binding. It is considerably more time efficient and requires less protein relative to column chromatography and other assays. Altogether, this provides a valuable tool to explore Ca2+/CaM signaling and proteins that interact with CaM.
Keywords: Molecular BIology, Issue 59, Calmodulin, calcium, IQ-motif, affinity chromatography, pull-down, Ca2+/Calmodulin-dependent Kinase II, neurogranin
Protocol
Refer to Figure 1 for a basic schematic of the procedure beginning with the homogenate. Estimated time from preparation of cellular extracts to elution of CaM-bound proteins is about six to seven hours.
1. Tissue preparation
Inject organotypic hippocampal slices with a virus containing a plasmid expressing the recombinant protein of interest (in this example, green fluorescent protein (GFP)-tagged Ng) and allow tissue to express protein overnight.
Approximately 12 to 18 hours after viral injection (depending on the viral expression time), prepare to collect the tissue. Add 1mL dissection buffer (10mM glucose, 4mM KCl, 26mM NaHCO3, 233mM sucrose, 5mM MgCl2, 1mM CaCl2, and 0.1% phenol-red, gassed with 5% CO2 95% O2 ) to a petri dish. Transfer cultured tissue/insert to petri dish and add 2mL dissection buffer to the insert to submerge the tissue.
Collect organotypic hippocampal tissue (between 5 and 10 slices) by gently scraping the tissue free of the insert membrane using a scalpel. Specifically removing a particular region of interest (e.g. CA1) is also an option. Transfer the suspended tissue to a 1.5mL microcentrifuge tube using an inverted Pasteur pipette.
Centrifuge samples at 1,500 rcf for 1 min to separate tissue from the dissection buffer. Carefully remove the supernatant by aspiration. Make sure not to disturb the pellet.
For each slice used, add 30 to 60 μL of homogenization buffer (150mM NaCl, 20mM Tris pH 7.5, 1mM DTT, 1μg/mL leupeptin, 1μg/mL chemostatin, 1μg/mL antipain, 1μg/mL pepstatin, and 1% Triton X-100) to the tissue and homogenize thoroughly with pestle.
In order to remove cellular debris, centrifuge the remaining homogenate at 1,100 rcf for 10 min and carefully remove the supernatant by aspiration while avoiding contamination from the pellet.
Take 10% of the supernatant as a sample of the input (sample 1). Store the remaining supernatant on ice during preparation of the CaM-sepharose beads for use in step 3.
Note: The tissue used here are organotypic hippocampal slices. However, one could use dissociated neurons or any other cell culturing system. In such a case, begin at step 1.4 after collecting your tissue in the appropriate manner.
2. Preparation of beads for pull-down
In handling the beads, it is important to salvage the beads and maximize the efficiency of the reactions by preventing the beads from drying on the sides of the tube. To do so, it is best to rotate your tubes on their side, allowing solution to wet the beads on the walls of the tube, immediately before centrifugation.
For each pull-down, pipette 400 μL of suspended Calmodulin-Sepharose beads into a 2 mL microcentrifuge tube with a relatively flat-bottom to maximize surface area and interaction of your solutions with the beads during your incubations.
Centrifuge beads at 21,000 rcf for 30 seconds and carefully remove the supernatant by aspiration. Make sure not to disturb the beads.
To wash the beads, add 100 μL of the respective homogenization buffer containing either 2 mM CaCl2 to those being used to pull-down Ca2+-CaM binding proteins and 2 mM EDTA (a known Ca2+ chelator) for beads pulling down ApoCaM binding proteins. Gently tap the tube to re-suspend beads and centrifuge at 1,500 rcf for 1 min. Carefully remove the supernatant by aspiration, making sure not to disturb the beads.
Note: For all the aspiration steps, it is recommended to use a pipette tip that has a fine opening (e.g. gel loading tips) to allow removal of solution without removing beads.
3. CaM-sepharose binding of proteins
Split your supernatant into two conditions containing an equal volume. Depending on your condition, add the appropriate amount of CaCl2 or EDTA to your supernatant up to 2 mM concentration for each.
Add supernatant from step 1.7 to beads washed in corresponding homogenization buffer. Gently tap the tube to mix.
Incubate samples at 4°C for 3 hrs on a shaker. Re-suspend beads every 30 min or so to increase efficiency of binding.
Centrifuge tube containing samples and beads at 1,500 rcf for 3 min.
Take 50μL of supernatant as a sample of unbound protein (sample 2) and carefully remove the remaining supernatant by aspiration and discard.
Wash beads three times as described in step 2.3 using 100μL of respective homogenization buffer.
4. Elution
Add 50μL of elution buffer (50mM Tris-HCl pH 7.5, and 150mM NaCl) containing the opposite condition (10mM CaCl2 or 10mM EDTA) to the beads. For example, samples that were homogenized and bound in buffer containing Ca2+ are eluted in elution buffer containing EDTA and vice-versa.
Optional: Warming the elution buffer to 37°C before adding to beads may enhance the yield.
Incubate solution with beads at room temperature for 30 min on a shaker. Mix the beads by gently tapping the tube about every 5 min.
Centrifuge beads at 1,500 rcf for 3 min and carefully remove the 50μL of supernatant for the bound protein (sample 3) by aspiration. Make sure not to disturb the beads.
Add protein loading buffer to all samples (i.e. homogenate, unbound and bound protein as well as those still bound to the beads).
Note: To maximize elution (especially in the case of inefficient elution), add 50μL of the corresponding elution buffer (e.g. adding EDTA containing buffer to beads bound in CaCl2) to the beads before heating samples to help elute any remaining bound protein and repeat steps described in 4.3 to remove remaining bound proteins.
5. SDS-PAGE and western blot
Conduct SDS-PAGE and analyze using western blot by probing for your protein of interest and probe for a protein known to bind CaM in the opposite condition as a positive control.
6. Representative Results
Figure 2B shows an example of a CaM-pull-down assay testing the CaM binding of GFP-tagged Ng compared to endogenous Ng. To do so, GFP-Ng was overexpressed in our organotypic hippocampal slices overnight and the tissue was homogenized. The homogenate was incubated with CaM-sepharose beads in the presence of either Ca2+ or EDTA. Homogenate input shows that the GFP-Ng was expressed in addition to endogenous Ng and Ca2+/CaM-dependent kinase II (CaMKII). As expected based on the known binding of endogenous Ng (illustrated in Fig. 2A), GFP-tagged Ng was eluted in the absence of Ca2+ (EDTA bound protein) and unbound in the presence of Ca2+ (Fig. 2B). In contrast, the control, CaMKII, was eluted only in the presence of Ca2+ (bound protein) and was unbound in its absence (EDTA). This shows that the CaM beads were functioning properly and the elutions were efficient. Most importantly, this shows that GFP-Ng binds to ApoCaM in a similar fashion to the endogenous form, suggesting that the GFP tag did not alter the function of our recombinant protein.
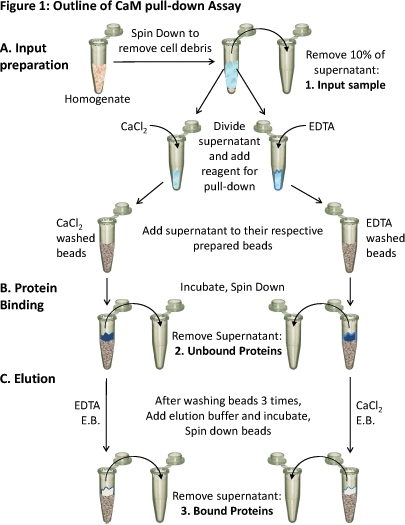 Figure 1. Outline of the CaM pull-down assay
(A) Tissue homogenate is spun down to remove cellular debris. About 10% of the supernatant is taken as a sample of the input (1). The remaining supernatant is divided equally for the different conditions and the appropriate reagents (CaCl2 or EDTA) are added to test binding in those conditions. Each supernatant (containing either CaCl2 or EDTA) is loaded onto the respectively prepared CaM-sepharose beads and (B) incubated to allow binding. Unbound proteins are removed (2) and (C) the bound proteins (3) are eluted off of the beads using elution buffer (E.B.) containing the opposite condition as the binding. The protein composition of these three protein samples can be analyzed using SDS-PAGE and western blot analysis.
Figure 1. Outline of the CaM pull-down assay
(A) Tissue homogenate is spun down to remove cellular debris. About 10% of the supernatant is taken as a sample of the input (1). The remaining supernatant is divided equally for the different conditions and the appropriate reagents (CaCl2 or EDTA) are added to test binding in those conditions. Each supernatant (containing either CaCl2 or EDTA) is loaded onto the respectively prepared CaM-sepharose beads and (B) incubated to allow binding. Unbound proteins are removed (2) and (C) the bound proteins (3) are eluted off of the beads using elution buffer (E.B.) containing the opposite condition as the binding. The protein composition of these three protein samples can be analyzed using SDS-PAGE and western blot analysis.
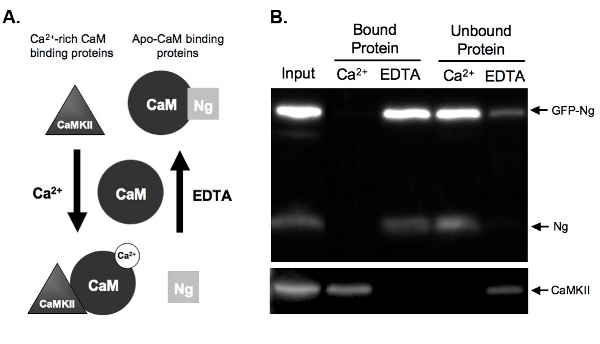 Figure 2.
A.) Schematic of Ca2+-dependent CaM binding and elution in pull-down assay Examples are given of two types of protein that bind CaM in a Ca2+-dependent manner.
Neurogranin (Ng) represents proteins that bind apo-CaM and CaMKII represents proteins that bind to Ca2+-rich CaM. CaM is shown in its dissociated state prior to incubation with the homogenate proteins. Once incubated under conditions of high Ca2+ concentrations (2 mM) or in the presence of a Ca2+ chelator, EDTA (2 mM), the proteins will bind to CaM accordingly. Ng binds CaM in the EDTA condition as there is little to no Ca2+ present, and would be eluted off the CaM-sepharose beads in the presence of Ca2+. CaMKII, however, would bind to CaM in the presence of high amounts of Ca2+ and would dissociate once the Ca2+ was chelated.
B.) Results from example CaM pull-down assay. This figure demonstrates the expected end result of a CaM-sepharose pull down with the samples probed for Ng and CaMKII. Both the endogenous Ng and GFP-Ng are present in the lanes of proteins bound to CaM in the presence of EDTA. No Ng is bound when samples are incubated with CaM in the presence of Ca2+, demonstrating that Ng only binds apo-CaM. Our positive control, CaMKII, on the other hand, binds to CaM only in the presence of Ca2+.
Figure 2.
A.) Schematic of Ca2+-dependent CaM binding and elution in pull-down assay Examples are given of two types of protein that bind CaM in a Ca2+-dependent manner.
Neurogranin (Ng) represents proteins that bind apo-CaM and CaMKII represents proteins that bind to Ca2+-rich CaM. CaM is shown in its dissociated state prior to incubation with the homogenate proteins. Once incubated under conditions of high Ca2+ concentrations (2 mM) or in the presence of a Ca2+ chelator, EDTA (2 mM), the proteins will bind to CaM accordingly. Ng binds CaM in the EDTA condition as there is little to no Ca2+ present, and would be eluted off the CaM-sepharose beads in the presence of Ca2+. CaMKII, however, would bind to CaM in the presence of high amounts of Ca2+ and would dissociate once the Ca2+ was chelated.
B.) Results from example CaM pull-down assay. This figure demonstrates the expected end result of a CaM-sepharose pull down with the samples probed for Ng and CaMKII. Both the endogenous Ng and GFP-Ng are present in the lanes of proteins bound to CaM in the presence of EDTA. No Ng is bound when samples are incubated with CaM in the presence of Ca2+, demonstrating that Ng only binds apo-CaM. Our positive control, CaMKII, on the other hand, binds to CaM only in the presence of Ca2+.
Discussion
The provided protocol utilizes CaM-sepharose beads to investigate the Ca2+-dependence of CaM-binding proteins. Many proteins bind CaM in a Ca2+-dependent manner. These interactions are of great importance given the number of CaM-binding proteins and their critical role in many signaling pathways. In this protocol, CaM-sepharose beads are used to separate CaM-binding proteins from tissue homogenate in the presence or absence of Ca2+. The results of this simple approach will further the understanding of how proteins may interact with CaM in a Ca2+-dependent manner. This approach differs from a commonly used technique in protein binding studies, column chromatography, in that the CaM-sepharose beads are in solution rather than fixed in a matrix.
Avoiding the use of a column is one advantage of this approach, because without the column the protocol is less time consuming, as there is no need to equilibrate a column or wait for gravity flow-through. Due to the virtue of its efficiency, the CaM pull-down requires significantly less tissue than column chromatography and simplifies sample preparation, as separation of cellular debris from the protein sample only requires a short centrifugation for the CaM-sepharose pull down described. Columns require protein samples that are free of any cellular debris, which may require additional purification steps. This approach also has advantages over other pull down methods, such as co-immunoprecipitation, which requires an antibody against the bait protein to capture it and any interacting proteins in the sample. The use of an antibody to capture the bait protein can be a limitation as poor interactions between the antibody and the protein during the experiment can lead to an inefficient pull down. These potential problems are avoided with the use of the described CaM-sepharose pull-down.
However, like the co-immunoprecipitation and column chromatography assays, the CaM-sepharose pull-down is limited to ex vivo CaM-binding. The results obtained may not reflect the in vivo reality of CaM interactions. For example, post-translational modifications often impact protein interactions. This is the case for neurogranin, whose interaction with CaM is prevented by PKC-mediated phosphorylation of its IQ domain5. Homogenizing tissue could alter post-translational modifications for example by allowing enzymes such as kinases or phosphatases to access target proteins which would normally be isolated from the enzymes within the cell. Disruption of localization and/or compartmentalization could also allow binding when the two proteins normally would not have a chance to interact in the cell. To minimize these reactions, it is important to store all samples on ice between preparation and loading. It is also for this reason that the incubation with the beads is done at 4°C. Phosphatase inhibitors or other enzyme inhibitors could also be added to the homogenization buffers to help limit their effects.
A positive control is important for this experiment to make sure that no significant errors occurred during the experiment. It can also ensure that differences in conditions were sufficient to cause conformational changes in CaM, allowing it to bind different proteins in the presence and absence of Ca2+. For example, if there is no signal for the protein of interest, it could be due to loading error or other potential errors. Probing for another protein known to bind in the other conditions (such as CaMKII in the example provided) can help resolve potential errors. Low Ca2+ or Ca2+ chelator (e.g. EDTA) concentrations can also interfere with expected results. EDTA has been used successfully but other Ca2+ chelators (e.g. EGTA) may be more effective if even higher concentrations are ineffective. Excessive CaM-binding protein can also lead to unexpected results as it may saturate the available CaM-sepharose beads, causing elution of the protein when it should be bound. This is seen in the shown example as a relatively small quantity of GFP-Ng is eluted in the EDTA condition. Quantification of protein before incubation with beads may help ameliorate this.
Proper preparation and handling of the CaM-sepharose beads throughout the experiment is also essential to success. Beads can easily be lost during the experiment, either inadvertently removed with supernatant to be discarded or stuck onto the sides and top of the 2.0 mL tube. This can be avoided by using caution while removing supernatant and ensuring thorough mixing immediately prior to centrifugation. Try not to let samples sit between mixing and centrifuging as it allows CaM-sepharose beads to dry on the sides of the tube. As a precautionary measure, resuspend beads and rotate solution around tube to wet beads immediately before centrifugation. Thorough immersion of the samples with the beads during both the binding and elution incubations is very important. Without suitable mixing of the sample with the beads, binding and elution will likely be inefficient.
This protocol is versatile and can easily be amended for other purposes. Performing this experiment with truncated or mutated proteins can reveal information about the region(s) and residues of the protein that are important for CaM binding and Ca2+-dependence, if observed. In the case of Ng, we were able to show that GFP-Ng, like the endogenous protein, binds CaM only in the absence of Ca2+ 8. To further explore this function of this binding, different mutations of this protein altering its IQ-domain help to understand the nature of Ng′s interaction with CaM as well as the function of post-translational modifications, which alter this interaction. We tested the importance of Ng-CaM binding using two different mutants: a phosphomimic aspartate in place of a serine (S36D or Ng-SD) and an IQ-less Ng to prevent binding to CaM. We also generated a mutant (Ng-SFAW) that has enhanced binding to CaM, staying bound even in the presence of Ca2+. Finally, we used a non-phosphorylatable mutant (Ng-SA) which binds to CaM in a Ca2+-dependent fashion similar to endogenous Ng but prevents phosphorylation from protein kinase C (PKC). The CaM pull-down assay helped test the functionality of these different mutants and helped show the importance of Ng-CaM binding in synaptic function8 and how Ng phosphorylation helps fine-tune synaptic plasticity15.
Additionally, proteins that bind to CaM-binding proteins could be eluted. This could provide further applications of this assay in determining proteins that indirectly bind CaM. It allows the study of these other interactions and other potential downstream effects of CaM and its binding partners.
In summary, the CaM pull-down assay provides an efficient and effective way to investigate Ca2+-dependence of CaM-protein interactions. This can be an important tool to study CaM-protein interactions and how different mutations or modifications may affect these interactions. This can be valuable in exploring the regulation of Ca2+/CaM signaling pathways. Furthermore, we can use this to explore pathologies caused by disruptions in Ca2+/CaM signaling.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Tiffany Cherry in her help in optimizing this protocol. This work was funded by National Institute of Aging (AG032320) as well as Advancing a Healthier Wisconsin.
References
- Vincenzi FF. Calmodulin in the regulation of intracellular calcium. Proc. West Pharmacol Soc. 1979;22:289–294. [PubMed] [Google Scholar]
- Cheung WY. Calmodulin plays a pivotal role in cellular regulation. Science. 1980;207:19–27. doi: 10.1126/science.6243188. [DOI] [PubMed] [Google Scholar]
- Zhang M, Tanaka T, Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- Alexander KA, Wakim BT, Doyle GS, Walsh KA, Storm DR. Identification and characterization of the calmodulin-binding domain of neuromodulin, a neurospecific calmodulin-binding protein. J. Biol. Chem. 1988;263:7544–7549. [PubMed] [Google Scholar]
- Huang KP, Huang FL, Chen HC. Characterization of a 7.5-kDa protein kinase C substrate (RC3 protein, neurogranin) from rat brain. Arch. Biochem. Biophys. 1993;305:570–580. doi: 10.1006/abbi.1993.1463. [DOI] [PubMed] [Google Scholar]
- Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002;513:107–113. doi: 10.1016/s0014-5793(01)03239-2. [DOI] [PubMed] [Google Scholar]
- Routtenberg A. Protein kinase C activation leading to protein F1 phosphorylation may regulate synaptic plasticity by presynaptic terminal growth. Behav. Neural. Biol. 1985;44:186–200. doi: 10.1016/s0163-1047(85)90184-0. [DOI] [PubMed] [Google Scholar]
- Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009;28:3027–3039. doi: 10.1038/emboj.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham DM. Myosin and adenosinetriphosphate in relation to muscle contraction. Biochim. Biophys. Acta. 1950;4:42–49. doi: 10.1016/0006-3002(50)90007-2. [DOI] [PubMed] [Google Scholar]
- Hathaway DR, Adelstein RS. Human platelet myosin light chain kinase requires the calcium-binding protein calmodulin for activity. Proc. Natl. Acad. Sci. U.S.A. 1979;76:1653–1657. doi: 10.1073/pnas.76.4.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Yamamoto H, Matsui K, Higashi K, Miyamoto E. Purification and characterization of a Ca2+- and calmodulin-dependent protein kinase from rat brain. J. Neurochem. 1982;39:1607–1617. doi: 10.1111/j.1471-4159.1982.tb07994.x. [DOI] [PubMed] [Google Scholar]
- Yang SD, Tallant EA, Cheung WY. Calcineurin is a calmodulin-dependent protein phosphatase. Biochem. Biophys. Res. Commun. 1982;106:1419–1425. doi: 10.1016/0006-291x(82)91272-4. [DOI] [PubMed] [Google Scholar]
- Huestis WH, Nelson MJ, Ferrell JEJ. Calmodulin-dependent spectrin kinase activity in human erythrocytes. Prog. Clin. Biol. Res. 1981;56:137–155. [PubMed] [Google Scholar]
- Yamniuk AP, Vogel HJ. Calmodulin's flexibility allows for promiscuity in its interactions with target proteins and peptides. Mol. Biotechnol. 2004;27:33–57. doi: 10.1385/MB:27:1:33. [DOI] [PubMed] [Google Scholar]
- Zhong L, Kaleka KS, Gerges NZ. Neurogranin phosphorylation fine-tunes long-term potentiation. Eur. J. Neurosci. 2011;33:244–250. doi: 10.1111/j.1460-9568.2010.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]