

Abstract
Objectives: Sleep disturbances commonly follow traumatic brain injury (TBI) and contribute to ongoing disability. However, there are no conclusive findings regarding specific changes to sleep quality and sleep architecture measured using polysomnography. Possible causes of the sleep disturbances include disruption of circadian regulation of sleep-wakefulness, psychological distress, and a neuronal response to injury. We investigated sleep-wake disturbances and their underlying mechanisms in a TBI patient sample.
Methods: This was an observational study comparing 23 patients with TBI (429.7 ± 287.6 days post injury) and 23 age- and gender-matched healthy volunteers on polysomnographic sleep measures, salivary dim light melatonin onset (DLMO) time, and self-reported sleep quality, anxiety, and depression.
Results: Patients with TBI reported higher anxiety and depressive symptoms and sleep disturbance than controls. Patients with TBI showed decreased sleep efficiency (SE) and increased wake after sleep onset (WASO). Although no significant group differences were found in sleep architecture, when anxiety and depression scores were controlled, patients with TBI showed higher amount of slow wave sleep. No differences in self-reported sleep timing or salivary DLMO time were found. However, patients with TBI showed significantly lower levels of evening melatonin production. Melatonin level was significantly correlated with REM sleep but not SE or WASO.
Conclusions: Reduced evening melatonin production may indicate disruption to circadian regulation of melatonin synthesis. The results suggest that there are at least 2 factors contributing to sleep disturbances in patients with traumatic brain injury. We propose that elevated depression is associated with reduced sleep quality, and increased slow wave sleep is attributed to the effects of mechanical brain damage.
Sleep disturbances are common following traumatic brain injury (TBI), reported by 30%–75% of individuals and contributing to ongoing disability.1–6 Reported sleep complaints include insomnia, hypersomnolence, and altered sleep-wake cycles.7 Understanding of changes to sleep following TBI is limited. Reduced sleep efficiency8 and increased sleep fragmentation post-TBI9–11 have been reported. Some studies have shown increased sleep onset latency (SOL),8,12 while others have reported no difference.9,10,13 Findings regarding sleep architecture are also inconsistent, with studies reporting no changes in patients with TBI,8 increased slow wave sleep,11 reduced REM sleep,11,13 increased REM in the second half of the night,14 no change to REM,8 or decreased REM onset latency.8,10
The mechanisms underlying sleep disturbances in patients with TBI are likely to be multifaceted. Injury-related damage to sleep-wake regulating centers and associated pathways or neurotransmitter systems is implicated as the cause of such disturbances.5,11,14 Anxiety and depression that frequently follow TBI are also likely contributing factors.10,15
The circadian (∼24-hour) pacemaker in the hypothalamic suprachiasmatic nuclei regulates the timing of sleep and several physiologic processes including pineal melatonin synthesis. Endogenous melatonin is involved in the circadian regulation of sleep-wakefulness.16 Circadian rhythm sleep disorders and delayed circadian rhythms have been reported in patients with mild TBI with insomnia.17
This study aimed to characterize sleep-wake disturbances following TBI and investigate their underlying mechanisms by assessing polysomnographic sleep, sleep quality, melatonin rhythm, and mood.
METHODS
Participants.
Participants were recruited from Epworth Hospital, Melbourne, Australia, where they had received rehabilitation following TBI. Potential participants were approached at 6 months post-injury. See appendix e-1 on the Neurology® Web site at www.neurology.org for inclusion/exclusion criteria.
Twenty-three patients with TBI and 23 age- and gender-matched healthy volunteers were recruited (table 1). Duration of posttraumatic amnesia (PTA), measured prospectively as an indicator of injury severity, showed that the majority of patients experienced a severe head injury.
Table 1 Participant characteristics
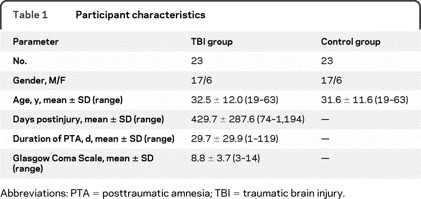
Preliminary data from the study (n = 10 per group) were published previously.11,18
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Monash University and Epworth Hospital Human Research Ethics Committees. Written informed consent was obtained from all participants.
Self-report measures.
Participants completed the Pittsburgh Sleep Quality Index (PSQI) to measure sleep quality, the Epworth Sleepiness Scale (ESS) to measure daytime sleepiness, and the Morningness Eveningness Questionnaire (MEQ) to assess preferred sleep-wake time. Anxiety and depression symptoms were measured using the Hospital Anxiety and Depression Scale (HADS) (see appendix e-1).
Procedure.
Participants attended the Monash University Sleep Laboratory for 2 overnight visits (≤7 days apart). The laboratory consisted of lightproof, sound-attenuated, and temperature-controlled bedrooms each with ensuite and kitchen.
Visit 1: Saliva collection and habituation.
On the first laboratory visit, participants arrived at approximately 17:00 hours. From 17:30 hours, a modified constant routine protocol was imposed19; participants remained in dim light (<10 lux; light levels were measured using a Lumacolor J17 luxmeter [Textronix, USA] by placing the sensor on the forehead of the participant in the angle of his or her gaze), posture and activity were controlled, and food intake was standardized. From 18:00 to 00:30 hours, participants provided half-hourly saliva samples using polyester swab Salivettes (Sarstedt, Germany). Immediately after collection samples were frozen (−20°C).
Radioimmunoassay of the saliva samples for melatonin concentration was conducted at the Department of Obstetrics and Gynaecology, University of Adelaide, Australia. As previously described,20 saliva (200 μL) was assayed in duplicate using reagents obtained from Buhlmann Laboratories (Allschwil Switzerland). Sensitivity of the assay was 4.3 pM. Intraassay and interassay coefficients of variation were <10 and <14%.
At approximately 19:00 hours, participants were fitted with face and scalp electrodes to facilitate polysomnographic recordings (see appendix e-1). Following the final saliva sample at 00:30 hours, participants were provided with an 8-hour sleep opportunity. The first laboratory visit served as an adaptation night to the sleep laboratory environment and as such polysomnography data from the first recording were not analyzed.
Visit 2: Polysomnographic sleep recording.
Participants arrived at the sleep laboratory approximately 2 hours before their habitual bedtime (determined by sleep-wake diary) for overnight polysomnographic monitoring including EEG, electrooculogram (EOG) (above and below cantomeatal plan), and EMG (submentalis) (S-Series Sleep Monitoring System, Compumedics Pty Ltd., Australia). Participants were instructed to get into bed 15 minutes prior to scheduled lights-out time and to remain in bed for approximately 8 hours.
Data analysis.
Polysomnography data were scored visually according to standard sleep staging criteria21 by experienced technicians blind to participant group. Awakenings were defined as any epoch with greater than 50% α or β low voltage EEG activity. Sleep recordings were evaluated for the following measures of sleep continuity and architecture: total sleep time, non-REM (NREM) stages (1, 2, slow wave sleep [SWS]) (%), REM (%), wake after sleep onset (WASO) (min), sleep efficiency (%), and sleep onset latency (min). Sleep efficiency was defined as the total sleep time divided by the time in bed, and sleep onset was defined as the first epoch of any stage other than awake.22
DLMO times were calculated for each participant according to a standard method.20 The mean of the first 3 saliva samples (18:00 hours, 18:30 hours, and 19:00 hours) plus 2 standard deviations was first calculated to give the threshold value. The first timepoint where a participant's salivary melatonin concentration rose above this threshold and remained above the threshold for 1 subsequent sample was taken as DLMO.
SPSS Statistics version 17.0 (SPSS Inc.) was used for all data analysis. Group differences were analyzed using independent group t tests, unless otherwise stated. Correlation analysis used Pearson r. Adjustments for multiple comparisons were not made.
RESULTS
Self-reported sleep-wake and psychological characteristics.
The TBI group reported higher PSQI scores than controls, t (26.92) = −4.35, p < 0.001, indicating poorer sleep quality, and more symptoms on the HADS anxiety and HADS depression subscales, t (29.91) = −3.98, p < 0.001, t (28.79) = −4.72, p < 0.001 (see table 2). After adjusting for HADS anxiety scores (analysis of covariance), the patients with TBI were still found to have elevated PSQI scores (F1,42 = 7.96, p = 0.007). Likewise, after adjusting for HADS depression scores, the patients with TBI were still found to have elevated PSQI scores (F1,42 = 8.04, p = 0.007).
Table 2 Self-reported sleep quality, sleep timing, and psychopathology measures
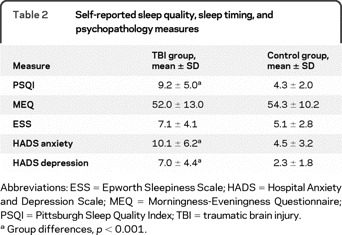
Although ESS scores were higher, on average, in the TBI group this difference in self-reported daytime sleepiness was not significant. The TBI and control groups did not differ significantly on MEQ scores, indicating no differences in preferred sleep time.
Dim light melatonin onset and melatonin production.
DLMO could not be determined for 9 patients and 9 controls due to sporadic levels or no apparent rise in level, leaving a final sample size of 14 per group (see figure). No significant difference was found in DLMO times between groups.
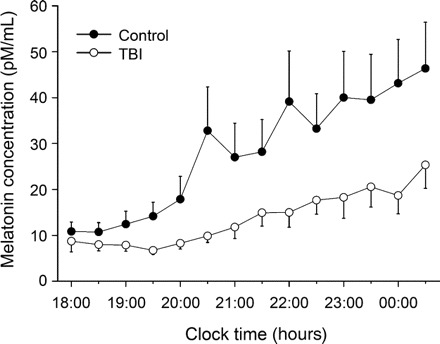
Figure Melatonin levels for traumatic brain injury (TBI) and control groups
Mean (±SE) salivary melatonin levels were calculated for patients with TBI and controls every half hour during the sampling period (18:00 hours to 00:30 hours). The control group had higher melatonin output across the sampling period than the TBI group (p = 0.031).
Based on the observed difference in melatonin levels between the 2 groups (see figure), we calculated total melatonin production during the sampling period for each participant, including those for whom DLMO could not be assessed. Using the trapezoid method, the area under each participant's melatonin curve (AUC) was calculated to estimate total melatonin production over the sampling period. The control group (AUC 361.7 ± 80.24) had higher melatonin production than the TBI group (AUC 171.3 ± 22.98), t (25.58) = 2.28, p = 0.031.
Polysomnographic sleep measures.
The TBI group had lower sleep efficiency (t[43] ≥2.33, p = 0.025) and longer WASO (t [31.06] = −3.18, p = 0.003) (see table 3). Trends were found for the TBI group to have less REM sleep (t[43] = 1.82, p = 0.075) and more SWS (t[43] = −1.73, p = 0.091). No group differences were found on other measures of sleep architecture.
Table 3 Polysomnographic sleep measures
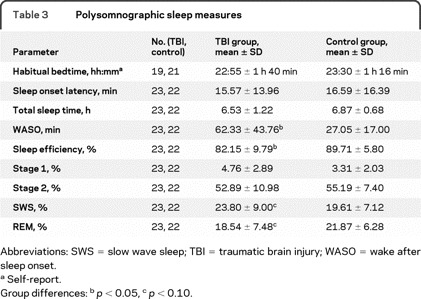
An association between injury severity and WASO was found in the patients with TBI (r = 0.56, n = 23, p = 0.006), with more severe injuries associated with longer WASO. There was an association between duration of PTA and sleep efficiency (r = −0.49, n = 23, p = 0.017). No association between duration of PTA and PSQI was found (r = −0.27, n = 22, p > 0.05).
Association between anxiety and depression and polysomnographic sleep measures.
Group differences in polysomnographic measures that reached or approached significance were reexamined while controlling for anxiety and depression scores (analysis of covariance), with α set at a more stringent 0.01 level due to violation of the homogeneity of variance assumption.23
After controlling for anxiety score, the TBI group continued to show longer WASO (F1,42 = 9.31, p = 0.004) and lower sleep efficiency (F1,42 = 7.37, p = 0.01). When depression score was included as a covariate, there was no group difference in WASO or sleep efficiency (F1,42 = 4.40, p = 0.042; F1,42 = 2.49, p = 0.12). Depression score was associated with WASO (r = 0.338, n = 45, p = 0.023), with depression score accounting for 6.5% of the variance in WASO, but was not associated with sleep efficiency (p > 0.01).
The TBI group was found to have higher levels of SWS after controlling for anxiety score (F1,42 = 7.67, p = 0.008) and depression score (F1,42 = 6.90, p = 0.012). In contrast, no group difference in REM sleep was found after controlling for anxiety or depression scores (p > 0.01).
Melatonin production and sleep measures, mood symptoms.
Melatonin level (AUC) was not associated with sleep efficiency or WASO, nor was it associated with anxiety or depression scores (p > 0.05). Melatonin level was associated with REM sleep (r = 0.35, n = 45, p = 0.017), accounting for 12% of the variance. Likewise, there was an association between melatonin level and NREM stage 2 sleep (r = −0.32, n = 25, p = 0.03).
DISCUSSION
TBI was associated with significant self-reported and objective sleep disturbance in patients who were on average 14 months postinjury. Specifically, patients with TBI reported lower sleep quality, and were shown to have lower sleep efficiency and increased WASO. These findings are consistent with previous reports of sleep disturbance in individuals with TBI.8–11 Patients with TBI were also found to have significantly elevated levels of depression and anxiety symptoms, consistent with previous studies.24 Importantly, the anxiety and depression measures (HADS) used do not include items on sleep, minimizing the possibility that these measures are confounded by disturbed sleep.
When the influence of anxiety and depression symptoms were statistically controlled the self-reported sleep quality remained lower in patients with TBI. This suggests that the subjective experience of poor sleep following TBI is associated with factors beyond mood disturbance. In contrast, when depression scores were controlled, the TBI group no longer showed poorer quality sleep on polysomnography measures. A significant association was observed between depression symptoms and sleep disturbance (WASO). These findings are consistent with the established link between depressive disorders and disturbed sleep,25,26 and previous reports in patients with TBI.15 Elevated rates of psychological symptoms and sleep disturbances are common in patients with TBI and do not appear to dissipate over time.27 We cannot rule out the possibility that presence of sleep disturbance leads to depression rather than occurring as a consequence of it.
While we found no significant difference between patients with TBI and controls in measures of sleep architecture, when the effects of either anxiety or depression scores were controlled, the TBI group showed significantly higher SWS. Elevated symptoms of psychological distress may therefore mask differences in sleep architecture. The higher level of SWS may be attributed to the effects of the mechanical brain damage caused by the injury. We speculate that the TBI group experiences higher sleep pressure due to endocrine imbalance, neural plasticity, global reaction to trauma, and diffuse damage to the homeostatic sleep system, which manifests as increased SWS.11
Consistent with our previous findings,18 the present study found no group differences in the self-reported sleep or DLMO times. When total production of melatonin during the collection period was assessed, the TBI group showed significantly lower melatonin production than controls. To our knowledge, melatonin production has not previously been assessed in individuals with TBI beyond the days immediately following injury, when melatonin is thought to have a neuroprotective role as an antioxidant in areas of damaged cerebral tissue.28 Patients with neurologically complete injuries to their lower cervical spinal cord (i.e., tetraplegia) show no detectable melatonin in plasma and lower sleep efficiency.29 Furthermore, pharmacologic suppression of melatonin secretion results in increased wake time and WASO.30 Exogenous melatonin administration has been shown to improve polysomnographic sleep measures in older individuals with low endogenous levels of melatonin.31 Although we did not find significant associations between melatonin level and sleep disturbance, we propose that studies investigating the efficacy of exogenous melatonin to improve sleep quality are warranted.
The present study also found a significant association between percentage REM sleep and melatonin production. REM sleep is strongly regulated by the circadian system32 and is temporally associated with high circulating levels of endogenous melatonin.33 Furthermore, exogenous melatonin increases REM sleep duration in healthy controls34 and in patients with abnormally lowered REM sleep.35 The absence of circulating levels of melatonin in tetraplegia patients is associated with increased latency to REM sleep.36 Based on these previous studies, it is hypothesized that the observed trend toward lower REM sleep in the TBI group is related to the reduced levels of melatonin, the attenuated melatonin levels possibly weakening the circadian system's ability to modulate REM sleep. Given that REM sleep is thought to play an important role in learning and memory consolidation,37 future studies may examine the possible relationship between REM sleep and cognitive deficits in the TBI population.
A somewhat paradoxical finding concerning sleep disturbance in the TBI population has been reported elsewhere: milder TBIs are associated with increased self-reported sleep disturbance as well as other postconcussional symptoms.7 We found a nonsignificant trend for those with milder injuries to self-report greater levels of sleep disturbance. We also found a significant relationship between objectively measured sleep disturbance (WASO, sleep efficiency) and injury severity. We suggest that people with more severe TBI are experiencing significant sleep disturbances, but they may underreport these changes, perhaps because of impaired self-awareness or because they do not perceive the sleep disturbance to be problematic relative to other disabilities.
This study was designed to assess the phase of the melatonin rhythm (DLMO) and not the 24-hour melatonin profile. It is possible that at a subset of patients with TBI show such profound circadian phase abnormalities that their DLMO was not captured during the assessment period, notwithstanding the fact that both groups reported average habitual bedtime of between approximately 23:00 to 23:30 hours. The possibility that undiagnosed sleep disorders may account for some of the observed differences in sleep cannot be ruled out, although questionnaires were used to exclude patients with high risk of obstructive sleep apnea.
Despite the widely accepted proposition that structural damage to the cerebral structures regulating sleep may cause sleep disturbance in patients with TBI, imaging studies have been unable to provide supporting evidence. Several studies (some single case) report no structural abnormalities on cerebral imaging (MRI or CT) despite significant sleep disturbance.12,17,38 Others have been unable to show associations between location of injury on CT or MRI scan with reports of sleep disturbance.3 Our own study indicates that cerebral damage associated with TBI may disrupt the neural structures regulating sleep-wakefulness, including synthesis of melatonin by the pineal gland, which may not reliably be revealed with nonfunctional imaging. Another possibility is that disrupted sleep in patients with TBI may impair neurogenesis and decrease cell proliferation thought to occur in adult brains, increasing further their susceptibility to cognitive impairment and mood disorders.39 A recent finding of reduced number of hypothalamic hypocretin (orexin) neurons of patients who died after severe TBI raises the possibility that loss of this wake-promoting neuropeptide may contribute to the hypersomnolence observed in patients with TBI.40
This study demonstrates that the sleep disturbances reported following TBI are evident in polysomnographic measures, in particular increased WASO and reduced sleep efficiency. Patients with TBI showed lower levels of melatonin production in the evening hours, indicating that the circadian regulation of melatonin synthesis was disrupted. Elevated levels of psychological distress, particularly depression, were found to be associated with reduced sleep quality. We suggest that the observed increase in SWS in patients with TBI (after controlling for anxiety and depression) may reflect the neural response to injury.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Julia Shekleton.
ACKNOWLEDGMENT
The authors acknowledge Athena Voultsios and Associate Professor David Kennaway from the University of Adelaide, who assisted with the assaying of melatonin samples. The authors also thank Dr. Tracey Sletten from Monash University for her assistance analyzing the melatonin data. The authors also thank Compumedics Pty Ltd., Australia, for providing the polysomnography equipment.
DISCLOSURE
J.A. Shekleton and Dr. Parcell report no disclosures. Dr. Redman has received research support from the National Health and Medical Research Council. J. Phipps-Nelson reports no disclosures. Dr. Ponsford has served on the Scientific Advisory Committee of the Victorian Neurotrauma Initiative; serves on the editorial boards of the Journal of the International Neuropsychological Society, Journal of Head Trauma Rehabilitation, Brain Injury, Neuropsychological Rehabilitation, NeuroRehabilitation, and Brain Impairment; receives royalties from the publication of Traumatic Brain Injury: Rehabilitation for Everyday Adaptive Living (Psychology Press, 1995) and Cognitive and Behavioral Rehabilitation: From Neurobiology to Clinical Practice (Guilford Press, 2004); receives research support from the NHMRC, the Victorian Neurotrauma Initiative, Monash University, and the Jack Brockhoff Foundation; and has provided medico-legal reports to numerous legal firms in relation to clients sustaining traumatic brain injury. Dr. Rajaratnam has received funding for travel from and served as a consultant (in capacity as an employee of Monash University) to Vanda Pharmaceuticals; has served as a paid expert witness or consultant to industry and government organizations on issues related to shift work, sleep, and/or circadian rhythms; and receives or has received research support as principal investigator or coinvestigator from Vanda Pharmaceuticals, Takeda Pharmaceutical Company Limited, ResMed Foundation, Respironics Sleep and Respiratory Research Foundation, Cephalon, Inc., Philips Lighting, the National Health and Medical Research Council, the Department of Homeland Security–Federal Emergency Management Agency (FEMA), Australia-India Council, NIH (NHLBI) R01 HL093279, the Royal Australasian College of General Practitioners/Centre of National Research on Disability and Rehabilitation Medicine, Centers for Disease Control and Prevention, and the National Institute of Justice; and has given expert testimony on behalf of New South Wales Nurses Association.
Supplementary Material
Glossary
- AUC
area under the curve
- DLMO
dim light melatonin onset
- EOG
electrooculogram
- ESS
Epworth Sleepiness Scale
- HADS
Hospital Anxiety and Depression Scale
- MEQ
Morningness Eveningness Questionnaire
- NREM
non-REM
- PSQI
Pittsburgh Sleep Quality Index
- PTA
posttraumatic amnesia
- SE
sleep efficiency
- SOL
sleep onset latency
- SWS
slow wave sleep
- TBI
traumatic brain injury
- WASO
wake after sleep onset.
Footnotes
Supplemental data at www.neurology.org
Study funding: Supported by the National Health and Medical Research Council (334002).
Disclosure: Author disclosures are provided at the end of the article.
Received December 17, 2009. Accepted in final form February 22, 2010.
REFERENCES
- 1.Cohen M, Oksenberg A, Snir D, Stern MJ, Groswasser Z. Temporally related changes of sleep complaints in traumatic brain injured patients. J Neurol Neurosurg Psychiatry 1992;55:313–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumann CR, Werth E, Stocker R, Ludwig S, Bassetti CL. Sleep-wake disturbances 6 months after traumatic brain injury: a prospective study. Brain 2007;130:1873–1883. [DOI] [PubMed] [Google Scholar]
- 3.Clinchot DM, Bogner J, Mysiw WJ, Fugate L, Corrigan J. Defining sleep disturbance after brain injury. Am J Phys Med Rehabil 1998;77:291–295. [DOI] [PubMed] [Google Scholar]
- 4.Hibbard MR, Uysal S, Sliwinski M, Gordon WA. Undiagnosed health issues in individuals with traumatic brain injury living in the community. J Head Trauma Rehabil 1998;13:47–57. [DOI] [PubMed] [Google Scholar]
- 5.Makley MJ, English JB, Drubach DA, Kreuz AJ, Celnik PA, Tarwater PM. Prevalence of sleep disturbance in closed head injury patients in a rehabilitation unit. Neurorehabil Neural Repair 2008;22:341–347. [DOI] [PubMed] [Google Scholar]
- 6.Ouellet MC, Beaulieu-Bonneau S, Morin CM. Insomnia in patients with traumatic brain injury: frequency, characteristics, and risk factors. J Head Trauma Rehabil 2006;21:199–212. [DOI] [PubMed] [Google Scholar]
- 7.Orff HJ, Ayalon L, Drummond SP. Traumatic brain injury and sleep disturbance: a review of current research. J Head Trauma Rehabil 2009;24:155–165. [DOI] [PubMed] [Google Scholar]
- 8.Williams BR, Lazic SE, Ogilvie RD. Polysomnographic and quantitative EEG analysis of subjects with long-term insomnia complaints associated with mild traumatic brain injury. Clin Neurophysiol 2008;119:429–438. [DOI] [PubMed] [Google Scholar]
- 9.Kaufman Y, Tzischinsky O, Epstein R, Etzioni A, Lavie P, Pillar G. Long-term sleep disturbances in adolescents after minor head injury. Pediatr Neurol 2001;24:129–134. [DOI] [PubMed] [Google Scholar]
- 10.Ouellet MC, Morin CM. Subjective and objective measures of insomnia in the context of traumatic brain injury: a preliminary study. Sleep Med 2006;7:486–497. [DOI] [PubMed] [Google Scholar]
- 11.Parcell DL, Ponsford JL, Redman JR, Rajaratnam SM. Poor sleep quality and changes in objectively recorded sleep after traumatic brain injury: a preliminary study. Arch Phys Med Rehabil 2008;89:843–850. [DOI] [PubMed] [Google Scholar]
- 12.Tobe EH, Schneider JS, Mrozik T, Lidsky TI. Persisting insomnia following traumatic brain injury. J Neuropsychiatry Clin Neurosci 1999;11:504–506. [DOI] [PubMed] [Google Scholar]
- 13.Schreiber S, Barkai G, Gur-Hartman T, et al. Long-lasting sleep patterns of adult patients with minor traumatic brain injury (mTBI) and non-mTBI patients. Sleep Med 2008;9:481–487. [DOI] [PubMed] [Google Scholar]
- 14.Frieboes RM, Muller U, Murck H, von Cramon DY, Holsboer F, Steiger A. Nocturnal hormone secretion and the sleep EEG in patients several months after traumatic brain injury. J Neuropsychiatry Clin Neurosci 1999;11:354–360. [DOI] [PubMed] [Google Scholar]
- 15.Rao V, Spiro J, Vaishnavi S, et al. Prevalence and types of sleep disturbances acutely after traumatic brain injury. Brain Inj 2008;22:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rajaratnam SW, Cohen DA, Rogers NL. Melatonin and melatonin analogs. Sleep Med Clin 2009;4:179–193. [Google Scholar]
- 17.Ayalon L, Borodkin K, Dishon L, Kanety H, Dagan Y. Circadian rhythm sleep disorders following mild traumatic brain injury. Neurology 2007;68:1136–1140. [DOI] [PubMed] [Google Scholar]
- 18.Steele DL, Rajaratnam SM, Redman JR, Ponsford JL. The effect of traumatic brain injury on the timing of sleep. Chronobiol Int 2005;22:89–105. [DOI] [PubMed] [Google Scholar]
- 19.Duffy JF, Dijk DJ. Getting through to circadian oscillators: why use constant routine protocols? J Biol Rhythms 2002;17:4–13. [DOI] [PubMed] [Google Scholar]
- 20.Voultsios A, Kennaway DJ, Dawson D. Salivary melatonin as a circadian phase marker: validation and comparison to plasma melatonin. J Biol Rhythms 1997;12:457–466. [DOI] [PubMed] [Google Scholar]
- 21.Rechtschaffen A, Kales A. A Manual of Standardized Terminology, Techniques, and Scoring System for Sleep States of Human Subjects. Washington, DC: Superintendent of Documents, US Government Printing Office; 1968
- 22.EEG arousals: scoring rules and examples: a preliminary report from the Sleep Disorders Atlas Task Force of the American Sleep Disorders Association. Sleep 1992;15:173–184. [PubMed] [Google Scholar]
- 23.Tabachnick BG, Fidell LS. Using Multivariate Statistics. 5th ed. Boston: Pearson/Allyn & Bacon; 2007
- 24.Kreutzer JS, Seel RT, Gourley E. The prevalence and symptom rates of depression after traumatic brain injury: a comprehensive examination. Brain Inj 2001;15:563–576. [DOI] [PubMed] [Google Scholar]
- 25.Plante DT, Winkelman JW. Polysomnographic features of medical and psychiatric disorders and their treatments. Sleep Med Clin 2009;4:407–419. [Google Scholar]
- 26.Mayers AG, Baldwin DS. The relationship between sleep disturbances and depression. Int J Psychiatry Clin Pract 2006;10:2–16. [DOI] [PubMed] [Google Scholar]
- 27.Parcell DL, Ponsford JL, Rajaratnam SM, Redman JR. Self-reported changes to nighttime sleep after traumatic brain injury. Arch Phys Med Rehabil 2006;87:278–285. [DOI] [PubMed] [Google Scholar]
- 28.Seifman MA, Adamides AA, Nguyen PN, et al. Endogenous melatonin increases in cerebrospinal fluid of patients after severe traumatic brain injury and correlates with oxidative stress and metabolic disarray. J Cereb Blood Flow Metab 2008;28:684–696. [DOI] [PubMed] [Google Scholar]
- 29.Zeitzer JM, Ayas NT, Shea SA, Brown R, Czeisler CA. Absence of detectable melatonin and preservation of cortisol and thyrotropin rhythms in tetraplegia. J Clin Endocrinol Metab 2000;85:2189–2196. [DOI] [PubMed] [Google Scholar]
- 30.Van Den Heuvel CJ, Reid KJ, Dawson D. Effect of atenolol on nocturnal sleep and temperature in young men: reversal by pharmacological doses of melatonin. Physiol Behav 1997;61:795–802. [DOI] [PubMed] [Google Scholar]
- 31.Haimov I, Lavie P, Laudon M, Herer P, Vigder C, Zisapel N. Melatonin replacement therapy of elderly insomniacs. Sleep 1995;18:598–603. [DOI] [PubMed] [Google Scholar]
- 32.Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci 1995;15:3526–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dijk DJ, Shanahan TL, Duffy JF, Ronda JM, Czeisler CA. Variation of electroencephalographic activity during non-rapid eye movement and rapid eye movement sleep with phase of circadian melatonin rhythm in humans. J Physiol 1997;505:851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cajochen C, Krauchi K, Mori D, Graw P, Wirz-Justice A. Melatonin and S-20098 increase REM sleep and wake-up propensity without modifying NREM sleep homeostasis. Am J Physiol 1997;272:R1189–1196. [DOI] [PubMed] [Google Scholar]
- 35.Kunz D, Mahlberg R, Muller C, Tilmann A, Bes F. Melatonin in patients with reduced REM sleep duration: two randomized controlled trials. J Clin Endocrinol Metab 2004;89:128–134. [DOI] [PubMed] [Google Scholar]
- 36.Scheer FA, Zeitzer JM, Ayas NT, Brown R, Czeisler CA, Shea SA. Reduced sleep efficiency in cervical spinal cord injury; association with abolished night time melatonin secretion. Spinal Cord 2006;44:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stickgold R. Sleep-dependent memory consolidation. Nature 2005;437:1272–1278. [DOI] [PubMed] [Google Scholar]
- 38.Boivin DB, James FO, Santo JB, Caliyurt O, Chalk C. Non-24-hour sleep-wake syndrome following a car accident. Neurology 2003;60:1841–1843. [DOI] [PubMed] [Google Scholar]
- 39.Meerlo P, Mistlberger RE, Jacobs BL, Heller HC, McGinty D. New neurons in the adult brain: The role of sleep and consequences of sleep loss. Sleep Med Rev 2009;13:187–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baumann CR, Bassetti CL, Valko PO, et al. Loss of hypocretin (orexin) neurons with traumatic brain injury. Ann Neurol 2009;66:555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.