

Abstract
Systemic lupus erythematosus (SLE) is a chronic, multi-organ inflammatory autoimmune disorder associated with high levels of circulating autoantibodies and immune complexes. We report that passive transfer of human SLE sera into mice expressing the uniquely human FcγRIIA and FcγRIIIB on neutrophils induces lupus nephritis and in some cases arthritis only when the mice additionally lack the CD18 integrin, Mac-1. The prevailing view is that Mac-1 on macrophages is responsible for immune complex clearance. However, disease permitted by the absence of Mac-1 is not related to enhanced renal immune-complex deposition or in situ C1q/C3 complement activation and proceeds even in the absence of macrophages. Instead, disease is associated with increased FcγRIIA-induced neutrophil accumulation that is enabled by Mac-1 deficiency. Intravital microscopy in the cremasteric vasculature reveals that Mac-1 mitigates FcγRIIA dependent neutrophil recruitment in response to deposited immune complexes. Our results provide direct evidence that human SLE immune-complexes are pathogenic, demonstrate that neutrophils are primary mediators of end organ damage in a novel humanized lupus mouse model, and identify Mac-1 regulation of FcγRIIA-mediated neutrophil recruitment as a key step in development of target organ damage.
INTRODUCTION
SLE is a systemic autoimmune disease that affects multiple organs including the kidney, joints and skin, with development of nephritis being one of the most serious complications of this disorder. SLE is characterized by a loss of tolerance to self-antigens and high titers of circulating autoantibodies (1). In mice, deletion of the common Fcγ-chain (γ−/−) that results in the loss of activating FcγRs, protects lupus prone NZB/NZW mice from spontaneously developing lupus nephritis (2, 3). Yet there is little direct evidence that immune complexes (ICs) are causative as there are no reports that the passive transfer of SLE sera reproduces disease. Moreover, autoantibodies to classical lupus antigens such as dsDNA are not sufficient for development of nephritis in lupus-prone mouse strains and renal biopsies of SLE patients without clinically evident nephritis can exhibit IC deposition. Thus factors in addition to ICs likely dictate the propensity to develop target organ damage the identification of which remains a central question in the field (4–6).
Human genome-wide association studies have shown that small variations in a large number of genes are associated with lupus. The current paradigm assumes that these traits modulate, through different pathways, the expression of disease in lupus. Variations in one of these genes is the leukocyte integrin Mac-1 (ITGAM, CD11b/CD18) (7), a member of the leukocyte CD18 integrin family of adhesion molecules that are obligate heterodimers of unique CD11 subunits paired with a common CD18 chain. Mac-1 has multiple ligands including complement fragment C3bi, and ICAM-1 on the endothelium (8) and the Mac-1 R77H polymorphism associated with lupus leads to a reduction in its binding to both these ligands (9). The question remains as to whether Mac-1, which typically promotes inflammation and host defense (10) plays a role in the pathogenesis of lupus. The prevailing view is that Mac-1 on macrophages clear circulating ICs (9), although there is no direct evidence of this. Likewise, polymorphisms in human FcγRIIA and FcγRIIIB are associated with lupus through mechanisms that remain largely unclear (7). The physiological roles of the uniquely human neutrophil FcγRs, which structurally differ from their mouse counterparts (11, 12), have been addressed in genetically engineered mice. Expression of the single polypeptide FcγRIIA, containing an immunotyrosine activating motif (ITAM), and the glycosyl-phosphatidyl-inositol (GPI)-anchored FcγRIIIB selectively on neutrophils of Fcγ-chain deficient mice (IIA+γ−/−, IIA+IIIB+γ−/−), restored susceptibility of γ−/− animals to progressive anti-glomerular basement membrane (GBM) nephritis and K/BxN serum-induced arthritis (13, 14). Although both human FcγRs promoted neutrophil accumulation, only FcγRIIA alone induced tissue injury likely through its established ability to promote neutrophil cytotoxic functions (13).
Difficulty in understanding the underlying mechanisms of SLE arises from the multifactorial influences on disease course including environmental, infectious and hormonal factors, the association of lupus with abnormalities at all levels of the immune system and the clinical heterogeneity of the disease (7). The disparate disease courses in SLE likely converge into a common route of end organ damage. Here, in a new model of lupus induced by the passive transfer of human SLE sera we provide direct evidence that SLE ICs are pathogenic, show a central role for neutrophils in target organ damage and demonstrate that two molecules, FcγRIIA and Mac-1 determine the capacity of ICs to cause tissue damage. In addition, these results coupled with the analysis of neutrophil behavior by intravital microscopy revealed a new regulatory role for Mac-1 in FcγRIIA mediated neutrophil recruitment following deposition of circulating ICs.
MATERIALS AND METHODS
Mice
Fcγ-chain deficient mice, human FcγRIIA and FcγRIIIB-expressing γ-chain deficient mice (IIA+γ−/− or IIA+IIIB+γ−/−), Mac-1-deficient mice (Mac-1−/−) and CD18-deficient mice (CD18−/−) were as described (13, 15–17). Mac-1 deficient mice were bred with IIA+γ−/− or IIA+IIIB+γ−/− mice to generate IIA+γ−/−Mac-1−/− and IIA+IIIB+γ−/−Mac-1−/− animals or with wild type mice to generate IIA+γ+/+Mac-1−/− and IIA+γ+/+ mice. Mice were bred in a specific pathogen- free facility. Age and gender matched mice 8–12 weeks old were used in all experiments. The Harvard Medical School Animal Care and Use Committee approved all procedures used in this study.
Passive transfer of human SLE sera or rabbit anti-GBM antibody
For the human SLE sera transfer model, mice were injected subcutaneously in both flanks with 2.5μg of human IgG (Jackson ImmunoResearch Labs) in complete Freund’s adjuvant (Thermo Scientific and Difco) on day -3. On days 0 and 2, 200μL of sterile serum was injected intravenously. For induction of nephrotoxic nephritis, mice were given rabbit IgG in CFA at day -3 in the flanks and rabbit αGBM antisera at day 0 as described (18) and urine and kidneys were collected at day 21.
Human sera samples
uman serum samples were obtained from the Lupus Clinic at the Beth Israel Deaconess Medical Center or Instituto Nacional de Ciencias Medicas y de la Nutricion Salvador Zubiran. Patients with SLE were diagnosed according to the classification criteria of the American College of Rheumatology (ACR). The inclusion criteria for SLE patients were patients who fulfilled ≥ 4 of 11 ACR criteria for SLE and that at the time of the first blood draw, had active disease (i.e. SLE disease activity index (SLEDAI) ≥6) and diagnosed with nephritis. Normal serum was obtained from healthy individuals matched for age, gender and ethnicity. At the time of serum collection, all individuals showed no signs or symptoms of infection. Informed consent was obtained from all patients and healthy donors under a BIDMC Institutional Review Board (IRB) approved protocol. Unless indicated, all experiments were performed using serum from SLE patient “A”.
Disease evaluation
Functional assessment of renal damage
Spot urine samples were collected and urine albumin and creatinine were evaluated by ELISA (Bethyl Labs) and a chemical assay (Cayman Chemical Company) respectively (13) and expressed as a ratio of urine albumin-to-creatinine.
Clinical scoring of arthritis
Mice were evaluated every other day after induction of disease. Inflammation of each limb was scored as reported previously (19): 0, no evident inflammation; 1, redness or swelling of one toe; 2, redness or swelling of >1 toe; 3, ankle or tarsal-metatarsal involvement; 4, redness or swelling of the entire paw.
Histological assessment of tissue injury
Tissue sections were blindly evaluated. Histological score included endocapillary proliferation, leukocyte infiltration and crescents, as previously described (20). For joint lesions, histological scores reflected leukocyte infiltration, synovial thickening, and cartilage and bone erosion.
Histological studies
Kidneys were fixed in formalin and paraffin-embedded, or frozen in OCT medium and 5μM sections were prepared. Toe specimens were fixed in 4% paraformaldehyde and decalcified with modified Kristensen’s solution. After dehydration, the tissues were embedded in paraffin and 5μm sections were made (21).
Immunohistochemistry
PAS (Periodic Acid-Schiff), H&E (Hematoxylin & Eosin) and dichloroacetate esterase (to identify neutrophils) on paraffin-embedded sections were performed as described (22). Neutrophils per 100 glomerular cross-sections were quantified. Immunohistochemistry on frozen sections was performed using a two-layer peroxidase method. Sections were immunostained with anti-F4/80 for macrophages (BioLegend), anti-CD3 (Serotech) for T cells, anti-C1q (Hycult Biotech) for C1q deposition and counterstained with Gill Hematoxylin No.2 (NewComerSupply Inc.). Tissue area occupied by macrophages or glomerular C1q deposition in four 20x consecutive fields was quantified using ImagePro. Sections of paraffin-embedded toes were stained with H&E. Immunohistochemistry was performed with anti-NIMP-R14 (Abcam) for neutrophil quantification, anti-F4/80 (BioLegend) for macrophages, anti-CD3 (Serotech) for T cells or anti-human IgG (Invitrogen). The total number of neutrophils in a toe cross-section was counted and divided by the total tissue area in each section, using ImagePro.
Immunofluorescence
Human and mouse IgG deposition were evaluated on frozen sections using anti-human IgG (Invitrogen) or anti-mouse IgG (Invitrogen) Alexa fluor 488- conjugated antibodies, respectively. Deposition of C3 was evaluated using a FITC-conjugated anti-C3 antibody (ICN/CAPPEL). Images of at least 15 glomeruli per mouse were captured and glomerular fluorescence intensity was measured using the acquisition and analysis software Metamorph (Molecular Devices). To evaluate human IgG subclasses, immunofluorescence was performed using anti-human IgG1, IgG2, IgG3, and IgG4 antibodies (The Binding site) followed by an anti-sheep DyLight-488 conjugated antibody (Jackson ImmunoResearch Laboratories Inc.).
Electron microscopy
Kidney samples were taken 10 days after injection of sera and processed using standard techniques.
Generation of a K562 stable cell line and analysis of binding to ICAM-1
The R77H mutation was introduced into human CD11b DNA by standard PCR techniques. K562 cells were transfected with WT or mutant (R77H) CD11b and CD18 plasmids by electroporation and confirmed by sequencing. Single cell sorting was used to select CD11b positive cells. Cells were cultured in RPMI 1640 media supplemented with 10% heat inactivated FBS, under constant selection using G418 (500μg/mL, Invivogen). CD11b expression was assessed by FACs analysis using anti-human CD11b-PE labeled antibody (BD Pharmingen). To evaluate K562 cell binding to ICAM-1Fc, cells were pre-activated with 1mM MnCl2 for 10min in HEPES buffer and drawn across a coverslip containing immobilized ICAM-1-Fc (15μg/mL, R&D) under 0.1dynes/cm2 shear flow. The number of cells bound in 4 different fields were quantified every 2 minutes over a 10 min period.
IgG depletion and quantification of human IgG
SLE serum was incubated with Protein A/G coated agarose beads (Thermo Scientific) or control agarose beads. Depletion was determined by measuring human IgG by ELISA (Bethyl Labs). Human immune complexes were quantified in human and mouse serum using an ELISA kit with C1q as the capture element (ALPCO Immunoassays). Due to cross-reactivity with mouse IgG, an anti-human HRP antibody (Bethyl Labs) was used as the secondary antibody for the quantification of human ICs in murine serum samples.
Macrophage depletion using clodronate liposomes
Mice were given an intraperitoneal injection of 500μL of liposomes containing clodronate or PBS (23) on day -5 before the first SLE injection, followed by 200μL every 5 days. Tissue was harvested at day 10 or 14 after SLE sera injection. Macrophage depletion was confirmed by immunohistochemistry of kidneys and spleen using anti-F4/80 antibody.
Intravital microscopy evaluation of Reverse Passive Arthus reaction in the cremaster muscle
Rabbit IgG anti-bovine serum albumin (BSA) antibody (200μg/300μL; Sigma-Aldrich, St. Louis, MO) was injected intrascrotally, followed by an intravenous injection of BSA (300μg/100μL; Sigma-Aldrich, St. Louis, MO). After 3hrs, leukocyte recruitment in the cremaster of anesthetized mice was evaluated by intravital microscopy as previously described (13). Four venules per mouse were analyzed. Leukocyte rolling velocities were measured by tracking single leukocytes (10/venule) over several frames and calculating the distance moved per unit time (μm/s). Adherent leukocytes were defined as cells remaining stationary for 30 seconds and were expressed as the number of cells/mm2 of venule.
MIP-2 was locally applied as previously described (24). Briefly, injections were done using a beveled glass pipette of 1mm outer diameter mounted on a manual micromanipulator (MM-33) (Warner Instruments, Hamden, CT). The tip was filled with 100nmol/L of MIP-2 (US Biologicals). The pipette was placed and the number of rollers across a perpendicular line and adherent cells (Ab) in a 200μm segment of the venule was recorded over a 1 minute period. Next, air pressure was applied to the pipette, to trigger the injection of MIP-2 (less than 1μL). Successful injection was verified by the presence of swelling of the interstitial tissue surrounding the pipette tip. The process of injection, and the following minute were recorded. The number of cells that were adherent after MIP-2 injection (Aa) was evaluated. The adherent cells, which is the percent of rollers that adhered in response to MIP-2 injection, was calculated using the following equation: (Aa-Ab)/#rollers X100.
Statistical analysis
Data are expressed as mean ± SEM. For proteinuria data, the line represents the median of the group. Differences were determined by Mann–Whitney test. For group analysis, one-way ANOVA was used for proteinuria data and two-way ANOVA was used for the cellular infiltration data. When significant differences were shown, data were subjected to Mann–Whitney test or Bonferroni test respectively for comparison between two mouse strains. P values <0.05 were considered significant.
RESULTS
Human SLE serum induces nephritis in mice that express neutrophil human FcγRs and lack Mac-1
Human SLE serum was obtained from patients that had active disease (SLEDAI≥6) and no clinical evidence of infection. Gender and race matched healthy individuals served as controls. Mice preimmunized with human IgG/CFA received two intravenous injections of SLE sera at day 0 and day 2. Nephritis was monitored by analyzing albumin leakage in the urine over a 21 day period. SLE serum from patient “A” caused marked proteinuria in mice that expressed human FcγRs (hFcγRs) only when the mice additionally lacked Mac-1 (IIA+γ−/− Mac-1−/− and IIA+IIIB+γ−/− Mac-1−/−) (Fig. 1A). Proteinuria peaked at day 14 (Fig. S1A). Mac-1 deficient mice expressing FcγRIIA alone, without FcγRIIIB exhibited significant proteinuria suggesting that it was sufficient for disease development (Fig. 1A). No proteinuria was observed in Mac-1-sufficient mice expressing hFcγRs (IIA+γ−/− or IIA+IIIB+γ−/−), Mac-1-deficient mice without hFcγRs (γ+/+Mac-1−/−) or wild-type animals (Fig. 1A). Surface expression of human FcγRs and the sister CD18 integrin, LFA-1 was similar in Mac-1 deficient and sufficient groups (Fig. S1B). Any effect of γ-chain deficiency superimposed on Mac-1 deficiency on disease susceptibility was ruled out as SLE serum induced nephritis in mice that express hFcγRIIA, lack Mac-1 and were γ-chain sufficient (Fig. 1B).
Fig 1. SLE sera induces nephritis in mice that express hFcγR and lack Mac-1.

A) Mice received two intravenous injections of SLE sera “A” on day 0 and 2 and urine albumin (normalized to creatinine) at day 14 was evaluated. The dashed line indicates the mean of proteinuria in mice not given SLE sera. Each data point represents one mouse and the solid line is the median of the group. B) Mice expressing the FcγRIIA in the presence of the γ-chain (IIA+γ+/+), and the same that are additionally Mac-1 deficient (IIA+γ+/+Mac-1−/−) were injected with SLE sera “A” and albuminuria was evaluated at day 7 and 14. Albuminuria in IIA+γ−/− Mac-1−/− mice given SLE sera was analyzed in parallel for comparison. Each dot represents one animal, and the line indicates the median of the group. ns= not significant. C) Murine anti-human IgG was quantified in renal tissue by immunofluorescence in indicated transgenic mice that were untreated (Unt) or 21 days following SLE sera “A” transfer (SLE). Wild-type mice given rabbit anti-GBM injection (WT αGBM) served as a positive control. Mean ±SEM are graphed. Dashed line is the average of pixel intensity in “Unt” mice. D) Representative images of renal sections (day 21) stained with Hematoxilin and Eosin (H&E) or Periodic Acid Schiff (PAS). The arrow indicates a glomerular crescent which was observed in ~5% of the glomeruli. A histological score was given and Mean ±SEM was graphed (n ≥ 5 mice per group). E) Electron microscopy of glomeruli: (i) IIA+IIIIB+γ−/− mice had normal glomerular architecture, (ii-iv) IIA+IIIIB+γ−/− Mac-1−/− mice exhibited mesangial expansion with increased cellularity (ii), neutrophils within capillary loops (iii) (x4730), mesangial electron-dense deposits (*) and lysosomal vacuoles containing protein reabsorption droplets (arrows) (iv) typically observed during proteinuria (x11100). en=endothelial, p=podocyte, m=mesangial cell, n=neutrophils. *p<0.05, **p<0.01 and ***p<0.001.
Mouse anti-human IgG was detected in the serum (Fig. S1C) and in the kidney (Fig. 1C) after SLE sera transfer but this was minimal compared to that in wild-type mice subjected to anti-glomerular basement membrane (nephrotoxic) nephritis (Fig. 1C), a widely used model of immune-mediated disease induced by preimmunization with rabbit IgG/CFA followed by transfer of rabbit anti-glomerular basement membrane serum (25). Importantly, mouse anti-human IgG levels in both the serum and renal tissue were similar in disease susceptible and non-susceptible mice (Fig. S1C and Fig. 1C). Although a role for mouse anti-human IgG and/or ICs formed from human IgG reacting with antigens in the mouse serum cannot be formally ruled out it is unlikely to be critical for disease development for the following two reasons. First, SLE sera induced nephritis in the absence of preimmunization with CFA/human IgG (data not shown) albeit disease was more variable. Second, human FcγRs and thus by inference, human IgG is required for development of nephritis. In the anti-GBM murine model rabbit IgG triggers a robust murine anti-rabbit reaction, nonetheless, tissue injury is largely independent of the humoral response (26). We propose that as is known for other autoimmune models, activation of the innate immune response by CFA in our SLE model may be important independent of the adaptive immune response (27).
Histopathological and electron microscopy analyses of renal tissue from human SLE sera-treated mice were undertaken. Congruent with proteinuria, glomerular damage and inflammation were observed only in hFcγR+γ−/−Mac-1−/− animals (Fig. 1D, E) and were associated with mesangial deposits of ICs (Fig. 1E). Together these data suggest that Mac-1 deficiency enables SLE-induced kidney damage. This may have relevance to human disease since the R77H polymorphism in the ITGAM gene (that encodes the CD11b chain of Mac-1) associated with SLE abrogates the capacity of Mac-1 expressed in a cell line, to bind its ligand ICAM-1 (Fig. 2).
Fig 2. The SLE associated R77H mutation of Mac-1 abrogates its capacity to bind ICAM-1 under shear flow.
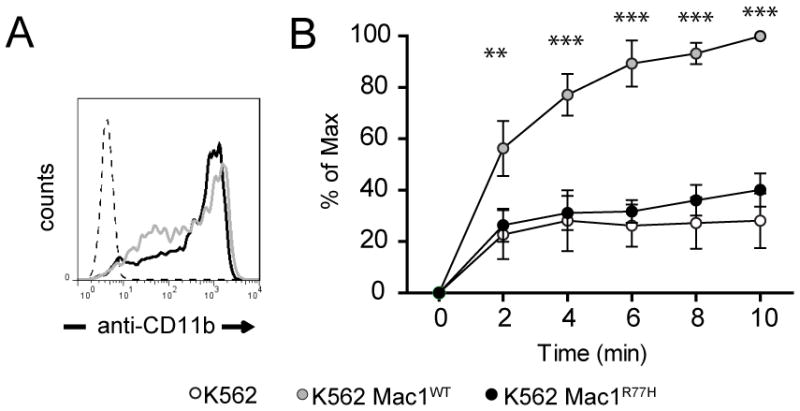
A) Flow cytometry analysis of K562 cells stably expressing Mac-1 wild-type (WT) or Mac-1 R77H. Similar levels of Mac-1 were observed on the surface of Mac-1 WT and Mac-1 R77H transfected cells. B) Binding of K562 cells expressing WT or the R77H Mac-1 variant to plate immobilized ICAM-1-Fc was evaluated under shear flow. Mac-1 R77H mutation prevented interaction with ICAM-1 compared to wild-type Mac-1. Mean ±SEM of 4 independent experiments is shown. **p<0.01, ***p<0.001.
Pathogenicity of SLE serum depends on the IgG fraction and its capacity to deposit in glomeruli
Sera collected from different SLE patients were tested in susceptible IIA+IIIB+γ−/− Mac-1−/− mice. Four out of six SLE sera induced nephritis, while sera from five healthy controls (normal serum), or heat aggregated human IgG (surrogate of ICs) failed to induce disease (Fig. 3A). Moreover, four out of five sera from SLE patients at an independent clinical center induced nephritis compared to normal controls (Fig. S1D). Nephritis was observed with multiple SLE serum samples from the same patient when treatment and disease activity varied but circulating ICs remained elevated. IgG depletion of SLE serum “A” abrogated its nephritis-inducing capacity (Fig. 3B), which suggests that ICs in the sera are required for disease development (Fig. 1A).
Fig 3. The pathogenicity of SLE serum correlates with the capacity of hIgG to deposit in the glomeruli.

A) IIA+IIIIB+γ−/− Mac-1−/− mice received sera from healthy donors (Normal, data is pooled from five donors), six SLE patients (denoted A–F), or heat aggregated human IgG (ICs). **p<0.01 and ***p<0.001 compared to mice that received normal sera. Each data point represents one mouse, and the line indicates the median. B) IIA+IIIIB+γ−/− Mac-1−/− mice were treated with SLE sera “A” subjected to sham or IgG depletion, and urine albumin was determined. C) Human IgG (hIgG) deposition in glomeruli was quantitated on day 21 in IIA+IIIIB+γ−/− Mac-1−/− mice injected with normal human sera, “non-pathogenic” (D, F) or “pathogenic” (A, E) SLE sera, or aggregated hIgG (ICs) (n ≥ 3 mice per group). Mean ±SEM is given and representative pictures of selected samples are shown. *p<0.05. D) Glomerular deposition of human IgG isotypes (1–4) was evaluated by immunofluorescence in IIA+IIIIB+γ−/− Mac-1−/− given normal or SLE sera “A” (SLE). Focal tubular basement membrane deposition (arrows) was detected in some cases.
The sera that caused disease, henceforth referred to as “pathogenic sera” (e.g. SLE sera “A” and “E”), were associated with glomerular deposits of human IgG that exhibited a granular pattern characteristic of ICs (Fig. 3C). In contrast, no human IgG deposition was observed when “non-pathogenic” sera were injected; these included SLE sera “D” and “F” and sera from healthy controls (Fig. 3C). All four human IgG isotypes, IgG 1-4 were observed in the glomeruli, predominantly as deposits in the mesangium (Fig. 3D). Specificity of the antibodies was confirmed by analyzing their reactivity against purified human IgG1-4 in western blots (data not shown). In the case of serum “A”, focal tubular basement membrane deposition of IgG was also detected (Fig. 3D), which is a pattern observed in human lupus nephritis. Sera pathogenicity (i.e. capacity of human ICs to deposit in glomeruli) did not correlate with the amount of ICs or IgG present in the sera (Table S1A), nor did it correlate with the patient type or lupus manifestations (Table S1B).
The susceptibility conferred by Mac-1 deficiency is not due to defects in IgG clearance
Levels of circulating (Fig. 4A) and deposited human IgG-ICs (Fig. 4B) as well as glomerular complement C1q and C3 deposition (Fig. 4B) were comparable between hFcγR expressing mice that were Mac-1 sufficient (IIA+IIIB+γ−/−) or deficient (IIA+IIIB+γ−/− Mac-1−/−), as were peripheral blood neutrophil and leukocyte counts (Table S1C).
Fig 4. Immune complex clearance and deposition are similar in susceptible and non-susceptible mice.
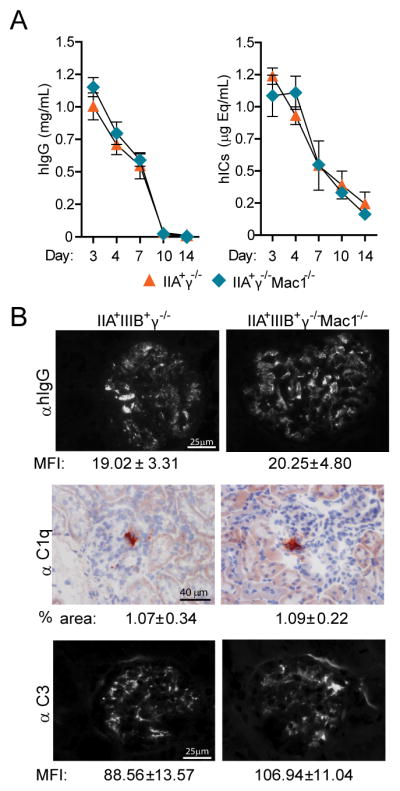
A) Serum was collected from animals at intervals up to day 14 after SLE sera “A” transfer. Circulating human IgG (hIgG) and human IgG-immune-complexes (hICs) were evaluated and the mean ±SEM were graphed. B) Kidneys were harvested at day 14 after SLE sera “A” transfer and renal sections were stained for hIgG, murine C1q, and complement C3. The mean fluorescence intensity (MFI, average pixel intensity, ±SEM) for glomerular hIgG and C3 and the percent of glomerular area positive for C1q (% Glm area ±SEM) were determined and found to be equivalent in both groups.
Passive transfer of human SLE serum induces arthritis in mice
Notably, two of the “pathogenic” SLE sera, “A” (Figure 5) and “E” (data not shown) caused arthritis in mice that affected primarily small joints, namely the distal interphalangeal and was evident at day 4. Patient “A” had clinical manifestations of arthritis, and Patient “E” did not have arthritis, but presented with Raynaud syndrome. There was no correlation between human symptoms and arthritis development as some sera from patients that clinically presented with nephritis and arthritis did not induce arthritis in mice (data not shown). Moreover, unlike nephritis, development of arthritis was more variable with different sera samples. As in the case of nephritis, only hFcγR expressing mice lacking Mac-1−/− developed arthritis (Fig. 5A). Histopathological analyses revealed intense joint inflammation with cellular infiltration associated with bone and cartilage destruction (Fig. 5B). Significant deposition of human IgG-IC within synovial blood vessels was observed in both disease-susceptible and non-susceptible mice (Fig. 5C). Neutrophils were abundant in the lesions (Fig. 5D) while macrophages and T cells were not detected (data not shown).
Fig 5. Human SLE sera cause arthritis in mice that express hFcγR and lack Mac-1.

Mice given SLE sera “A” were evaluated for the development of arthritis. A) Inflammation of each limb was assessed and a clinical score was given as described in Methods. As shown in representative pictures, mostly small joint involvement (arrows) was observed. *p<0.05 compared to IIA+IIIIB+γ−/− and IIA+γ−/− given SLE sera. B) Representative images of histological sections stained with H&E (upper panels) with magnification of boxed area (lower panels) are shown for indicated mice. Bone (b) erosion and inflammatory cell infiltration (*) were apparent only in IIA+IIIIB+γ−/− Mac-1−/− mice. The histological score in affected limbs of IIA+IIIIB+γ−/− Mac-1−/− and similar digits in IIA+IIIIB+γ−/− mice included leukocyte infiltration, synovial thickening, and cartilage and bone erosion. C) Tissue hIgG staining by immunohistochemistry showed intravascular IC deposition (arrows) in susceptible and non-susceptible mice treated with SLE sera. D) Neutrophils in joint sections harvested at indicated days after SLE serum injection were quantitated. Robust neutrophil accumulation was observed only in IIA+IIIIB+γ−/− Mac-1−/−. Representative pictures of neutrophil infiltrates (arrow) adjacent to damaged articular surfaces as well as periarticular soft tissue are shown. All data are mean ±SEM. For B and C, *p<0.05.
Marked glomerular neutrophil infiltration is observed in susceptible mice while macrophages do not contribute to disease
Susceptible mice (hFcγR+γ−/− Mac-1−/−) exhibited abundant glomerular neutrophil infiltration at day 10 that significantly exceeded that in non-susceptible mice (Fig. 6A). In contrast, a mild interstitial macrophage and T cell infiltration was observed in both groups of mice (Fig. 6A). Thus Mac-1 deficiency appears to selectively enable neutrophil accumulation. To distinguish if the Mac-1-expressing cell responsible for resistance to nephritis corresponded to circulating (i.e. monocytes or neutrophils) or resident cells, we generated bone marrow chimeras. Bone marrow from IIAγ−/− or IIAγ−/− Mac-1−/− mice was transferred into lethally irradiated γ−/− Mac-1−/− or γ−/− mice, respectively (i.e. IIAγ−/− → γ−/− Mac-1−/− and IIAγ−/− Mac-1−/− → γ−/−), and 3 months later SLE serum was administered. IIAγ−/− Mac-1−/− bone marrow provided susceptibility to nephritis when transferred into non-susceptible γ−/− mice suggesting that a lack of Mac-1 on hFcγRIIA expressing circulating cells allows the development of disease (Fig. S2A). Immunohistochemistry in tissue of γ−/− Mac1−/− recipients of IIAγ−/− bone marrow revealed Mac-1 positive macrophages in the renal interstitium following human SLE serum transfer, suggesting that SLE serum induces renal infiltration of blood monocytes and their subsequent differentiation into macrophages (Fig. S2A). To determine if monocytes/macrophages contribute to renal injury, these populations were depleted prior to SLE serum transfer by administering clodronate liposomes, which are internalized by these cells and lead to their apoptosis (Fig. S2B). Macrophage-depleted IIAγ−/− Mac-1−/− mice remained highly susceptible to nephritis (Fig. 6B) suggesting that macrophages do not significantly contribute to renal injury. Moreover, macrophage-depletion in disease non-susceptible (IIAγ−/−) mice did not result in proteinuria or glomerular neutrophil infiltration at day 10 (Fig. 6C), suggesting that Mac-1 deficiency in monocytes/macrophages does not enable disease.
Fig 6. Disease in susceptible mice is associated with neutrophil accumulation; macrophages do not participate in disease development.

A) Kidneys were harvested from mice at day 21 after injection of SLE serum “A”. nd= not determined. Dotted line represents the mean value obtained in susceptible mice given normal human serum and harvested at day 10 (top panel) or day 14 (middle and bottom panels). Neutrophil, macrophage and T cell infiltration were quantified by immunohistochemistry. Mice treated with anti-GBM serum are included as a positive control. Mean ±SEM is shown. *p<0.05, **p<0.01 compared to IIA+IIIIB+γ−/− injected with SLE sera. n=3–7 mice per group. nd= not determined. Animals were treated with liposomes that contained clodronate or PBS and proteinuria was evaluated at indicated times in IIA+γ−/− Mac-1−/− (B) and IIA+γ−/− (C) mice. ns= not significant, *p<0.05.
Mac-1 deficiency modifies the response of neutrophils towards tissue deposited immune complexes in vivo
To elucidate the mechanism(s) by which Mac-1 regulates FcγRIIA mediated neutrophil accumulation, we conducted intravital microscopy of the cremaster muscle that allows the real time visualization of neutrophil behavior within the vessel wall. The Reverse Passive Arthus (RPA) reaction, induced by the intravenous administration of soluble BSA and an intrascrotal injection of anti-BSA results in IC deposition within and outside the cremasteric vessels (13, 28). The RPA induces neutrophil rolling that is P- and E-selectin dependent (28), while the slowing of the rolling velocity and adhesion is dependent on murine FcγRs (29) and FcγRIIA (13). Following induction of the RPA, neutrophils in the venules of IIAγ−/− Mac-1−/− mice rolled significantly slower than in IIAγ−/− mice (Fig. 7A). This was not a result of differences between the two groups in expression of LFA-1 on circulating neutrophils or neutrophils recruited to the cremaster (Fig. S2C). The slow rolling required FcγRIIA, as it was not observed in γ−/− Mac-1−/−, and γ−/− mice (Fig. 6B). The downmodulatory effect of Mac-1 was selective for ICs as the rolling velocity induced by an intrascrotal injection of TNFα was actually increased in IIAγ−/− Mac-1−/− mice compared to IIAγ−/− mice (data not shown) as previously described (30). In the RPA, the observed slower neutrophil rolling in IIAγ−/− Mac-1−/− mice was not associated with an increase in the number of adherent cells compared to IIAγ−/− mice (Fig. 7C). E-selectin mediated activation of LFA-1 has been shown to slow neutrophil rolling velocity, and in turn lead to efficient firm adhesion only in response to a local chemoattractant stimulus, Macrophage Inflammatory Protein-2 (MIP-2) (24). Accordingly, the local microinjection of MIP-2 favored adhesion of neutrophils in IIAγ−/− Mac-1−/− compared to IIAγ−/− animals subjected to the RPA (Fig. 7D). IC induced slow rolling and MIP-2 induced neutrophil adhesion is likely LFA-1 dependent as these steps were markedly diminished in CD18 deficient mice (Fig. 7E, F).
Fig 7. Mac-1 deficiency slows the rolling velocity, and increases the chemoattractant induced adhesion of neutrophils following the Reverse Passive Arthus reaction.

The RPA was induced in the cremaster and 3hrs later, the mice were prepared for intravital microscopy and neutrophil rolling velocity was measured in hemodynamically similar venules of A) IIA+γ−/− Mac-1−/− and IIA+γ−/− mice (n=33 venules in 6 mice per group), and B) γ−/− Mac-1−/− and γ−/− mice (n=13 venules in 3 mice per group). Cumulative histograms of rolling velocities are shown. C) The number of adherent neutrophils in mice analyzed in panel A was determined. D) Mice were subjected to the RPA, the cremaster was prepared for intravital microscopy and a local injection of MIP-2 was given as depicted in the diagram. The increase in neutrophil adhesion after local MIP-2 injection (Δ of adherent cells) in IIA+γ−/− and IIA+γ−/− Mac-1−/− mice was calculated (n=15–16 venules in 6–7 mice per group). E) Cumulative histogram of rolling velocities of WT and CD18−/− neutrophils, and F) Δ of adherent cells upon MIP-2 injection are shown (n=6 venules in 3 mice per group). For C, D and F, mean ±SEM are graphed. **p<0.01, ***p<0.001.
In summary, FcγRIIA dependent slow rolling, which correlates with efficient leukocyte adhesion in response to MIP-2 is regulated by Mac-1, with a deficiency in this integrin resulting in a further decrease in rolling velocity and subsequent enhanced responsiveness of neutrophils to a local chemotactic stimulus.
DISCUSSION
In this study, we show in a novel lupus model that IC deposition and organ damage are not inextricably linked but that additional genetic factors dictate the propensity for target organ damage. In particular, we demonstrate that the neutrophils response to deposited ICs and subsequent tissue injury, following human lupus sera transfer is modulated by a functional association between an integrin and an IgG receptor. The role of Mac-1 in attenuating FcγRIIA mediated neutrophil recruitment in the context of deposited ICs, is unanticipated from previous work that have established Mac-1 largely as a positive regulator of neutrophil influx and inflammation (8, 31, 32). Our data predict that the R77H Mac-1 variant, which leads to reduced binding to its ligands, may have functional consequences for SLE patients and offer a mechanism by which Mac-1 dysfunction may contribute to end organ injury in SLE.
Disease susceptibility permitted by the absence of Mac-1 was not related to alterations in IC handling presumably by macrophages. Moreover, Mac-1 on macrophages and indeed macrophages themselves did not influence disease susceptibility. Thus this cell type, abundant in SLE lesions (33) is not essential for target organ injury in our model at least in the time frame of our experiments. Instead Mac-1 deficiency likely predisposes to in disease development due to unrestrained FcγRIIA mediated neutrophil accumulation in the kidney. The reported physical interaction of Mac-1 with human FcγRs on the cell surface (34), and the sharing of intracellular ITAM-based signaling cascades by these two receptors (35) led us to the intriguing possibility that Mac-1 on neutrophils, in cis, modulates neutrophil FcγRIIA activity and function.
FcγRIIA on neutrophils clearly has the capacity to induce tissue injury in vivo (13, 14). However, in the context of ICs deposits in the kidney and joints, the regulation of FcγRIIA mediated neutrophil recruitment appears to be a key step in conferring disease susceptibility. Recent work suggests that binding of FcγRIIA to ICs and function may be regulated. In vitro, the G-protein coupled receptor agonists C5a and fmlp enhances (14, 36) while co-engagement of FcγRIIB inhibits (37) FcγRIIA function. How is FcγRIIA regulated by Mac-1? Clues were provided by intravital microscopy. Slowing of the velocity of rolling cells improves their efficiency of firm adhesion (30) and in the RPA, FcγRIIA plays an important role in mediating slow rolling and leukocyte arrest (13). Additionally, CD18 integrins are required as slow rolling and MIP-2 mediated adhesion in the RPA is abrogated in CD18 deficient mice. We postulate that FcγRIIA promotes the activation of LFA-1. A deficiency in Mac-1 heightens FcγR activity towards ICs and subsequent LFA-1 activation that in turn leads to further deceleration of the rolling velocity and increased responsiveness to MIP-2. Mac-1 may be responsible for mitigating FcγRIIA mediated signaling or limiting FcγRIIA mobility or clustering, the molecular details of which require further investigation. Human FcγRIIA appears to functionally interact with murine Mac-1 in cis suggesting that this process uses conserved features of human and mouse Mac-1.
The observed decrease in neutrophil accumulation in Mac-1 deficient mice in other models of IC mediated disease including acute anti-GBM, thrombotic GN and bullous pemphigoid (22, 38, 39) suggest differential roles for Mac-1 in distinct models of IC-based diseases. Complex biological circuits predict dual and sometimes opposing roles for the same receptor that is cell type and context dependent. For example, incomplete phosphorylation of the ITAM following engagement of B or T cell receptors by low affinity/valency ligands favors inhibitory over activating signaling by these immune receptors (40). Moreover, unsustained FcαRI clustering markedly inhibits whereas sustained FcαRI aggregation promotes cell activation through differential recruitment of the tyrosine kinase Syk versus an inhibitory protein phosphatase SHP-1 (41, 42). We posit that Mac-1 relays similar context dependent inhibitory and activating signals, a hypothesis that is supported by the finding that although the R77H Mac-1 polymorphism is a susceptibility factor for lupus (9) it does not predispose to rheumatoid arthritis (43), another IC mediated disease. Interestingly, FcγRIIIB may also play context dependent roles in IC mediated inflammation. While low expression of FcγRIIIB is associated with SLE (44), high FcγRIIIB levels increase susceptibility to anti-neutrophil cytoplasmic antibody (ANCA)–associated systemic vasculitides, a disease associated with in situ rather than soluble ICs (45, 46). This concept is recapitulated in our mouse models. Human SLE serum induced nephritis was reduced in mice expressing both human FcγRIIA and FcγRIIIB versus FcγRIIA alone, which contrasts with the greater proteinuria following anti-GBM nephritis in mice expressing both hFcγRs compared with mice expressing FcγRIIA alone (13).
Our studies suggest that neutrophils are the primary cellular link between IgG and target organ damage in our model. Neutrophils are present in renal SLE lesions (47, 48), precede macrophage infiltration (47) and are appreciated as a sign of disease severity (49). Importantly, recent studies identified a subset of neutrophils in SLE patients that synthesize type I IFNs, induce endothelial cell damage (50), release neutrophil extracellular traps (NETs) that are potentially autoantigenic, and drive IFNα production by dendritic cells (51, 52). SLE sera transfer results in the development of lesions that recapitulate several features of lupus nephritis. The mice obviously lack the imbalances in the adaptive immune response and therefore chronicity of the disease. However like SLE patients, where development of lupus nephritis is not associated with autoantibodies of singular IgG specificity (53) a spectrum of IgG1-4 subtypes were found deposited in the glomerulus with the capacity of SLE sera to induce proteinuria being directly correlated with their ability to deposit in the glomerulus. Histological features similar to human disease include mesangial hypercellularity and mesangial deposits (mainly IgG), C3 deposition, endocapillary and extracapillary proliferation (crescents) and neutrophils. The lack of the interstitial component, thickening of the membrane or podocyte effacement is likely because the immune deregulation in SLE patients is much more profound than in our model. Renal injury in our model is unlikely a consequence simply of an immune response against heterologous human IgG as normal human serum, some SLE patient serum and heat aggregated human IgG failed to induce nephritis and there was a lack of significant murine anti-human IgG antibody deposition in the glomeruli.
Indiscriminate immunosuppression remains the prevailing therapy in SLE with significant toxicity, and the expectations from therapeutic targeting of the aberrant immune system have remained unfulfilled (4). Our data in a humanized mouse model indicate that deposition of circulating autoantibodies is not sufficient for target organ damage. Rather, regulation of neutrophil FcγRIIA by Mac-1 fundamentally influences IC-mediated end organ damage. Moreover, our humanized passive SLE sera transfer model will aid in delineating additional mechanisms specifically driving lupus induced tissue injury and serve as a preclinical platform to test new therapeutics targeted at preventing lupus end organ damage.
Supplementary Material
Acknowledgments
We thank Dr. Isaac Stillman (BIDMC, Boston, MA) for help in histologic interpretations, Dr. Klaus Ley (La Jolla Institute for Allergy and Immunology, La Jolla, CA) for technical advice in IVM and Lorna Andersson, Elizabeth Thayer and Lina Du (Brigham and Women’s Hospital, Boston MA) for technical assistance.
Nonstandard abbreviations
- IC
immune complex
- ITAM
immunoreceptor tyrosine activating motif
Footnotes
The work was supported by NIH RO1 HL065095 and AR050800 (T.N.M), AI42269 (G.T.), K23 AR055672 (V.K.), CONACyT and Fundación México en Harvard (F.R.S.), the Arthritis Foundation (N.T.), Japan Society for the Promotion of Science Postdoctoral Fellowship for Research Abroad (H.N.) and the Novartis Foundation (T.E.).
References
- 1.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349:1526–1533. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunol Lett. 2005;100:56–67. doi: 10.1016/j.imlet.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 3.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–290. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 4.Wu T, Mohan C. Three pathogenic determinants in immune nephritis--anti-glomerular antibody specificity, innate triggers and host genetics. Front Biosci. 2007;12:2207–2211. doi: 10.2741/2223. [DOI] [PubMed] [Google Scholar]
- 5.Bagavant H, Fu SM. New insights from murine lupus: disassociation of autoimmunity and end organ damage and the role of T cells. Curr Opin Rheumatol. 2005;17:523–528. doi: 10.1097/01.bor.0000169361.23325.1e. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen C, Limaye N, Wakeland EK. Susceptibility genes in the pathogenesis of murine lupus. Arthritis Res. 2002;4(Suppl 3):S255–263. doi: 10.1186/ar583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crispin JC, Liossis SN, Kis-Toth K, Lieberman LA, Kyttaris VC, Juang YT, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: recent advances. Trends Mol Med. 2010;16:47–57. doi: 10.1016/j.molmed.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ross GD, Vetvicka V. Review: CR3 (CD11b, CD18): a phagocyte and NK cell membrane receptor with multiple ligand specificities and functions. Clin Exp Immunol. 1993;92:181–184. doi: 10.1111/j.1365-2249.1993.tb03377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nath SK, Han S, Kim-Howard X, Kelly JA, Viswanathan P, Gilkeson GS, Chen W, Zhu C, McEver RP, Kimberly RP, Alarcon-Riquelme ME, Vyse TJ, Li QZ, Wakeland EK, Merrill JT, James JA, Kaufman KM, Guthridge JM, Harley JB. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 10.Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–294. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 11.Hogarth PM. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol. 2002;14:798–802. doi: 10.1016/s0952-7915(02)00409-0. [DOI] [PubMed] [Google Scholar]
- 12.Mayadas TN, Tsokos GC, Tsuboi N. Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. 2009;120:2012–2024. doi: 10.1161/CIRCULATIONAHA.108.771170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuboi N, Asano K, Lauterbach M, Mayadas TN. Human neutrophil Fcgamma receptors initiate and play specialized nonredundant roles in antibody-mediated inflammatory diseases. Immunity. 2008;28:833–846. doi: 10.1016/j.immuni.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuboi N, Ernandez T, Li X, Nishi H, Cullere X, Mekala D, Hazen M, Kohl J, Lee DM, Mayadas TN. Regulation of human neutrophil Fcg receptor IIA by C5a receptor promotes inflammatory arthritis in mice. Arthritis Rheum. 2011;63:467–478. doi: 10.1002/art.30141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 16.Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 17.Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenkranz AR, Mendrick DL, Cotran RS, Mayadas TN. P-selectin deficiency exacerbates experimental glomerulonephritis: a protective role for endothelial P-selectin in inflammation. J Clin Invest. 1999;103:649–659. doi: 10.1172/JCI5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, Ochaion A, Weinberger A, Fishman P. Antiinflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
- 20.Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, Bordes C, Viallard JF, Goulvestre C, Pellegrin JL, Weil B, Moreau JF, Batteux F, Blanco P. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci Transl Med. 2010;2:47ra63. doi: 10.1126/scitranslmed.3001001. [DOI] [PubMed] [Google Scholar]
- 21.Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, Fitzgerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum-transfer arthritis. Arthritis Rheum. 2008;58:1354–1365. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- 22.Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med. 1997;186:1853–1863. doi: 10.1084/jem.186.11.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 24.Ley K, Allietta M, Bullard DC, Morgan S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ Res. 1998;83:287–294. doi: 10.1161/01.res.83.3.287. [DOI] [PubMed] [Google Scholar]
- 25.Fu Y, Du Y, Mohan C. Experimental anti-GBM disease as a tool for studying spontaneous lupus nephritis. Clin Immunol. 2007;124:109–118. doi: 10.1016/j.clim.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Rosenkranz AR, Knight S, Sethi S, Alexander SI, Cotran RS, Mayadas TN. Regulatory interactions of alphabeta and gammadelta T cells in glomerulonephritis. Kidney Int. 2000;58:1055–1066. doi: 10.1046/j.1523-1755.2000.00263.x. [DOI] [PubMed] [Google Scholar]
- 27.Billiau A, Matthys P. Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukoc Biol. 2001;70:849–860. [PubMed] [Google Scholar]
- 28.Norman MU, Van De Velde NC, Timoshanko JR, Issekutz A, Hickey MJ. Overlapping roles of endothelial selectins and vascular cell adhesion molecule-1 in immune complex-induced leukocyte recruitment in the cremasteric microvasculature. Am J Pathol. 2003;163:1491–1503. doi: 10.1016/S0002-9440(10)63506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stokol T, O’Donnell P, Xiao L, Knight S, Stavrakis G, Botto M, von Andrian UH, Mayadas TN. C1q governs deposition of circulating immune complexes and leukocyte Fcgamma receptors mediate subsequent neutrophil recruitment. J Exp Med. 2004;200:835–846. doi: 10.1084/jem.20040501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 31.Jones SL, Brown EJ. Functional cooperation between Fcg receptors and complement receptors in phagocytes. In: van de Winkel JGJ, Capel PJA, editors. Human IgG Fc Receptors. R.G. Landes Company; 1996. pp. 149–163. [Google Scholar]
- 32.Li X, Utomo A, Cullere X, Choi MM, Milner DA, Jr, Venkatesh D, Yun SH, Mayadas TN. The beta-Glucan Receptor Dectin-1 Activates the Integrin Mac-1 in Neutrophils via Vav Protein Signaling to Promote Candida albicans Clearance. Cell Host Microbe. 2011;10:603–615. doi: 10.1016/j.chom.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Lee PY, Reeves WH. Monocyte and macrophage abnormalities in systemic lupus erythematosus. Arch Immunol Ther Exp (Warsz) 2010;58:355–364. doi: 10.1007/s00005-010-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petty HR, Worth RG, Todd RF., 3rd Interactions of integrins with their partner proteins in leukocyte membranes. Immunol Res. 2002;25:75–95. doi: 10.1385/IR:25:1:75. [DOI] [PubMed] [Google Scholar]
- 35.Mocsai A, Abram CL, Jakus Z, Hu Y, Lanier LL, Lowell CA. Integrin signaling in neutrophils and macrophages uses adaptors containing immunoreceptor tyrosine-based activation motifs. Nat Immunol. 2006;7:1326–1333. doi: 10.1038/ni1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagarajan S, Venkiteswaran K, Anderson M, Sayed U, Zhu C, Selvaraj P. Cell-specific, activation-dependent regulation of neutrophil CD32A ligand-binding function. Blood. 2000;95:1069–1077. [PubMed] [Google Scholar]
- 37.Syam S, Mero P, Pham T, McIntosh CA, Bruhns P, Booth JW. Differential recruitment of activating and inhibitory Fc gamma RII during phagocytosis. J Immunol. 2011;184:2966–2973. doi: 10.4049/jimmunol.0900016. [DOI] [PubMed] [Google Scholar]
- 38.Liu Z, Zhao M, Li N, Diaz LA, Mayadas TN. Differential roles for beta2 integrins in experimental autoimmune bullous pemphigoid. Blood. 2006;107:1063–1069. doi: 10.1182/blood-2005-08-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hirahashi J, Hishikawa K, Kaname S, Tsuboi N, Wang Y, Simon DI, Stavrakis G, Shimosawa T, Xiao L, Nagahama Y, Suzuki K, Fujita T, Mayadas TN. Mac-1 (CD11b/CD18) links inflammation and thrombosis after glomerular injury. Circulation. 2009;120:1255–1265. doi: 10.1161/CIRCULATIONAHA.109.873695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waterman PM, Cambier JC. The conundrum of inhibitory signaling by ITAM-containing immunoreceptors: potential molecular mechanisms. FEBS Lett. 2010;584:4878–4882. doi: 10.1016/j.febslet.2010.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pasquier B, Launay P, Kanamaru Y, Moura IC, Pfirsch S, Ruffie C, Henin D, Benhamou M, Pretolani M, Blank U, Monteiro RC. Identification of FcalphaRI as an inhibitory receptor that controls inflammation: dual role of FcRgamma ITAM. Immunity. 2005;22:31–42. doi: 10.1016/j.immuni.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Kanamaru Y, Pfirsch S, Aloulou M, Vrtovsnik F, Essig M, Loirat C, Deschenes G, Guerin-Marchand C, Blank U, Monteiro RC. Inhibitory ITAM signaling by Fc alpha RI-FcR gamma chain controls multiple activating responses and prevents renal inflammation. J Immunol. 2008;180:2669–2678. doi: 10.4049/jimmunol.180.4.2669. [DOI] [PubMed] [Google Scholar]
- 43.Phipps-Green AJ, Topless RK, Merriman ME, Dalbeth N, Gow PJ, Harrison AA, Highton J, Jones PB, Stamp LK, Harrison P, Wordsworth BP, Merriman TR. No evidence for association of the systemic lupus erythematosus-associated ITGAM variant, R77H, with rheumatoid arthritis in the Caucasian population. Rheumatology (Oxford) 2009;48:1614–1615. doi: 10.1093/rheumatology/kep254. [DOI] [PubMed] [Google Scholar]
- 44.Niederer HA, Clatworthy MR, Willcocks LC, Smith KG. FcgRIIB, FcgRIIIB, and systemic lupus erythematosus. Ann N Y Acad Sci. 2009;1183:69–88. doi: 10.1111/j.1749-6632.2009.05132.x. [DOI] [PubMed] [Google Scholar]
- 45.Aitman TJ, Dong R, Vyse TJ, Norsworthy PJ, Johnson MD, Smith J, Mangion J, Roberton-Lowe C, Marshall AJ, Petretto E, Hodges MD, Bhangal G, Patel SG, Sheehan-Rooney K, Duda M, Cook PR, Evans DJ, Domin J, Flint J, Boyle JJ, Pusey CD, Cook HT. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 46.Willcocks LC, Lyons PA, Clatworthy MR, Robinson JI, Yang W, Newland SA, Plagnol V, McGovern NN, Condliffe AM, Chilvers ER, Adu D, Jolly EC, Watts R, Lau YL, Morgan AW, Nash G, Smith KG. Copy number of FCGR3B, which is associated with systemic lupus erythematosus, correlates with protein expression and immune complex uptake. J Exp Med. 2008;205:1573–1582. doi: 10.1084/jem.20072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Segerer S, Henger A, Schmid H, Kretzler M, Draganovici D, Brandt U, Noessner E, Nelson PJ, Kerjaschki D, Schlondorff D, Regele H. Expression of the chemokine receptor CXCR1 in human glomerular diseases. Kidney Int. 2006;69:1765–1773. doi: 10.1038/sj.ki.5000337. [DOI] [PubMed] [Google Scholar]
- 48.Mayadas TN, Rosetti F, Ernandez T, Sethi S. Neutrophils: game changers in glomerulonephritis? Trends Mol Med. 2010;16:368–378. doi: 10.1016/j.molmed.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Molad Y, Cronstein BN. Neutrophils (Polymorphonuclear Leukocytes) in Systemic Lupus Erythematosus. In: Tsokos GC, Smolen JS, editors. Systemic lupus erythematosus: a companion to Rheumatology. Mosby, Inc., an affiliate of Elsevier Inc; Philadelphia PA: 2007. [Google Scholar]
- 50.Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184:3284–3297. doi: 10.4049/jimmunol.0902199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra20. doi: 10.1126/scitranslmed.3001201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA-Peptide Complexes in Systemic Lupus Erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bagavant H, Fu SM. Pathogenesis of kidney disease in systemic lupus erythematosus. Curr Opin Rheumatol. 2009;21:489–494. doi: 10.1097/BOR.0b013e32832efff1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.