

Abstract
Twenty-one new 4-substituted diarylaniline compounds (DAANs) (Scheme 2, series 13, 14, and 15) were designed, synthesized, and evaluated against wild-type and drug resistant HIV-1 viral strains. As a result, approximately a dozen new DAANs showed high potency with low nano- to sub-nanomolar EC50 values ranging from 0.2 to 10 nM. The three most promising compounds 14e, 14h, and 15h exhibited high potency against wild-type and drug-resistant viral strains with EC50 values at the sub-nanomolar level (0.29–0.87 nM), and were comparable to or more potent than the new NNRTI drug riplivirine (2) in the same assays. Drug-like physicochemical property assessments revealed that the most active DAANs (EC50 <10 nM) have better aqueous solubility (>1–90 μg/mL at pH 7.4 and pH 2) and metabolic stability in vitro than 2, as well as desirable log P values (<5) and polar surface area (PSA) (<140 Å2). These promising results warrant further development of this novel compound class as potential potent anti-AIDS clinical trial candidates.
INTRODUCTION
Since the first case of acquired immunodeficiency syndrome (AIDS) was reported by the U. S. in 1981, the scientific progress in HIV/AIDS research has been extraordinary, especially in the development of antiretroviral therapy (ART) that has proven to be life-saving to millions of people. Recent scientific evidence has demonstrated that ART is also effective at preventing infection,1,2 thus offering an unprecedented opportunity to control the AIDS pandemic. Therefore, the discovery and development of novel highly potent anti-HIV drugs remains imperative. Among current anti-HIV drugs, non-nucleoside reverse transcriptase inhibitors (NNRTIs) are used in combination as key components in the highly active antiretroviral therapy (HAART).3,4 Currently, five NNRTI drugs have been approved by the US FDA for AIDS therapy, including three first-generation compounds, nevirapine, delavirdine, and efavirenz, and two second-generation compounds, eravirine (1)5 and rilpivirine (2)6 (Figure 1). NNRTI drugs have the advantages of high potency and low toxicity, but rapid viral drug-resistance to the first-generation NNRTI drugs has limited their clinical uses. Fortunately, the new NNRTI drugs 1 and2 overcome the deficiency of early NNRTI drugs. Both 1 and 2 possess extremely potent antiviral activity with low nano- or sub-nanomolar EC50 values against wild-type and a broad spectrum of mutated NNRTI resistant strains. There is also a higher genetic barrier for HIV-1 to evolve resistance to the two new NNRTIs. 7, 8 Drugs 1 and 2 belong to the same structural family, diarylpyrimidines (DAPYs). Rilpivirine is three-fold more potent than etravirine and is used in a low oral dose (25 mg/tablet) once-daily. These successes greatly encourage continuing research to discover and develop additional novel next-generation NNRTI drugs with diverse molecular scaffolds for more efficacious therapy and potential AIDS prevention.
Figure 1.

Next-generation NNRTI drugs and DAAN lead 3.
During our prior studies,9,10 we discovered a series of diarylaniline (DAAN) compounds with low nanomolar anti-HIV potency against wild-type and HIV-1 RT-resistant viral strains, as well as a new chemo-type scaffold and simplified synthesis. As an example, lead 3 (Figure 1) exhibited sub-nanomolar EC50 values against wild-type (EC50 0.63 nM) and RT multi-resistant (0.9 nM) viral strains, which were comparable to those of rilpivirine (0.52 nM and 0.49 nM, respectively) in the same assays. However, further physical chemistry studies revealed that 3 possessed poor aqueous solubility (0.29 μg/mL) and low permeability (Paap < 2 × 10−6 cm/s) in a Caco-2 assay, which might limit molecular absorption and cause low oral bioavailability in vivo. Therefore, further lead optimization of DAANs is necessary to surmount these deficiencies.
Our further optimization strategy is shown in Figure 2. Based on prior SAR results, the two known pharmacophores of DAANs, the para-cyanoaniline moiety (A-ring) and the crucial amino group (NH2) on the central phenyl ring (B-ring) ortho to the NH group linking the B and A rings were maintained. With regard to the structural difference between drugs 1 and 2, 11 one modifiable point appeared to be the para-substituent (R2) on the trisubstituted phenoxy ring (C-ring). We postulated that a hydrophobic, flexible, and more linear substituent at this position might insert into the narrow tunnel on the NNRTI binding site and interact with the reserved W229 to enhance antiviral potency against wild-type and resistant viral strains. While early molecular modeling results on DAANs supported the importance of the NH2 on the central phenyl (ring B) for high anti-HIV activity, the results also revealed that the nitro group (R1) opposite the amino group on the central phenyl ring (B-ring) created a small electrostatic interaction with the positively charged amino acid K172 on the NNRTI binding site. 9 Thus, R1 presents another opportunistic position that we hypothesized could lead to improvement in molecular physicochemical properties and related drug-like properties without loss of anti-HIV potency. Accordingly, a series of new DAAN analogues with different R1 substituents on the B-ring, either ionizable, polar groups or H-bond donors and acceptors, were synthesized and evaluated. Herein, we report the synthesis, anti-HIV activity, physicochemical properties, and some drug-like properties in vitro of new 4-substituted 1,5-diarylaniline analogues (DAANs). Their structure-activity relationship (SAR) and structure-property relationship (SPR) correlations are also discussed.
Figure 2.

Leads DAANs, known SAR, and further optimization strategy
Chemistry
We designed a short synthetic route (see Schemes 1 and 2) that would allow the rapid and simultaneous optimization of the two variable points (i.e., R1 and R2) to produce three (13, 14, 15) new series of DAAN compounds. Our approach was to first prepare building blocks 7–9 (Scheme 1). 2,4-Dichloro-5-trifloromethyl-nitrobenzene (4) is commercially available. 2,4-dichloro-5-nitrobenzenesulfonamide (5) was prepared from 2,4-dichloronitrobenzene by treatment with chlorosulfuric acid, followed by addition of ammonia hydroxide solution. Nitration of 2,4-dichlorobenzoic acid with concentrated HNO3 and H2SO4, followed by esterification with methanol in the presence of H2SO4 produced methyl 2,4-dichloro-5- nitrobenzoate (6). Subsequently, coupling of 4, 5, or 6 with 4-cyanoaniline in DMF in the presence of excess cesium carbonate provided the corresponding intermediate 4-substituted 5-chloro-N-(4-cyanophenyl)-2- nitroanilines 7–9, respectively. Consistent with our previous results, the 4-cyanoaniline moiety preferred to attack the chloride at the ortho-position to the nitro group.9 The 1H NMR signals of the linker NH in 7–9 were shifted down-field to δ 9.76–9.84 ppm due to a neighboring chelating effect with the adjacent nitro group.
Scheme 1.

a) ClSO3H, 0–140 °C; b) NH4OH/THF, 0 °C, 0.5 h; c) HNO3/H2SO4, 2h; d) H2SO4/MeOH, reflux, 2 h; e) t-BuOK/DMF, r.t. 1 h.
Scheme 2.

a) K2CO3/DMF, 190 °C, MW, 15 min or traditional heating at 130 °C, 5 h; b) (EtO)2P(O)CH2CN, t-BuOK/THF, 0 °C – r.t., 3 h; c) aq NaOH/THF/MeOH, r.t., 4 h; d) (i) SOCl2/CH2Cl2, reflux, 3 h; (ii) NH4OH/THF, 0 °C, 0.5 h; e) (i) SOCl2/CH2Cl2, reflux, 3 h; (ii) NH2CH3 in THF, 0 °C, 0.5 h; f) (i) SOCl2/CH2Cl2, reflux, 3 h; (ii) N2H4·H2O, r.t, 1 h; g) CH3COCH3, MeOH/aq NaOH (10%), 0 °C – rt, 1 h; h) LiBH4, THF/MeOH, 1 h; i) Na2S2O4/NH4OH, THF/H2O (v/v 1:1), r.t.; j) H2/Pd-C, EtOH, 2–4 h; k) CH(OEt)3, HCl (1N), C2H5OC2H5/DMF, r.t. 3 h.
As shown in Scheme 2, intermediates 7–9 were subsequently coupled with 4-substituted 3,5-dimethylphenols in DMF in the presence of potassium carbonate, either under microwave irradiation at 190 °C for 10–15 min or by traditional heating at 130 °C for 5 h, to afford corresponding 2,4-diarylnitrobenzene compounds 10a–c and 11c–d with the required three-phenyl ring scaffold. Furthermore, Aldol condensation of 10a with acetone generated compound 11a. Wittig-Honer reaction of aldehyde 10a–10c with diethyl cyanomethyl phosphonate generated corresponding cyanovinyl compounds 12a–12c. Compound 12c was hydrolyzed under basic condition to yield acid 12d. Compound 12d was treated with thionyl chloride and the resulting acid chloride was treated with: (i) ammonia or (ii) methylamine or (iii) hydrazine to generate corresponding compounds 12e or 12f or 12g respectively. The reduction of the ester group in 10c and 12c with lithium borohydride (LiBH4) produced hydroxymethyl compounds 11e and 12h, respectively. The preparation of intermediate 12i was reported previously.10 Finally, the nitro group on the central ring in 11a and 12 was treated with the mild reducing reagent sodium hydrosulfite to produce corresponding target DAAN compounds 13a and series 14 (a–f, h). Catalytic hydrogenation of 12 with hydrogen (30–40 p.s.i.) in the presence of Pd/C (5–10%) in anhydrous ethanol afforded the new series 15 DAAN compounds, in which the nitro group on the central ring and the double bond of the para-cyanovinyl group on the phenoxy moiety (C-ring) were reduced. By using the same catalytic hydrogenation, 5-substituted 2,4-diaryl-nitrobenzenes 11a and 11c–e were converted target series 13 (b–e). To confirm the importance of the NH2 on the B-ring for anti-HIV activity, 1,6-diaryl-benzoimidazole compounds 16a and 16b were prepared by cyclization of 14f and 15f, respectively, with triethyl orthoformate.12. The successful synthesis of 16a and 16b further validated the structures of intermediates 7–9 as shown and described above.
RESULTS AND DISCUSSION
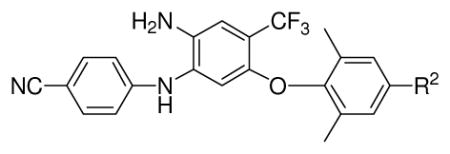
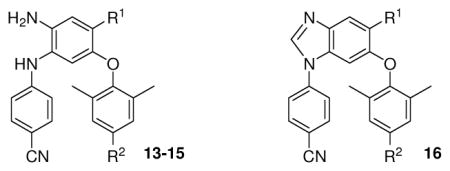
Initially, we evaluated the 4-trifluoromethyl DAAN compounds (13a–d, 14a, and 15a) in which the nitro group (R1) on the B-ring of lead 3 had been replaced with a strong electron-withdrawing trifluoromethyl group, and various para-substituents (R2) were present on the C-ring. All six compounds exhibited high potency against wild-type HIV-1 replication with low nanomolar EC50 values ranging from 1.3 to 13.8 nM and selective indexes (SI) between 789 and 6454 (Table 1). These results showed that the R1 substituent on the central phenyl ring could be modified without losing high anti-HIV activity. However, the introduced trifluoromethyl group resulted in log P values greater than 5 and did not improve aqueous solubility (< 1 μg/mL) over a broad pH range (2.0–7.4) (Table 1). Consequently, R1 on the B-ring was changed to other substituents in an attempt to achieve potential drug candidates with more desirable drug-like properties and high anti-HIV potency. Newly synthesized series 14 and 15 DAANs with various R1 groups on the B-ring and either a para-cyanovinyl or para-cyanoethyl R2 group, respectively, on the C-ring were evaluated against wild-type HIV-1 replication. Except for 14b, 15b, 14d, and 15d, the compounds in these two series exhibited very high antiviral potency with low to sub-nanomolar EC50 values (0.39–10 nM). Among them, the two most potent compounds 14h and 15h, with a 4-hydroxymethyl group (R1), exhibited sub-nanomolar EC50 values of 0.53 nM and 0.39 nM, respectively, which were comparable with or slight better than that of 2 in the same assay. Three other pairs of analogues, 14c and 15c with a 4-ester (COOCH3), 14e and 15e with 4-amide (CONH2), and14f and 15f with 4-methylamide (CONHCH3), also displayed high potency with low nanomolar EC50 values ranging from 0.87 to 5.72 nM, regardless of whether R2 on the C-ring was para-cyanovinyl (14) or para-cyanoethyl (15). In contrast, the presence of 4-sulfamoyl (SO2NH2, 14b & 15b) or 4-carboxylic acid (COOH, 14d & 15d) on the B-ring resulted in substantially reduced antiviral activity (EC50 94–380 nM), suggesting that a very polar substituent at the 4-position might impede the interaction between the inhibitor(s) and the NNRTI binding site. In a comparison of 4-amide-DAAN 15e, 4-amino-DAAN 15i, and 4-hydrazine-DAAN 15g, compound 15e (EC50 1.39 nM) was 5 and 13-fold more potent than 15i (7.30 nM) and 15g (EC50 19.1), respectively, indicating that the distance between the amino group in the R1 substituent and the central phenyl ring might influence the antiviral activity. Therefore, we postulated that an electron-withdrawing R1 group with appropriate volume and length on the B-ring might be necessary to provide additional interaction(s) within the NNRTI binding pocket for maintaining or enhancing antiviral potency. However, when the two neighboring amino groups on the central phenyl ring of the active compound 14f and 15f were cyclized, both products 16a and 16b lost antiviral potency (EC50 > 220 nM). This result supported our previous hypothesis that an amino group on the central B-ring ortho to the aniline moiety is crucial for achieving high potency. Finally, DAAN 13e, with two hydroxymethyl groups (R1 and R2), exhibited 20-fold lower activity (EC50 10.0 nM) than the most active compounds 14h and 15h, in which R2 was para-cyanovinyl or para-cyanoethyl, respectively. This finding is consistent with our previous SAR results,10 that a hydrophobic and linear para-R2 on the C-ring is favored for antiviral activity.
Table 1.
Structures, antiviral activity, and physicochemical properties of 4-trifluoromethyl diarylanilines 13a–d, 14a, and 15a
| |||||||
|---|---|---|---|---|---|---|---|
| R2 | NL4-3 (wt) in TZM-bl cells | Aqueous solubility | Log P | ||||
|
| |||||||
| EC50 (nM)a | CC50 (μM)b | SIc | pH 7.4 | pH 2.0 | |||
| 13a | CH=CHCOCH3 | 10.9 ± 2.58 | >8.6 | >789 | -- | -- | -- |
| 13b | CH2CH2COCH3 | 6.85 ± 1.73 | 11.1 | 1620 | 0.16 | 0.28 | >5 |
| 13c | CN | 3.79 ± 0.83 | 13.3 | 3509 | 0.26 | 0.96 | >5 |
| 13d | NH2 | 13.8 ± 4.37 | 15.8 | 1145 | -- | -- | -- |
| 14a | CH=CHCN | 9.38 ± 2.45 | 15.0 | 1599 | 0.24 | 0.42 | >5 |
| 15a | CH2CH2CN | 1.13 ± 0.31 | > 7.1 | > 6454 | 0.24 | 0.33 | >5 |
| 3 | 0.63 ± 0.13 | >24.4 | >38730 | 0.29 | 0.29 | >5 | |
| 2 | 0.52 ± 0.14 | 19.4 | 37305 | ||||
Concentration of compound that causes 50% inhibition of viral replication, presented as mean ± standard deviation (SD), performed at least in triplicate.
XTT assay was used to determine the CC50 value that causes cytotoxicity to 50% cells.
SI (Selectivity index) is the ratio of CC50/EC50.
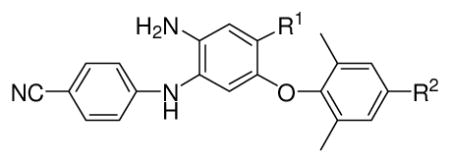
Considering them to be next-generation NNRTIs, we chose new DAANs with EC50 values less than 5 nM to test against three NNRTI resistant HIV-1 mutants RT-multi-drug-resistant (MRTDR), K101E, and E138K. It is reported that K101E and E138K are two of the mutations that confer resistance to new drugs 1 and 2.13 Interestingly, the newly synthesized active DAANs exhibited potent activity against the drug resistant viral strains with low nanomolar EC50 values. The most active compounds 14h and 15h were slightly more potent than 2 and three- to four-fold more potent than 3 against these mutated viral strains, with the notable exception that 15h was now more resistant against the RTMDR strain.
To evaluate their potential as drug candidates, five pairs of new active DAANs (EC50 < 10 nM) were further assessed for their physicochemical and drug-like properties as shown in Table 4. We first measured their aqueous solubility by using an HPLC/UV method, in parallel with 2 and 3. All tested DAANs, except 14a and 15a, showed improved solubility at both pH 7.4 and pH 2.0 compared with 3, and were generally more soluble in acidic than neutral conditions, because of the presence of a free amine group. Notably, the most active compound 15h had aqueous solubility of 89.8 μg/mL at pH 2.0 and 18.8 μg/mL at pH 7.4, comparable to and better than 2 (86 μg/mL at pH 2.0 and 0.24 μg/mL at pH 7.4) in the same assay (lit. 20 ng/mL at pH 7.0).14 In addition, 15c, 15f, 14h, and 13e also showed moderate solubility ranging from 20.9–44.7 μg/mL under acidic conditions, which would enhance molecular absorbability in the stomach. A comparison of the 14 and 15 series showed that the 15 series compounds were generally more soluble than the corresponding 14 series compounds, suggesting that the greater flexibility of the cyanoethyl group (R2) on the C-ring might contribute to better aqueous solubility. Additionally, 15 series compounds had slightly or significantly lower melting points than corresponding 14 series compounds. Next, we assessed the lipophilicity of the active DAANs by measuring their octanol/water (aqueous buffer pH 7.4) partition coefficients by an HPLC/UV method. The log P values of 14c/15c, 14e/15e, 14f/15f, and 14h/15h ranged from 2.65–4.15 and, consistent with aqueous solubility, were also improved compared with 14c/15c, 2, and 3. The pattern was also similar to the predicted clog D values. Subsequently, molecular PSA (polar surface area) values were calculated by using DS software (3.0) as a prediction of oral bioavailability. The values for all compounds fell in a desirable range of 88.13–138 (a criterion <140 Å2).15 Furthermore, metabolic stability of these active DAANs was evaluated by a human liver microsome incubation assay, in parallel with 2. Most of the active DAANs displayed better stability than 2 (t1/2 34.2 min); however, the most potent 4-hydroxymethyl DAANs 14h and 15h appeared to be less stable (t1/2 30.2 and 20.9 min, respectively) and had higher clearance (CL 0.23 and 0.33, respectively) than 2 and others in the same assay.
Table 4.
Structures and drug-like properties of active DAANs 14 and 15 (EC50 <10 nM)
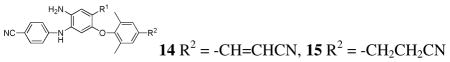
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R1 | EC50 (nM) | Aqueous solubility (μg/mL) a
|
Mp °C | logP a | clogD b | PSAb | HLM c
|
|||
| pH 2.0 | pH 7.4 | t1/2 min | CL mL/min/mg | |||||||
| 14a | CF3 | 9.38 | 0.42 | 0.24 | 224–6 | >5 | 7.09 | 94.86 | 45.0 | 0.15 |
| 15a | CF3 | 1.13 | 0.33 | 0.24 | 172–4 | >5 | 6.49 | 94.86 | 55.7 | 0.12 |
| 14c | CO2CH3 | 2.74 | 1.75 | 1.30 | 240–2 | 4.15 | 5.06 | 121.16 | 283 | 0.02 |
| 15c | CO2CH3 | 4.32 | 21.8 | 1.21 | 236–8 | 4.15 | 4.46 | 121.16 | 186 | 0.04 |
| 14e | CONH2 | 0.87 | 2.09 | 0.63 | 290–2 | 3.53 | 4.38 | 137.94 | 35.0 | 0.20 |
| 15e | CONH2 | 1.39 | 9.11 | 0.06 | 274–6 | 2.65 | 3.79 | 137.94 | 42.0 | 0.16 |
| 14f | CONHCH3 | 5.72 | 8.63 | 1.32 | 112–4 | 4.12 | 3.93 | 123.96 | 44.8 | 0.15 |
| 15f | CONHCH3 | 2.73 | 44.7 | 8.28 | 110–2 | 2.65 | 3.33 | 123.96 | 55.7 | 0.12 |
| 14h | CH2OH | 0.53 | 20.9 | 3.23 | 186–8 | 4.14 | 3.55 | 115.09 | 30.2 | 0.23 |
| 15h | CH2OH | 0.39 | 89.8 | 18.8 | 115–7 | 3.68 | 2.95 | 115.09 | 20.9 | 0.33 |
| 3 | NO2 | 0.63 | 0.29 | 0.29 | 252–4 | >5 | 5.88 | 140.67 | 47.8 | 0.15 |
| 2 | 0.52 | 86.8 | 0.24d | 246–8 | >5 | 3.62 | 97.41 | 34.2 | 0.20 | |
Measured at pH 7.4 in triplicate. A general solubility guideline for human oral absorption is <10 μg/mL low; 10–60 μg/mL moderate, >60 μg/mL high.16
Predicted by using software ACD or DS 3.0, respectively.
Human liver microsome incubation assay, data from at least two experiments in parallel.
20 ng/mL at pH 7.0 reported in reference 14.
CONCLUSION
Based on our current optimization strategy, 21 new DAAN analogues (series 13, 14, and 15) were synthesized by modifying R1 on the central phenyl ring (B-ring) and R2 on the phenoxy ring (C-ring). Seventeen of the new target compounds showed significant anti-HIV activity (EC50 < 20 nM). The most promising compounds 14e, 14h, and 15h exhibited high potency against wild-type and drug-resistant viral strains with EC50 values at a subnanomolar level (0.29–0.87 nM), and were comparable or more potent than new NNRTI drug 2 in the same assays. The modifications of the R1 substituent on the central B-ring provided the following new structure-activity relationship (SAR) correlations for DAANs: (1) R1 is modifiable to improve molecular drug-like properties without loss of potency. (2) R1 as H-bond donor and/or acceptor is favorable for anti-HIV potency and drug-like properties, and (3) the presence of a too polar R1, such as COOH or SO2NH2, greatly decreases potency. In addition, current results also supported our previous SAR: (1) a favorable R2 substituent on the C-ring should be hydrophobic, flexible and more linear, such as either a para-cyanovinyl or para-cyanoethyl; (2) the amino (NH2) group on the central ring is crucial for achieving high potency (see 16a and 16b). The assessment of drug-like physicochemical properties for four pairs of active DAANs (EC50 < 6 nM) indicated that introducing a suitable R1 on the central phenyl ring can indeed improve molecular physicochemical properties, such as aqueous solubility, log P values, PSA, and metabolic stability, which will benefit drug ADME properties. The current results hold great promise for the identification of potential new drug candidates with high potency and desirable drug-like properties. Further studies on pharmacokinetic properties in vivo are ongoing and will be reported later.
EXPERIMENTAL SECTION
Chemistry
Melting points were measured with an RY-1 melting apparatus without correction. The proton nuclear magnetic resonance (1H NMR) spectra were measured on a JNM-ECA-400 (400 MHz) spectrometer using tetramethylsilane (TMS) as the internal standard. The solvent used was CDCl3 unless otherwise indicated. Mass spectra were measured on an ABI Perkin-Elmer Sciex API-150 mass spectrometer with electrospray ionization, and the relative intensity of each ion peak is presented as percent (%). The purities of target compounds were ≥95%, measured by HPLC analyses, which were performed on an Agilent 1100 HPLC system with a UV detector and a Grace Alltima HP C18 column (100 × 2.1 mm, 3 μm) eluting with a mixture of solvents A and B (condition 1: acetonitrile/water 70:30, flow rate 1.0 mL/min; condition 2: MeOH/water 70:30, flow rate 0.8 mL/min; UV 254 nm). The microwave reactions were performed on a microwave reactor from Biotage, Inc. Thin-layer chromatography (TLC) and preparative TLC plates used silica gel GF254 (200–300 mesh) purchased from Qingdao Haiyang Chemical Company. Medium-pressure column chromatography was performed using a CombiFlash companion purification system. All chemical reagents and solvents were obtained from Beijing Chemical Works or Sigma-Aldrich, Inc. NADPH, MgCl2, KH2PO4, K2HPO4 and reference compound propranolol were purchased from Sigma-Aldrich. HPLC-grade acetonitrile for LC-MS analysis was purchased from VWR. Pooled human liver microsomes (Lot No# 28831) were purchased from BD biosciences (Woburn, MA).
2,4-Dichloro-5-nitrobenzene-1-sulfonyl Chloride
Chlorosulfonic acid (30 mL) cooled to 0 °C was added dropwise into 2,4-dichloro-nitrobenzene (9.6 g, 50 mmol) with stirring. Then the solution was allowed to warm to rt, followed by heating at 140 °C for 2 h. After cooling to rt, ice-water was added dropwise to the mixture. A solid precipitated, and was filtered and washed with cold water. The collected crude product was re-crystallized from EtOAc to give 10.6 g of pure sulfonyl chloride in 73% yield, white solid, mp 116–118 °C; 1H NMR (DMSO-d6) δppm 7.95 (1H, s, ArH-3), 8.44 (1H, s, ArH-5).
2,4-Dichloro-5-nitrobenzenesulfonamide (5)
A solution of the above sulfonyl chloride (290 mg, 1 mmol) in THF was added to aqueous ammonia solution (25%, 1 mL, excess) at 0 °C (ice-bath) and stirred at the same temperature for an additional 0.5 h. The mixture was then poured into ice-water and let stand overnight. The precipitated white solid was filtered and washed with cold water to provide 207 mg of 5, 77% yield, white powder, mp 176–178 °C; 1H NMR (DMSO-d6) δ ppm 8.02 (2H, s, SO2NH2), 8.27 (1H, s, ArH-3), 8.58 (1H, s, ArH-5).
2, 4-Dichloro-5-nitrobenzoic Acid
2, 4-Dichlorobenzoic acid (191 mg, 1 mmol) was dissolved completely in H2SO4 (98%, 5 mL) with a mechanical stirrer at 0 °C. Then the solution was added dropwise to HNO3 (con., 0.5 mL) with stirring, and the temperature was kept below 5 °C. The mixture was stirred at rt for 2 h with monitoring by TLC to reaction finish. The mixture was slowly poured into ice-water with stirring and let stand overnight. The precipitated solid was collected and washed with cold water to neutral pH. After drying, 215 mg of 2,4-dichloro-5-nitrobenzoic acid was obtained in 91% yield, white powder, mp 148–150 °C; 1H NMR (DMSO-d6) δ ppm 8.15 (1H, s, ArH-3), 8.50 (1H, s, ArH-5).
Methyl 2,4-Dichloro-5-nitrobenzoate (6)
A solution of 2,4-dichloro-5-nitrobenzoic acid (235 mg, 1 mmol) in MeOH (10 mL) in the presence of H2SO4 (conc., 2 mL) was refluxed for 2 h. The mixture was poured into ice-water and let stand overnight. The precipitated white solid was collected, and washed with cold water to neutral. After drying, 215 mg of 6 was obtained in 86% yield, white powder, mp 46–48 °C; 1H NMR δ ppm 3.98 (3H, s, CH3), 7.71 (1H, s, ArH-3), 8.49 (1H, s, ArH-5).
General Preparation of 4-Substituted 5-Chloro-N1-(4-cyanophenyl)-2-nitroanilines (7–9)
To a solution of 5-substituted 2,4-dichloro-nitrobenzene (1 equiv) and para-cyanoaniline (1.1 equiv) in DMF (3 mL) was slowly added potassium tert-butoxide (2 equiv) at 0 °C (ice-water bath) with stirring and then for an additional 1 h at room temperature. The reaction was monitored by TLC until completion. The mixture was poured into ice-water and pH adjusted to 6 with aq HCl (5%). The solid crude product was collected, washed with cold water, and purified by CombiFlash medium-pressure column chromatography system.
5-Chloro-N1-(4-cyanophenyl)-4-trifluoromethyl-2-nitroanilines (7)
Starting with 260 mg (1 mmol) of commercially available 2,4-dichloro-5-nitrobenzotrifluoride (4) and 130 mg (1.1 mmol) of para-cyanoaniline to afford 281 mg of 7, yield 82% yield, yellow solid, mp 180–182 °C; 1H NMR δ ppm 7.40 (1H, s, ArH-6), 7.41 (2H, d, J = 8.4 Hz, ArH-2′,6′), 7.78 (2H, d, J = 8.4 Hz, ArH-3′,5′), 8.61 (1H, s, ArH-3), 9.78 (1H, s, NH); MS m/z (%) 359.2 (M+NH4, 100).
5-Chloro-N1-(4-cyanophenyl)-4-sulfonamide-2-nitroanilines (8)
Yield 33%, starting with 271 mg (1 mmol) of 5 and 130 mg (1.1 mmol) of para-cyanoaniline to afford 116 mg of 8, yellow solid, mp 236–238 °C; 1H NMR δ ppm 5.09 (2H, s, NH2), 7.39 (1H, s, ArH-6), 7.41 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.79 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.99 (1H, s, ArH-3), 9.84 (1H, s, NH); MS m/z (%) 375.1 (M+Na, 100).
5-Chloro-N1-(4-cyanophenyl)-4-methoxycarbonyl-2-nitroanilines (9)
Yield 57%, starting with 250 mg (1 mmol) of 6 and 130 mg (1.1 mmol) of para-cyanoaniline to afford 189 mg of 9, yellow solid, mp 198–202 °C; 1H NMR δ ppm 3.95 (3H, s, CH3), 7.38 (2H, d, J = 8.4 Hz, ArH-2′,6′), 7.42 (1H, s, ArH-6), 7.77 (2H, d, J = 8.4 Hz, ArH-3′,5′), 8.92 (1H, s, ArH-3), 9.76 (1H, s, NH); MS m/z (%) 354.3 (M+Na, 100).
General Coupling Reaction of Diphenylether (under microwave irradiation) for 10a, 10c, 11c, and 11d
A mixture of 4-substituted N1-(4-cyanophenyl)-5-chloro-2-nitroaniline (1 equiv, 7 or 9) and para-substituted 2,6-dimethylphenol (1.2 equiv) in DMF (3 mL) in the presence of anhydrous potassium carbonate (2 equiv) was irradiated under microwave conditions with stirring at 190 °C for 15 min. The mixture was poured into ice-water, adjusted to neutral pH with aq HCl (5%), and extracted with EtOAc three times. After removal of organic solvent under reduced pressure, crude product was purified by PTLC or flash column chromatography (gradual eluant: petroleum ether/CH2Cl2).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-formylphenoxy)-4-trifluoromethyl-2-nitroaniline (10a)
Starting with 171 mg (0.5 mmol) of 7 and 90 mg (0.6 mmol) of 2,6-dimethyl-para-formylphenol to afford 182 mg of 10a, yield 80%, yellow solid, mp 192–194 °C; 1H NMR δ ppm 2.21 (6H, s, 2 × CH3), 6.24 (1H, s, ArH-6), 7.08 (2H, d, J = 8.4 Hz, ArH-2′,6′), 7.51 (2H, d, J = 8.4 Hz, ArH-3′,5′), 7.67 (2H, s, ArH-3″,5″), 8.68 (1H, s, ArH-3), 9.92 (1H, s, NH), 9.97 (1H, s, CHO); MS m/z (%) 456.4 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-formylphenoxy)-4-methoxycarbonyl-2-nitroaniline (10c)
Starting with 166 mg (0.5 mmol) of 9 and 90 mg (0.6 mmol) of 2,6-dimethyl-para-formylphenol to afford 150 mg of 10c, yield 67%, yellow solid, mp 220–222 °C; 1H NMR δ ppm 2.22 (6H, s, 2 × CH3), 3.97 (3H, s, OCH3), 6.23 (1H, s, ArH-6), 7.06 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.48 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.65 (2H, s, ArH-3″,5″), 9.02 (1H, s, ArH-3), 9.88 (1H, s, NH), 9.96 (1H, s, CHO); MS m/z (%) 446.1 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyano-2″,6″-dimethylphenoxy)-4-trifluoromethyl-2-nitroaniline (11c)
Starting with 171 mg (0.5 mmol) of 7 and 88 mg (0.6 mmol) of para-cyano-2,6-dimethylphenol to afford 150 mg of 11c, yield 66%, yellow solid, mp 182–184 °C; 1H NMR δ ppm 2.17 (6H, s, 2 × CH3), 6.21 (1H, s, ArH-6), 7.09 (2H, d, J = 8.4 Hz, ArH-2′,6′), 7.44 (2H, s, ArH-3″,5″), 7.56 (2H, d, J = 8.4 Hz, ArH-3′,5′), 8.67 (1H, s, ArH-3), 9.92 (1H, s, NH); MS m/z (%) 453.2 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-nitrophenoxy)-4-trifluoromethyl-2-nitroaniline (11d)
Starting with 171 mg (0.5 mmol) of 7 and 100 mg (0.6 mmol) of 2,6-dimethyl-para-nitrophenol to afford 215 mg of 11d, yield 91%, yellow solid, mp 240–242 °C; 1H NMR δ ppm 2.24 (6H, s, 2 × CH3), 6.22 (1H, s, ArH-6), 7.10 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.55 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.02 (2H, s, ArH-3″,5″), 8.69 (1H, s, ArH-3), 9.92 (1H, s, NH); MS m/z (%) 473.3 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-formylphenoxy)-4-sulfamoyl-2-nitroaniline (10b)
A mixture of 353 mg (1 mmol) of 8 and 180 mg (1.2 mmol) of 2,6-dimethyl-para-formylphenol in DMF in the presence of anhydrous potassium carbonate (2 equiv) was heated at 130 °C for 5 h. The mixture was poured into ice-water, adjusted to neutral pH with aq HCl (5%), and extracted with EtOAc three times. After removal of organic solvent under reduced pressure, crude product was purified by PTLC or medium-pressure column (gradual eluant: petroleum ether/CH2Cl2) to afford 310 mg of 10b, yield 67%, yellow solid, mp 178–180 °C; 1H NMR (DMSO-d6) δ ppm 2.26 (6H, s, 2 × CH3), 6.35 (1H, s, ArH-6), 7.49 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.56 (2H, s, ArH-3″,5″), 7.82 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.24 (1H, s, ArH-3), 9.76 (1H, s, NH), 9.88 (1H, s, -CHO); MS m/z (%) 465.2 (M−1, 100).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-(3-oxobut-1-enyl)phenoxy)-4-trifluoromethyl-2-nitroaniline (11a)
To a solution of 10a (228 mg, 0.5 mmol) in acetone (10 mL) and MeOH (3 mL) was added 10% aq NaOH (2 mL) slowly at 0 °C (ice-water bath) with stirring for about 10 min and then for additional 1 h at room temperature. After the reaction was completed, the mixture was poured into ice-water, and the pH adjusted with 5% aq. HCl to neutral. The solid crude product was collected, washed with water, and purified on a flash silica gel column (eluent: CH2Cl2/MeOH = 100/1) to afford 138 mg of 11a, 56% yield, yellow solid, mp 190–192 °C. 1H NMR δ ppm 2.14 (6H, s, 2 × CH3), 2.42 (3H, s, COCH3), 6.27 (1H, s, ArH-6), 6.68 (1H, s, J = 16.4 Hz, =CH), 7.09 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.30 (2H, s, ArH-3″,5″), 7.43 (1H, s, J = 16.4 Hz, CH=), 7.51 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.66 (1H, s, ArH-3), 9.89 (1H, s, NH); MS m/z (%) 496.5 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-hydroxymethylphenoxy)-4-hydroxymethyl-2-nitroaniline (11e)
To a solution of LiBH4 (5 mmol) in THF (5 mL) was added dropwise 223 mg (0.5 mmol) of 10c in THF (5 mL) at r.t. The mixture was stirred for 15 min and then 1–2 mL MeOH was added, and stirring continued for 1 h. The mixture was poured into ice-water, pH was adjusted to 2–3 with 5% aq HCl, and the resulting solution extracted with EtOAc three times. After removal of organic solvent in vacuo, crude product was purified by PTLC or medium-pressure column (eluent: PE/EtOAc = 3/1) to afford 178 mg of 11e, 85% yield, yellow solid, mp 216–218 °C; 1H NMR δ ppm 2.13 (6H, s, 2 × CH3), 4.66 and 4.88 (each 2H, d, CH2O), 6.28 (1H, s, ArH-6), 7.03 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.12 (2H, s, ArH-3″,5″), 7.47 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.38 (1H, s, ArH-3), 9.70 (1H, s, NH); MS m/z (%) 442.4 (M+Na, 100).
General Preparation of 4-Substituted N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-2-nitroaniline (12a–12c)
To a solution of diethylcyanomethyl phosphonate [(EtO)2P(O)CH2CN, 266 mg, 1.5 mmol] in THF (15 mL) was added t-BuOK (168 mg, 1.5 mmol) at 0 °C (ice-water bath) with stirring for 30 min. The solution of an aldehyde (10a, 10b, or 10c, 1 mmol) in THF (15 mL) was added dropwise into the above mixture at rt and stirring continued for about 3 h until the reaction was completed. The mixture was poured into water and extracted with EtOAc three times. After removal of solvent under reduced pressure, the crude product was purified by a silica gel column using the medium-pressure system of CombiFlash companion (gradual eluant: MeOH / CH2Cl2, 0–4.5%).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-trifluoromethyl-2-nitroaniline (12a)
Starting with 455 mg (1 mmol) of 10a and 266 mg (1.5 mmol) of (EtO)2P(O)CH2CN to afford 416 mg of pure 12a, 87% yield, yellow solid, mp 248–250 °C; 1H NMR δ ppm 2.15 (6H, s, 2 × CH3), 5.85 (1H, d, J = 16.4 Hz, =CH), 6.27 (1H, s, ArH-6), 7.10 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.21 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.4 Hz, CH=), 7.52 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.67 (1H, s, ArH-3), 9.91 (1H, s, NH); MS m/z (%) 501.1 (M+Na, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-sulfamoyl-2-nitroaniline (12b)
Starting with 10b (466 mg, 1 mmol) and 266 mg (1.5 mmol) of (EtO)2P(O)CH2CN to afford 218 mg of 12b, yield 45%, yellow solid, mp 198–200 °C; 1H NMR (DMSO-d6) δ ppm 2.16 (6H, s, 2 × CH3), 6.38 (1H, d, J = 16.4 Hz, =CH), 6.54 (1H, s, ArH-6), 7.32 (2H, s, ArH-3″,5″), 7.51 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.52 (1H, d, J = 16.4 Hz, =CHCN), 7.85 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.25 (1H, s, ArH-3), 9.79 (1H, s, NH); MS m/z (%) 488 (M−1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-methoxycarbonyl-2-nitroaniline (12c)
Starting with 445 mg (1 mmol) of 10c and 266 mg (1.5 mmol) of (EtO)2P(O)CH2CN to afford 412 mg of 12c, 88% yield, yellow solid, mp 260–262 °C; 1H NMR δ ppm 2.16 (6H, s, 2 × CH3), 3.96 (3H, s, OCH3), 5.84 (1H, d, J = 16.4 Hz, =CH), 6.26 (1H, s, ArH-6), 7.08 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.20 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.4 Hz, CH=), 7.50 (2H, d, J = 8.8 Hz, ArH-3′,5′), 9.00 (1H, s, ArH-3), 9.87 (1H, s, NH); MS m/z (%) 469.4 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-carboxy-2-nitroaniline (12d)
To a solution of 12c (234 mg, 0.5 mmol) in THF (15 mL) and MeOH (10 mL) was slowly added NaOH aqueous solution (80 mg NaOH in 4 mL of water) at rt with stirring for 4 h. The mixture was poured into ice-water, pH adjusted to 2–3 with 5% aq HCl, and solid was collected and washed with cold water to neutral. The crude product was purified on a silica gel column using a CombiFlash system (gradual eluant: MeOH/CH2Cl2, 0–4.5%) to afford 204 mg of pure 12d in 90% yield, yellow solid, mp 262–265 °C; 1H NMR (DMSO-d6) δ ppm 2.06 (6H, s, 2 × CH3), 5.95 (1H, s, ArH-6), 6.44 (1H, d, J = 16.4 Hz, =CH), 7.22 ( 2H, d, J = 8.8 Hz, ArH-2′,6′), 7.47 (2H, s, ArH-3″,5″), 7.58 (1H, d, J = 16.4 Hz, CH=), 7.65 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.72 (1H, s, ArH-3), 9.79(1H, s, NH), 13.21 (1H, s, COOH); MS m/z (%) 453.4 (M−1, 100).
Preparation of Amidated Compounds (12e–g)
To a solution of 12d (0.5 mmol) in CH2Cl2 (5 mL) was added dropwise SOCl2 (0.5 mL) at rt and then heated at reflux for 3 h. After removal of solvent, the obtained brown solid was dissolved in dry THF (5 mL), followed by addition of an amine aqueous solution (1 mL, excess, NH3, methylamine, or hydrazine) with stirring for an additional 30 min at 0 °C (ice-bath) until the reaction was completed. The mixture was poured into ice-water and pH was adjusted to neutral with 5% aq. HCl. The solid was collected, washed with cold water, and dried. The crude product was purified on a flash silica gel column (gradual eluant: MeOH/CH2Cl2, 0–4.5%) using a CombiFlash syetem.
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-carbamoyl-2-nitroaniline (12e)
Starting with 227 mg of 12d (0.5 mmol) in CH2Cl2 (5 mL), SOCl2 (0.5 mL) was added dropwise to obtain acyl chloride (brown solid), which was dissolved in dry THF (5 mL) and added to aqueous ammonia solution (1 mL) with stirring for 30 min at 0 °C (ice-bath) until the reaction was completed. After general work-up as described above, 198 mg of 12e was obtained in 87% yield, yellow solid, mp 278–280 °C; 1H NMR δ ppm 2.16 (6H, s, 2 × CH3), 5.88 (1H, d, J = 16.4 Hz, =CH), 6.29 (1H, s, ArH-6), 7.08 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.24 (2H, s, ArH-3″,5″), 7.33 (1H, d, J = 16.4 Hz, CH=), 7.50 (2H, d, J = 8.8 Hz, ArH-3′,5′), 9.30 (1H, s, ArH-3), 9.82 (1H, s, NH); MS m/z (%) 454.3 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-methylcarbamoyl-2-nitroaniline (12f)
Starting with 12d (227 mg, 0.5 mmol) with SOCl2 (0.5 mL), followed by dry THF (5 mL) and aqueous methylamine solution (25–30%, 1 mL) at ice-bath with stirring for 30 min. After the same work-up as above, 140 mg of pure 12f was obtained in 60% yield, yellow solid, mp 218–220 °C; 1H NMR δ ppm 2.17 (6H, s, 2 × CH3), 3.08 (3H, d, CH3), 5.88 (1H, d, J = 16.4Hz, =CH), 6.28 (1H, s, ArH-6), 7.07 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.25 (2H, s, ArH-3″,5″), 7.34 (1H, d, J = 16.4Hz, CH=), 7.49 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.50 (1H, NMe), 9.29 (1H, s, ArH-3), 9.78 (1H, s, NH); MS m/z (%) 468.3 (M+1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-hydrazide-2-nitroaniline (12g)
Starting with 12d (140 mg, 0.30 mmol) with SOCl2 (0.50 mL), followed by dry THF (3 mL) and dropped slowly into hydrazine hydrate aqueous solution (85%, 1 mL) at rt with stirring for 1 h. After the same work-up as above, crude product was purified on a flash silica gel column (gradual eluant: MeOH/CH2Cl2, 0–4.5%) to afford 116 mg of pure 12g in 80% yield, yellow solid; 1H NMR δ ppm 2.13 (6H, s, 2×CH3) 5.82 (1H, d, J = 16.4 Hz, =CHCN), 6.30 (1H, s, ArH-6), 6.98 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.21 (2H, s, ArH-3″, 5″), 7.33 (1H, d, J = 16.4 Hz, =CHAr), 7.53 (2H, d, J = 8.8 Hz, ArH-3′,5′), 9.20 (H, s, ArH-3), 9.81 (1H, s, NH); MS m/z (%) 469 (M + 1, 100).
N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-hydroxymethyl-2-nitroaniline (12h)
The preparation was the same as that of 11e. Starting with 234 mg (0.5 mmol) of 12c, reduction with LiBH4 afforded 100 mg of pure 12h in 91% yield, yellow solid, mp 232–234 °C; 1H NMR δ ppm 2.15 (6H, s, 2 × CH3), 4.89 (2H, s, OCH2), 5.85 (1H, d, J = 16.4 Hz, =CH), 6.27 (1H, s, ArH-6), 7.05 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.21 (2H, s, ArH-3″,5″), 7.28 (1H, d, J = 16.4 Hz, CH=), 7.47 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.41 (1H, s, ArH-3), 9.70 (1H, s, NH); MS m/z (%) 439.3 (M−1, 100).
General Preparation of 4-Substituted 1,5-Diarylbenzene-1,2-diamines (13a, 14a–f, and 14h)
To a solution of a diaryl-nitrobenzene (1 equiv) in THF and water (30 mL, v/v 1:1) was added aqueous ammonia solution (25%) and sodium hydrosulfite (10 equiv) successively. The mixture was stirred at rt for 2 h monitored by TLC (CH2Cl2/MeOH 60:1) until reaction was completed. The mixture was poured into ice-water and extracted with EtOAc several times. After removal of organic solvent under reduced pressure, crude product was purified on a flash column chromatograph (gradual eluant: MeOH/CH2Cl2, 0–4.5%) with a CombiFlash separation system to obtain pure target compound.
(E)-N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-(3-oxobut-1-enyl)phenoxy)-4-trifluoromethylbenzene-1,2-diamine (13a)
Starting with 495 mg (1 mmol) of 11a to afford 316 mg of 13a, yield 68%, brown solid, mp > 300 °C; 1H NMR δ ppm 2.14 (6H, s, 2 × CH3), 2.37 (3H, s, COCH3), 5.71 (1H, s, NH), 6.21 (1H, s, ArH-6), 6.64 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.65 (1H, s, J = 16.4 Hz, =CH), 7.16 (1H, s, ArH-3), 7.27 (2H, s, ArH-3″,5″), 7.42 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.43 (1H, s, J = 16.4Hz, CH=); MS m/z (%) 466.2 (M+1, 100); HPLC purity 97.9%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-trifluoromethylbenzene-1,2-diamine (14a)
Starting with 478 mg (1 mmol) of 12a to afford 112 mg of 14a, yield 25%, white solid, mp 224–226 °C; 1H NMR δ ppm 2.15 (6H, s, 2 × CH3), 5.68 (1H, s, NH), 5.80 (1H, d, J = 16.8 Hz, =CH), 6.18 (1H, s, ArH-6), 6.63 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.16 (1H, s, ArH-3), 7.18 (2H, s, ArH-3″,5″), 7.27 (1H, d, J = 16.8 Hz, CH=), 7.43 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 449 (M+1, 100); HPLC purity 97.3%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-sulfamoylbenzene-1,2-diamine (14b)
Starting with 489 mg (1 mmol) of 12b to afford 184 mg of 14b in 40% yield, brown solid, mp 135–137 °C; 1H NMR (DMSO-d6) δ ppm 2.14 (6H, s, 2 × CH3), 4.59 (2H, s, NH2), 6.37 (1H, d, J = 16.8 Hz, =CH), 6.78 (1H, s, ArH-6), 6.93 (1H, d, J = 16.8 Hz, CH=), 6.95 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.30 (2H, s, ArH-3″,5″), 7.61 (2H, d, J = 8.8 Hz, ArH-3′,5′), 8.20 (1H, s, ArH-3); MS m/z (%) 460.3 (M+1,100); HPLC purity 95.8%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-methoxycarbonylbenzene-1,2-diamine (14c)
Starting with 468 mg (1 mmol) of 12c to afford 280 mg of 14c, yield 64%, brown solid, mp 240–242 °C. 1H NMR δ ppm 2.16 (6H, s, 2 × CH3), 3.93 (3H, s, OCH3), 5.78 (1H, s, NH), 5.80 (1H, d, J = 16.8 Hz, =CH), 6.20 (1H, s, ArH-6), 6.67 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.17 (2H, s, ArH-3″,5″), 7.31 (1H, d, J = 16.8 Hz, CH=), 7.42 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.45 (1H, s, ArH-3); MS m/z (%) 439.3 (M+1, 100); HPLC purity 96.6%.
(E)-4-Carboxy-N1-(4′-cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)benzene-1,2-diamine (14d)
Starting with 454 mg (1 mmol) of 12d to afford 148 mg of 14d, yield 35%, brown solid, mp 226–228 °C; 1H NMR δ ppm 2.19 (6H, s, 2 × CH3), 5.84 (1H, d, J = 16.8 Hz, =CH), 6.04 (1H, s, NH), 6.27 (1H, s, ArH-6), 6.75 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.22 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.8 Hz, CH=), 7.44 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.72 (1H, s, ArH-3); MS m/z (%) 423.2 (M−1, 100); HPLC purity 95.6%.
(E)-4-Carbamoyl-N1-(4′-cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)benzene-1,2-diamine (14e)
Starting with 453 mg (1 mmol) of 12e to afford 266 mg of 14e, yield 63%, white solid, mp 290–292 °C; 1H NMR (DMSO-d6) δ ppm 2.10 (6H, s, 2 × CH3), 4.76 (2H, s, NH2), 6.00 (1H, s, ArH-6), 6.39 (1H, d, J = 16.4 Hz, =CH), 6.63 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.47 (2H, s, ArH-3″,5″), 7.47 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.56 (1H, d, J = 16.4 Hz, CH=), 7.60 (1H, s, ArH-3), 8.20 (1H, s, NH); MS m/z (%) 424.2 (M+1, 100); HPLC purity 98.2%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-methylcarbamoylbenzene-1,2-diamine (14f)
Starting with 467 mg (1 mmol) of 12f to afford 135 mg of 14f, yield 31%, brown solid, mp 112–114 °C; 1H NMR δ ppm 2.16 (6H, s, 2 × CH3), 3.07 (3H, d, NCH3), 5.80 (1H, s, NH), 5.83 (1H, d, J = 16.8 Hz, =CH), 6.18 (1H, s, ArH-6), 6.65 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.21 (2H, s, ArH-3″,5″), 7.32 (1H, d, J = 16.8 Hz, CH=), 7.41 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.81 (1H, s, ArH-3); MS m/z (%) 438.4 (M+1, 100); HPLC purity 100.0%.
(E)-N1-(4′-Cyanophenyl)-5-(4″-cyanovinyl-2″,6″-dimethylphenoxy)-4-hydroxymethylbenzene-1,2-diamine (14h)
Starting with 440 mg (1 mmol) of 12h to afford 332 mg of 14h, yield 81%, brown solid, mp 186–188 °C. 1H NMR δ ppm 2.13 (6H, s, 2 × CH3), 4.87 (2H, s, ArCH2O), 5.50 (1H, s, NH), 5.79 (1H, d, J = 16.8 Hz, =CH), 6.03 (1H, s, ArH-6), 6.55 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.94 (1H, s, ArH-3), 7.17 (2H, s, ArH-3″,5″), 7.30 (1H, d, J = 16.8 Hz, CH=), 7.40 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 411.3 (M+1, 100); HPLC purity 99.9%.
General Procedure for Catalytic Hydrogenation to Prepare 4-Substituted 1,5-Diarylbenzene-1,2-diamines (13b–e and 15a–i)
A solution of a diaryl-nitrobenzene (1 equiv) in 30 mL of anhydrous EtOH in the presence of excess Pd/C (5%) was shaken with hydrogen gas under 30–40 p.s.i. until the hydrogen was no longer absorbed (ca. 2 h). The catalyst was filtered from the solution and washed with EtOH several times (ca. 20 mL). After the solvent was removed under reduced pressure, the residue was purified by flash column chromatography (gradual eluant: MeOH/CH2Cl2, 0–4.5%) to obtain pure desired product.
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-(3-oxobutyl)phenoxy)-4-trifluoromethylbenzene-1,2-diamine (13b)
Starting with 495 mg (1 mmol) of 11a to afford 234 mg of 13b, yield 50%, brown solid, mp 94–96 °C, 1H NMR δ ppm 2.07 (6H, s, 2 × CH3), 2.14 (3H, s, COCH3), 2.73 (2H, t, J = 7.2 Hz, CH2CO), 2.80 (2H, t, J = 7.2 Hz, ArCH2), 6.21 (1H, s, ArH-6), 6.64 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.88 (2H, s, ArH-3″,5″), 7.14 (1H, s, ArH-3), 7.43 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 468.4 (M+1, 100); HPLC purity 97.4 %.
5-(4″-Cyano-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-trifluoromethylbenzene-1,2-diamine (13c)
Starting with 452 mg (1 mmol) of 11c and to afford 329 mg of 13c, yield 78%, white solid, mp 240–242 °C; 1H NMR (DMSO-d6) δ ppm 2.09 (6H, s, 2 × CH3), 4.93 (2H, s, NH2), 5.76 (1H, s, NH) 6.06 (1H, s, ArH-6), 6.65 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.18 (1H, s, ArH-3), 7.50 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.69 (2H, s, ArH-3″,5″); MS m/z (%) 423 (M+1, 100); HPLC purity 99.9 %.
5-(4″-Amino-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-trifluoromethylbenzene-1,2-diamine (13d)
Starting with 472 mg (1 mmol) of 11d to afford 316 mg of 13c, yield 77%, brown solid, mp 194–196 °C; 1H NMR δ ppm 2.02 (6H, s, 2 × CH3), 5.71 (1H, s, NH), 6.30 (1H, s, ArH-6), 6.39 (2H, s, ArH-3″,5″), 6.65 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.12 (1H, s, ArH-3), 7.43 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 413.4 (M+1, 100); HPLC purity 96.2 %.
N1-(4′-Cyanophenyl)-5-(2″,6″-dimethyl-4″-hydroxymethylphenoxy)-4-hydroxymethylbenzene-1,2-diamine (13e)
Starting with 419 mg (1 mmol) of 11e to afford 237 mg of 13e, yield 61%, brown solid, mp 190–192 °C; 1H NMR δ ppm 2.12 (6H, s, 2 × CH3), 3.52 (2H, s, NH2), 4.63 and 4.87 (each 2H, s, CH2O), 5.48 (1H, s, NH), 6.06 (1H, s, ArH-6), 6.54 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.92 (1H, s, ArH-3), 7.08 (2H, s, ArH-3″,5″), 7.39 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 390.3 (M+1, 100); HPLC purity 99.4 %.
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-trifluoromethylbenzene-1,2-diamine (15a)
Starting with 478 mg (1 mmol) of 12a to afford 140 mg of 15a, yield 31%, brown solid, mp 172–174 °C; 1H NMR δppm 2.11 (6H, s, 2 × CH3), 2.61 (2H, t, J = 7.2 Hz, CH2CN), 2.87 (2H, t, J = 7.2 Hz, ArCH2), 5.72 (1H, s, NH), 6.20 (1H, s, ArH-6), 6.64 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.94 (2H, s, ArH-3″,5″), 7.14 (1H, s, ArH-3), 7.44 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 451.1 (M+1, 100); HPLC purity 99.7 %.
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-sulfamoylbenzene-1,2-diamine (15b)
Starting with 489 mg (1 mmol) of 12b to afford 263 mg of 15b, yield 57%, brown solid, mp 140–142 °C; 1H NMR δ ppm 2.13 (6H, s, 2 × CH3), 2.64 (2H, t, J = 6.8 Hz, CH2), 2.86 (2H, t, J = 6.8 Hz, ArCH2), 6.14 (1H, s, NH), 6.33 (1H, s, ArH-6), 6.88 (1H, s, ArH-3), 6.93 (2H, s, ArH-3″,5″), 7.06 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.57 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 460.2 (M−1, 100); HPLC purity 100.0%
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-methoxycarbonylbenzene-1,2-diamine (15c)
Starting with 468 mg (1 mmol) of 12c to afford 207 mg of 15c, yield 47%, white solid, mp 236–238 °C; 1H NMR δ ppm 2.12 (6H, s, 2 × CH3), 2.61 (2H, t, J = 7.2 Hz, CH2), 2.87 (2H, t, J = 7.2 Hz,, ArCH2), 3.93 (3H, s, OCH3), 5.90 (1H, s, NH), 6.23 (1H, s, ArH-6), 6.69 (2H, d, J = 9.2 Hz, ArH-2′,6′), 6.93 (2H, s, ArH-3″,5″), 7.43 (2H, d, J = 9.2 Hz, ArH-3′,5′), 7.45 (1H, s, ArH-3); MS m/z (%) 441.5 (M+1, 15); HPLC purity 100.0%.
4-Carboxy-5-(4″-cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)benzene-1,2-diamine (15d)
Starting with 454 mg (1 mmol) of 12d to afford 141 mg of 15d, yield 33%, white solid, mp 234–236 °C; 1H NMR δ ppm 2.16 (6H, s, 2 × CH3), 2.64 (2H, t, J = 7.2 Hz, CH2CN), 2.91 (2H, t, J = 7.2 Hz, ArCH2), 6.08 (1H, s, NH), 6.31 (1H, s, ArH-6), 6.76 (2H, d, J = 8.8 Hz, ArH-2′,6′), 7.01 (2H, s, ArH-3″,5″), 7.46 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.73 (1H, s, ArH-3); MS m/z (%) 425.2 (M−1, 100); HPLC purity 94.8%.
4-Carbanoyl-N1-(4′-cyanophenyl)-5-(4″-cyanoethyl-2″,6″-dimethylphenoxy)benzene-1,2-diamine (15e)
Starting with 453 mg (1 mmol) of 12e to afford 378 mg of 15e, yield 84%, brown solid, mp 274–276 °C; 1H NMR δ ppm 2.14 (6H, s, 2 × CH3), 2.63 (2H, t, J = 7.2 Hz, CH2CN), 2.89 (2H, t, J = 7.2 Hz, ArCH2), 5.90 (1H, s, NH), 6.23 (1H, s, ArH-6), 6.70 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.99 (2H, s, ArH-3″,5″), 7.43 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.79 (1H, s, ArH-3), 5.93, 7.85 (CONH2); MS m/z (%) 426.4 (M+1, 100); HPLC purity 100.0%.
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-methylcarbanoylbenzene-1,2-diamine (15f)
Starting with 467 mg (1 mmol) of 12f to afford 289 mg of 15f, yield 65.9%, brown solid, mp 110–112 °C; 1H NMR δ ppm 2.10 (6H, s, 2 × CH3), 2.63 (2H, t, J = 7.2 Hz, CH2CN), 2.89 (2H, t, J = 7.2 Hz, ArCH2), 3.06 (3H, d, J = 4.8 Hz, NCH3), 5.85 (1H, s, NH), 6.21 (1H, s, ArH-6), 6.67 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.98 (2H, s, ArH-3″,5″), 7.42 (2H, d, J = 8.8 Hz, ArH-3′,5′), 7.82 (1H, s, ArH-3), 7.97 (1H, d, J = 4.8 Hz, CONH); MS m/z (%) 440.4 (M+1, 100); HPLC purity 96.5 %.
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-hydrazide-2-aminoaniline (15g)
Starting with 100 mg (0.21 mmol) of 12g to afford 70 mg of pure 15g, yield 75%, white solid; 1H NMR δ ppm 2.11 (6H, s, 2 × CH3), 2.61 (2H, t, J = 6.8 Hz, CH2CN), 2.86 (2H, t, J = 6.8 Hz, CH2Ar), 6.21 (1H, s, ArH-6), 6.30 (1H, s, NH), 6.48 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.98 (2H, s, ArH-3″, 5″), 7.00 (1H, s, ArH-3), 7.43 (1H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 441 (M + 1, 100).
5-(4″-Cyanoethyl-2″,6″-dimethylphenoxy)-N1-(4′-cyanophenyl)-4-hydroxymethylbenzene-1,2-diamine (15h)
Starting with 440 mg (1 mmol) of 12h to afford 264 mg of 15h, yield 64%, brown solid, mp 115–117 °C, 1H NMR δ ppm 2.10 (6H, s, 2 × CH3), 2.61 (2H, t, J = 7.2 Hz, CH2CN), 2.87 (2H, t, J = 7.2 Hz, ArCH2), 4.86 (2H, s, CH2O), 5.54 (1H, s, NH), 6.04 (1H, s, ArH-6), 6.55 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.91 (1H, s, ArH-3), 6.93 (2H, s, ArH-3″,5″), 7.40 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 413.4 (M+1, 100); HPLC purity 99.1 %.
5-(4″-Cyanoethyl-2″,6″-dimethyl)phenoxy-N1-(4′-cyanophenyl)benzene-1,2,4-triamine (15i)
Starting with 100 mg (0.22 mmol) of 12i, which was reported previously,10 to afford 60 mg of 15i, yield 70%, white solid, mp 118–20 °C, 1H NMR δ ppm 2.11 (6H, s, 2 × CH3), 2.61 (2H, t, J = 7.2 Hz, CH2CN), 2.86 (2H, t, J = 7.2 Hz, CH2Ar), 5.36 (1H, s, NH), 5.94 (1H, s, ArH-6), 6.30 (1H, s, ArH-3), 6.48 (2H, d, J = 8.8 Hz, ArH-2′,6′), 6.91 (2H, s, ArH-3″, 5″), 7.37 (2H, d, J = 8.8 Hz, ArH-3′,5′); MS m/z (%) 398 (M + 1, 100); HPLC purity 100.0 %.
(E)-1-(4-Cyanophenyl)-6-(4-(2-cyanovinyl)-2,6-dimethylphenoxy)-N-methyl-1H-benzo[d]imidazole-5-carboxamide (16a)
Hydrochloric acid (1N, 0.5 mL) was added to a solution of 14f (20 mg, 0.046 mmol) in triethyl orthoformate (0.1 mL, excess), diethyl ether, and DMF (2 mL) with stirring. The mixture was stirred at rt for 3 h under a nitrogen atmosphere, poured into water, adjusted to pH 6–7, and stirred for an additional 15 min. The white solid was collected and washed with water until neutral to yield 17 mg of 16a in 85% yield, mp 296–298 °C; 1H NMR δ ppm 2.18 (6H, s, 2×CH3), 3.12 (3H, d, J = 4.8 Hz, NCH3), 5.88 (1H, d, J = 16.8 Hz, =CHCN), 6.43 (1H, s, ArH-7), 7.27 (2H, s, ArH-3″, 5″), 7.36 (1H, d, J = 16.8 Hz, ArCH=), 7.42 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.80 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 8.09 (1H, s, ArH-4), 8.89 (1H, s, ArH-2). MS m/z (%): 448 (M+1,100). HPLC 96.2%.
6-(4-(2-Cyanoethyl)-2,6-dimethylphenoxy)-1-(4-cyanophenyl)-N-methyl-1H-benzo[d]imidazole-5-carboxamide (16b)
The same reaction and conditions as for the preparation of 16a. Starting with 15f (20 mg, 0.046 mmol) with triethyl orthoformate (0.1 mL, excess) to afford 16.5 mg of 16b, yield 81%, white solid, mp 300–302 °C; 1H NMR δ ppm 2.14 (6H, s, 2×CH3), 2.68 (2H, t, J = 6.8 Hz, CH2CN), 2.92 (2H, t, J = 6.8 Hz, ArCH2), 3.11 (3H, d, J = 4.8 Hz, NHCH3), 6.47 (1H, s, ArH-7), 7.05 (2H, s, ArH-3″, 5″), 7.40 (2H, d, J = 8.8 Hz, ArH-3′, 5′), 7.81 (2H, d, J = 8.8 Hz, ArH-2′, 6′), 8.09 (1H, s, ArH-4), 8.90 (1H, s, ArH-2); MS m/z (%) 450 (M+1,100); HPLC 100%.
Assessment of Inhibitory Activity on HIV-1 Replication in TZM-bl Cells
Inhibition of HIV-1 infection was measured as reduction in luciferase gene expression after a single round of virus infection of TZM-bl cells as described previously.17 Briefly, 200 TCID50 of virus (NL4-3) was used to infect TZM-bl cells in the presence of various concentrations of compounds. Two days after infection, the culture medium was removed from each well and 100 μL of Bright Glo reagent (Promega, Luis Obispo, CA) was added to the cells for measurement of luminescence using a Victor 2 luminometer. The effective concentration (EC50) against HIV-1 strains was defined as the concentration that caused a 50% reduction of luciferase activity (Relative Light Units) compared to virus control wells.
Assessment of In Vitro Cytotoxicity in TZM-bl Cells
The in vitro cytotoxicity of compounds on TZM-bl cells was measured using a colorimetric 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay.17 Briefly, 100 μL of the test compounds at graded concentrations were added to equal volumes of cells (5 × 105/mL) in wells of a 96-well plate. After incubation at 37 °C for 4 days, 50 μL of XTT solution (1 mg/mL) containing 0.02 μM of phenazine methosulphate (PMS) was added. After 4 h, the absorbance at 450 nm was measured with an ELISA reader. The CC50 (concentration for 50% cytotoxicity) values were calculated using the CalsuSyn computer program as described above.
Aqueous Solubility Determination
Solubility was measured separately at pH 7.4 and pH 2.0 by using an HPLC-UV method. Test compounds were initially dissolved in DMSO at 10 mg/mL. Ten microliters of this stock solution was spiked into either pH 7.4 phosphate buffer (1.0 mL) or 0.01 M HCl (approximately pH 2.0, 1 mL) with the final DMSO concentration being 1%. The mixture was stirred for 4 h at rt, and then concentrated at 3000 rpm for 10 min. The saturated supernatants were transferred to other vials for analysis by HPLC-UV. Each sample was performed in triplicate. For quantification, a model 1200 HPLC-UV (Agilent) system was used with an Agilent TC-C18 column (250 × 4.6 mm, 5 μm) and gradient elution of acetonitrile (ACN) in water, starting with 0% of ACN, which was linearly increased up to 70% over 10 min, then slowly increased up to 98% over 15 min. The flow rate was 1.0 mL/min and injection volume was 15 μL. Aqueous concentration was determined by comparison of the peak area of the saturated solution with a standard curve plotted peak area versus known concentrations, which were prepared by solutions of test compound in ACN at 50 μg/mL, 12.4 μg/mL, 3.125 μg/mL, 0.781μg/mL, and 0.195 μg/mL.
Log P Measurement
Using above same DMSO stock solution (10 mg/mL), 20 μL of this solution was added into n-octane (1 mL) and water (1 mL). The mixture was stirred at rt for 24 h and then stood overnight. Each solution (ca. 0.5 mL) was transferred from two phases respectively into other vials for HPLC analysis. The instrument and conditions were the same as those for solubility determination. Log P value was calculated by the peak area ratios in n-octane and in water.
Microsomal Stability Assay
Stock solutions of test compounds (1 mg/mL) were prepared by dissolving the pure compound in DMSO and stored at 4 °C. Before assay, the stock solution was diluted with ACN to 0.1 mM concentration. For measurement of metabolic stability, all compounds were brought to a final concentration of 1 μM with 0.1 M potassium phosphate buffer at pH 7.4, which contained 0.1 mg/mL human liver microsomes and 5 mM MgCl2. The incubation volumes were 300 μL, and reaction temperature was 37 °C. Reactions were started by adding 60 μL of NADPH (final concentration of 1.0 mM) and quenched by adding 600 μL of ice-cold ACN to stop the reaction at 5, 15, 30, and 60 min time points. Samples at 0 min time point were prepared by adding 600 μL ice-cold ACN first, followed by 60 μL NADPH. Incubations of all samples were conducted in duplicate. After quenching, all samples were centrifuged at 12,000 rpm for 5 min at 0 °C. The supernatant was collected, and 20 μL of the supernatant was directly injected onto a Shimadzu LC-MS-2010 system with an electrospray ionization source (ESI) for further analysis. The following controls were also conducted: 1) positive control incubation containing liver microsomes, NADPH, and reference compound propranolol; 2) negative control incubation omitting NADPH; and 3) baseline control containing only liver microsomes and NADPH. The peak heights of test compounds at different time points were converted to percentage of remaining, and the peak height values at initial time (0 min) served as 100%. The slope of the linear regression from log percentage remaining versus incubation time relationships (−k) was used to calculate in vitro half-life (t1/2) value by the formula of in vitro t1/2 = 0.693/k, regarded as first-order kinetics. Conversion to in vitro CLint (in units of ml/min/mg protein) was calculated by the formula15: CLint = (0.693/in vitro t1/2) × (ml incubation/mg microsomes). The HPLC-MS analysis was carried out on Shimadzu LCMS-2010 with an electrospray ionization source (ESI). An Alltima C18 column (5 μm, 150 mm × 2.1 mm) was used for HPLC with a gradient elution at a flow rate of 0.3 mL/min. The elution condition was ACN (B) in water (A) at 30% for 0–2 min, 85% for 2–6 min, 100% for 6–9 min, and 30% for 9–12 min. The MS conditions were optimized to detector voltage +1.6 kV, acquisition mode selected ion monitoring (SIM) of the appropriate molecular weights of the testing compounds. The CDL and heat block temperature was 200 °C and neutralizing gas flow was 1.5 L/min. Samples were injected by auto-sampler. Electrospray ionization was operated in the positive and negative mode.
Supplementary Material
Table 2.
Structures and antiviral activity of diarylanilines 14–16 and 13ea
| |||||
|---|---|---|---|---|---|
| R1 | R2 | NL4-3 (wt)
|
|||
| EC50(nM)b | CC50(μM)c | SId | |||
| 14b | SO2NH2 | CH=CHCN | 93.7 ± 24.0 | >21.8 | >233 |
| 15b | SO2NH2 | CH2CH2CN | 380 ± 67 | 11.1 | 29.2 |
| 14c | COOCH3 | CH=CHCN | 2.74 ± 0.71 | >22.8 | >8321 |
| 15c | COOCH3 | CH2CH2CN | 4.32 ± 1.54 | >22.7 | >5255 |
| 14d | COOH | CH=CHCN | 230 ± 38 | 14.9 | 65 |
| 15d | COOH | CH2CH2CN | 96.0 ± 21 | 14.3 | 149 |
| 14e | CONH2 | CH=CHCN | 0.87 ± 0.28 | >23.6 | >27126 |
| 15e | CONH2 | CH2CH2CN | 1.39 ± 0.33 | >9.4 | >6762 |
| 14f | CONHCH3 | CH=CHCN | 5.72 ± 0.94 | >9.2 | >1608 |
| 15f | CONHCH3 | CH2CH2CN | 2.73 ± 0.41 | >9.1 | >3330 |
| 15g | CONHNH2 | CH2CH2CN | 19.1 ± 5.68 | >22.7 | >1188 |
| 14h | CH2OH | CH=CHCN | 0.53 ± 0.13 | 16.3 | 30755 |
| 15h | CH2OH | CH2CH2CN | 0.39 ± 0.11 | >23.8 | 61026 |
| 15i | NH2 | CH2CH2CN | 7.30 ± 1.26 | >25 | >3906 |
| 13e | CH2OH | CH2OH | 10.0 ± 2.06 | 15.9 | 1590 |
| 16a | CONHCH3 | CH=CHCN | >220 | >890 | __ |
| 16b | CONHCH3 | CH2CH2CN | >220 | >890 | __ |
| 3 | NO2 | CH=CHCN | 0.63 ± 0.13 | >24.4 | >38730 |
| 2 | 0.52 ± 0.14 | 19.4 | 37305 | ||
Against HIV-1 NL3-4 (wild-type) virus in TZM-bl cell lines.
Concentration of compound that causes 50% inhibition of viral replication, presented as mean ± standard deviation (SD), and performed at least in triplicate.
XTT assay was used to determine the CC50 value that causes cytotoxicity to 50% cells.
SI (Selectivity index) is the ratio of CC50/EC50.
Table 3.
Antiviral activity against wild-type and mutated viral strains of selected active DAANs
| ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | EC50 (nM)a (Fold Change)b
|
||||
| NL4-3 | RTMDRc | K101Ed | E138Kd | |||
| 14e | CONH2 | CH=CHCN | 0.87 | 1.8 ± 0.5 (2.1) | 8.3 ± 3.3 (9.5) | 9.0 ± 2.6 (10.3) |
| 14h | CH2OH | CH=CHCN | 0.53 | 0.4 ± 0.1 (0.7) | 3.4 ± 1.0 (6.4) | 3.9 ±0.9 (7.4) |
| 15c | COOCH3 | CH2CH2CN | 4.32 | 4.1 ± 0.7 (0.9) | 14 ± 4.3 (3.2) | 7.5 ± 2.7 (1.7) |
| 15f | CONHCH3 | CH2CH2CN | 2.73 | 8.0 ± 4.3 (2.9) | 14 ± 4.8 (5.1) | 10 ± 3.9 (3.7) |
| 15h | CH2OH | CH2CH2CN | 0.39 | 1.8 ± 0.5 (4.6) | 3.4 ± 1.3 (8.7) | 2.9 ± 1.2 (7.4) |
| 3 | NO2 | CH=CHCN | 0.63 | 0.9 ± 0.3 (1.4) | 11 ± 3.6 (17.5) | 12 ± 3.9 (19.0) |
| 2 | 0.52 | 0.5 ± 0.1 (0.9) | 5.7 ± 1.4(11.0) | 5.2 ± 1.6 (10.0) | ||
Experiments performed at least in triplicate and data presented as means± SD.
resistant fold change.
HIV-1 RTMDR (obtained from AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH), which contains mutations in RT amino acid residues L74, M41L, V106A, and T215Y, is resistant to AZT, ddI, nevrapine, and other noncucleoside RT inhibitors.
mutated amino acids in the HIV-RT non-nucleoside binding pocket (NNBP) confer resistance to 2.
Acknowledgments
This investigation was supported by grant 30930106 from the National Natural Science Fund of China (NSFC) awarded to L. Xie and U.S. NIH grants awarded to C. H. Chen (AI65310) and K. H. Lee (AI33066).
ABBREVIATIONS USED
- ACN
acetonitrile
- ART
antiretroviral therapy
- DAAN
diarylaniline
- DAPY
diarylpyrimidine
- HAART
highly active antiretroviral therapy
- LiBH4
lithium borohydride
- PMS
phenazine methosulphate
- PSA
polar surface area
- SPR
structure-property relationship
- PMS
XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide
Footnotes
The authors declare no competing financial interest.
References
- 1.Anthony S. Fauci AIDS: let science inform policy. Science. 2011;333(6038):13. doi: 10.1126/science.1209751. [DOI] [PubMed] [Google Scholar]
- 2.Shattock RJ, Warren M, McCormack S, Hankins C. Turning the tide against HIV. Science. 2011;333(6038):42–43. doi: 10.1126/science.1206399. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann GR, Cooper DA. Antiretroviral therapy of HIV-1 infection: established treatment strategies and new therapeutic options. Curr Opin Microbiol. 2000;3:508–514. doi: 10.1016/s1369-5274(00)00131-4. [DOI] [PubMed] [Google Scholar]
- 4.Vella S, Palmisano L. Antiretroviral therapy: state of the HAART. Antiviral Res. 2000;45:1–7. doi: 10.1016/s0166-3542(99)00068-6. [DOI] [PubMed] [Google Scholar]
- 5.Pecora Fulco P, McNicholl IR. Etravirine and rilpivirine: nonnucleoside reverse transcriptase inhibitors with activity against human immunodeficiency virus type 1 strains resistant to previous nonnucleoside agents. Pharmacotherapy. 2009;29 (3):281–294. doi: 10.1592/phco.29.3.281. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents. 2009;33:307–320. doi: 10.1016/j.ijantimicag.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 7.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, Andries K, Pauwels R, de Bethune MP. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J Virol. 2005;79:12773–12782. doi: 10.1128/JVI.79.20.12773-12782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin BJ, Jiang XK, Lu H, Tian XT, Barbault F, Huang L, Qian K, Chen CH, Huang R, Jiang S, Lee KH, Xie L. Diarylaniline derivatives as a distinct class of HIV-1 non-nucleoside reverse transcriptase inhibitors. J Med Chem. 2010;53:4906–4916. doi: 10.1021/jm1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian XT, Qin BJ, Wu ZY, Wang XF, Lu H, Morris-Natschke SL, Chen CH, Jiang S, Lee KH, Xie L. Design synthesis and evaluation of diarylpyridines diarylanilines as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. J Med Chem. 2010;53:8287–8297. doi: 10.1021/jm100738d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Corte BL. From 4, 5, 6, 7-Tetrahydro-5-methylimidazo[4, 5, 1-jk](1, 4) benzodiazepin- 2(1H)-one (TIBO) to Etravirine (TMC125): fifteen years of research on non-nucleoside inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2005;48:1689–1696. doi: 10.1021/jm040127p. [DOI] [PubMed] [Google Scholar]
- 12.Piersanti G, Giorgi L, Bartoccini F, Tarzia G, Minetti P, Gallo G, Giorgi F, Castorina M, Ghirardi O, Carminati P. Synthesis of benzo[1,2-d;3,4-d′] diimidazole and 1H-pyrazolo[4,3-b]pyridine as putative A2A receptor antagonists. Org Biomol Chem. 2007;5:2567–2571. doi: 10.1039/b707599e. [DOI] [PubMed] [Google Scholar]
- 13.Azijn H, Tirry I, Vingerhoets de Bethune J, MP, Kraus G, Boven K, Jochmans D, Craenenbroeck EV, Picchio G, Rimsky LT. TMC278 a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) active against wild-type NNRTI-resistant HIV-1. Antimicro Agents Chemother. 2010;54:718–727. doi: 10.1128/AAC.00986-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen PA, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune MP, Pauwels R, Das K, Clark AD, Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2, 6-dimethylphenyl]amino]-2-pyrimidinyl]amino]b enzonitrile (R278474, Rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]
- 15.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 16.Kerns EH, Di L. Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. Vol. 65 Academic Press; 2008. [Google Scholar]
- 17.Dang Z, Lai W, Qian K, Lee KH, Chen CH, Huang L. Betulinic acid derivatives as human immunodeficiency virus type 2 (HIV-2) inhibitors. J Med Chem. 2009;52:7887–7891. doi: 10.1021/jm9004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.