

Abstract
Micropipette manipulation measurements quantified the pre-steady state binding kinetics between cell pairs mediated by Xenopus cleavage stage cadherin. The time-dependence of the intercellular binding probability exhibits a fast forming, low probability binding state, which transitions to a slower forming, high probability state. The biphasic kinetics are independent of the cytoplasmic region, but the transition to the high probability state requires the third extracellular domain EC3. Deleting either EC3 or EC3–5, or substituting Trp2 for Ala reduces the binding curves to a simple, monophasic rise in binding probability to a limiting plateau, as predicted for a single site binding mechanism. The two stage cadherin binding process reported here directly parallels previous biophysical studies, and confirms that the cadherin ectodomain governs the initial intercellular adhesion dynamics.
The cadherin family of adhesion proteins mediates cell-cell interactions in all solid tissues (1). These calcium-dependent cell surface glycoproteins are critical for morphogenesis and for directing the segregation of cells into distinct tissues during development. In addition to their mechanical role as adhesion molecules, they are also signaling proteins that influence cytoskeletal reorganization, cell migration, and proliferation through interactions with other cadherins and possibly with other cell surface receptors.
Classical cadherins are the most extensively studied of the cadherin superfamily. The proteins comprise an extracellular region, and single-pass transmembrane domain, and a cytoplasmic domain (2). The extracellular region embeds the adhesive and selectivity functions of the protein. It folds into five structurally homologous extracellular (EC)4 domains numbered 1–5 from the N-terminal domain (3). The cytoplasmic domain mediates signaling through interactions with catenins (1).
Several approaches have been used to investigate cadherin recognition, binding, and signal transduction. Sequence exchange and cell aggregation studies mapped the specificity-determining region to the first extracellular domain EC1 (4). For this reason, this domain has been the focus of the majority of mechanistic studies of cadherin adhesion and binding specificity. In the crystal structure of the soluble N-terminal domain (EC1) of neural cadherin, the Trp2 (W2) residue was docked in a hydrophobic pocket of the adjacent EC1 domain (5). This reciprocal Trp2 exchange is referred to as a “strand dimer.” The structure of the ectodomain of Xenopus cleavage stage cadherin (C-cadherin) similarly exhibited this Trp2 exchange, but between anti-parallel EC1 domains (3). Electron tomography images of desmosomal cadherins in mouse epidermis also suggested that similar interactions form in tissue, although the images contain a wide variety of other configurations and possible interactions (6). Studies showing that W2A and W2G mutations eliminate cell adhesion, also suggest that the docked Trp2 side chain forms the sole adhesive interface (7, 8).
Other biophysical measurements, however, identified additional cadherin bonds, which involve other regions of the cadherin ectodomain than EC1. Surface force measurements first identified additional domain interactions (9, 10). In addition to adhesion between EC1 domains, Zhu et al. (11) mapped a second, stronger bond to the third EC domain (EC3). Other classical cadherins exhibit similar behavior (12). Cell adhesion studies using flow assays also implicated additional domains in adhesion (13). Similar to the surface force measurements, single bond rupture measurements demonstrated that the outer EC12 fragment forms two relatively weak bonds with fast dissociation kinetics. However, the full-length extracellular fragment EC1–5 forms two stronger bonds with slow dissociation kinetics, in addition to the weak, fast EC12 bonds (14, 15). The population of strong bonds also increases at the expense of the weak bonds with increasing protein contact times (15). These findings are not consistent with a simple, one site binding mechanism.
A recent proposal that Trp2 is an allosteric regulator of global cadherin adhesive activity may reconcile the multi-bond model with the Trp2 requirement for adhesion. Prakasam et al. (16) showed that the W2A mutation both abrogates the weak EC12-mediated bond and substantially attenuates the strong, EC3-dependent binding. Tsuji et al. (17) similarly reported weak residual binding between ectodomain dimers with the W2A mutation, and showed that this mutation disrupts lateral C-cadherin dimers on the cell surface. Prakasam et al. (16) postulated that W2 mediates the EC1 bond and allosterically regulates the activity of other domains, e.g. EC3 in the extracellular segment. Nevertheless, the translation of these force measurements at the molecular level to adhesion at the cell level has not yet been demonstrated.
Quantitative biophysical studies of cadherins have been based primarily on measurements of soluble, cadherin extracellular domains (3–5, 9, 10, 15, 16, 18–22). The underlying assumption that the truncated, soluble ectodomain accurately models the full-length, membrane-bound protein is untested. In the case of integrins, for example, allosteric coupling between the cytoplasmic and extracellular regions underlies outside-in and inside-out signaling (23–25). This is decoupled in soluble fragments, so that mutations are required to lock-in either the active or inactive integrin configuration (24, 26, 27). There is also evidence for inside-out signaling in the cadherin family. In Xenopus embryos, chemokines can activate cleavage stage cadherin, independent of changes in cadherin expression (28, 29). Src kinase activation correlates with the disruption of (cadherin-mediated) cell-cell junctions (30, 31). Apparent differences between adhesion by soluble ectodomains and membrane-bound cadherin also suggest some influence from the cytoplasmic domain (32). There is, however, no direct comparison of the binding properties of soluble, recombinant, and membrane-bound cadherin.
Only a few techniques allow quantitative comparisons between recombinant, soluble protein, and the cell surface forms. Comparing the trajectories of cells in flow chambers with those of protein-decorated beads could test this (33–35). One could also compare the bond rupture forces between soluble ectodomains (15) with those between soluble ectodomains and membrane-bound cadherin (32).
Such comparisons are possible with the micropipette manipulation technique, which quantifies binding between individual cell pairs bearing complementary receptors and ligands (36, 37). Live cells with surface-bound receptors and ligands are aspirated into opposite micropipettes, and brought into contact for a defined period. Typically, at least one of the cells is a red blood cell (RBC). Adhesion causes the RBC to distort during separation, and to then recoil at bond rupture. The RBC distortion gives the adhesion strength (37). Alternatively, the kinetic rates and two-dimensional affinities of the receptor-ligand bonds are quantified from the dependence of the binding probability on the cell contact time (36). The binding probability is the number of detected binding events divided by the total number of cell-cell contacts. Because these micropipette measurements are also used to quantify the binding kinetics between soluble protein fragments immobilized on RBCs, they enable quantitative comparisons of the properties of recombinant proteins with their membrane bound forms. Micropipette measurements have been used to study interactions between Fcγ receptors, selectins, integrins, and CD8 with their respective receptors (36, 38–44).
Here, we describe micropipette binding probability measurements of the pre-steady state kinetics of intercellular adhesion mediated by Xenopus cleavage stage cadherin. The binding probability curves exhibit complex kinetics characterized by a fast, low probability binding state and a slower forming, high probability binding state. This kinetic behavior contrasts with the simple, monophasic rise to a limiting binding probability, predicted by a single site binding model. Studies with isolated extracellular domains tested the impact of the cytoplasmic domain on the cadherin binding dynamics. In addition, by deleting either the EC3 or EC3–5 domains we identified protein segments required for this complex kinetic behavior. The biphasic kinetics exhibited by the full-length extracellular region directly parallels prior biophysical studies, and confirms that the nanomechanical properties of the recombinant cadherin ectodomain govern the initial dynamics of cell-cell contact formation.
EXPERIMENTAL PROCEDURES
Cell Lines, Proteins, and Plasmids
The pEE14 plasmid containing the full-length Xenopus C-cadherin cDNA, the pEE14 plasmid containing the cDNA encoding the hexahistidine-tagged C-cadherin extracellular domain with the W2A mutation, and the CHO cell line expressing the full-length C-cadherin with the W2A mutation were generously provided by B. Gumbiner (University of Virginia).
To generate cell lines expressing the W2A mutant of soluble CEC1–5-His6 and the full-length C-cadherin, CHO-K1 cells were stably transfected with the plasmids encoding the different C-cadherin constructs. Cells were transfected using Lipofectamine 2000, according to the manufacturer’s protocols (Invitrogen). The cells were cultured in Glasgow MEM medium supplemented with 10% dialyzed fetal calf serum, and selected with 25 μM methionine sulfoximine (MSX) as described (13). For the secreted proteins, the clone with highest protein production rate was identified by Western blot. The mouse monoclonal anti-His antibody (Upstate, Charlottesville, VA) and goat anti-mouse HRP conjugated antibody (Santa Cruz Biotechnology, Santa Cruz, CA) were used to detect the protein.
Prior to the micropipette experiment, the cells were detached from the flask with Hank’s Balanced Salt Solution (HBSS) (Invitrogen, Carlsbad, CA), with 0.01% trypsin and 1 mM CaCl2 for 15 min (45). The cells were incubated in phosphate buffered saline (PBS) with 5 mM EDTA and 1% bovine serum albumin (BSA) at 4 °C for at least 30 min, and then used in micropipette assays within 12 h. Prior to use, the EDTA was removed and replaced with 2 mM Ca2+.
The production and purification of the soluble C-cadherin extracellular domain deletion mutants EC1245-Fc and EC12-Fc, CEC1–5-His6, and W2A CEC1–5-His6 are described else-where (11, 13). The protein purity was assessed by SDS-PAGE. The aggregation activity of each protein was further characterized with bead aggregation assays (16).
Sample Configuration and Preparation in the Micropipette Manipulation Assays
Fig. 1A exemplifies the configuration of the cells in the micropipette manipulation experiment. In this case, the CHO cell on the left expresses the full-length, wild-type C-cadherin (C-CHO) (Fig. 1B). The RBC on the right is modified with anti-hexahistidine antibody, which in turn captured soluble hexahistidine-tagged C-cadherin ectodomain fragments (CEC1–5His6). In other cases, the capture antibody was anti-human IgG, and the C-cadherin fragment was C-terminally fused to a human Fc tag (Fig. 1B). In a third configuration (not shown), both cells were modified RBCs.
FIGURE 1.

A, configuration of the CHO cells and modified red blood cell in the micropipette measurements. The CHO cell (left) is aspirated into a 7-μm diameter glass pipette. The CHO expresses the full-length cadherin, including the cytoplasmic, transmembrane, and extracellular regions. The red blood cell (right) is aspirated into a 1.3-μm diameter pipette. The RBC, which is activated with CrCl3 binds capture antibody to the cell surface. The capture antibodies used were either monoclonal anti-His6 or monoclonal anti-human-Fc antibodies. These in turn capture the hexahistidine-tagged or Fc-tagged soluble cadherin ectodomains. B, scheme illustrating the relative protein configurations on the opposing cells. The full-length C-cadherin on the C-CHO (top) faces the recombinant cadherin ectodomain bound to the RBC surface (bottom). This illustrates the CEC1–5 Fc captured by the monoclonal anti-human Fc antibody, which is covalently bound to a glycoprotein on the RBC.
The monoclonal anti-hexahistidine (Upstate) or anti-human immunoglobin G Fc domain antibodies (Sigma-Aldrich), were used to capture cadherin on the RBC surface. The antibodies were attached chemically to red blood cell surfaces, using CrCl3 coupling chemistry (46, 47).
RBCs were obtained from the peripheral blood of a healthy donor, which was collected in sterile Vacutainers (BD Biosciences, San Jose, CA) containing EDTA, using a protocol approved by the Institutional Review Board of Georgia Institute of Technology (38). After centrifugation, the RBCs were collected and washed with 0.9% NaCl, and then resuspended in red blood cell storage solution EAS 45 (48). About 108 red blood cells (in 100 μl of EAS 45) were collected for the CrCl3 coupling reaction. The cells were resuspended in 250 μl saline (0.85% NaCl, w/v) containing 1 μg capture antibody. To these cells were added 250 μl of CrCl3 solution (in 0.02 M acetate and 0.85% NaCl, w/v). However, to vary the surface density of capture antibodies, we varied the final concentrations of CrCl3 between 10−6 and 10−4 % (w/v). After 5 min, the reaction was quenched with 500 μl of PBS containing 5 mM EDTA and 1% BSA. The thus modified RBCs were stored in EAS 45 buffer at 4 °C. The RBCs can be thus stored for up to 3 weeks without significant hemolysis.
To couple the different Xenopus C-cadherin extracellular fragments to the antibody-functionalized RBC surface, 1 μg of hexahistidine or human Fc-tagged C-cadherin fragments were incubated with 2 × 105 antibody-labeled RBCs at 4 °C for 1 h in 100 μl of phosphate-buffered saline, supplemented with 5 mM EDTA and 1% BSA. The cells were then pelleted and rinsed, in order to remove unbound cadherin. The thus modified cells were used for micropipette measurements.
Quantifying the Cadherin Surface Density on RBCs and CHO Cells
The cadherin density on the cells was determined by fluorescence-activated cell sorting (FACS). Calibrated fluorescein isothiocyanate (FITC)-labeled standard beads (Bangs Laboratories, Fishers, IN) were used as a reference. Monoclonal anti-C-cadherin antibody (1 μg) (Santa Cruz Biotechnology) was used to stain the C-cadherin-labeled cells, which were stored in phosphate-buffered saline with 0.5 mM EDTA and 1% BSA at 4 °C for 30 min. Cells were then stained with 1 μg of FITC-conjugated anti-goat IgG antibody in phosphate-buffered saline with 0.5 mM EDTA and 1% BSA at 4 °C for 30 min. The fluorescence intensity of the labeled cells was quantified with a BD LSR flow cytometer (BD Biosciences, San Jose, CA) as described (38). The fluorescence intensity for each population of the five standard beads was used to determine the fluorescence calibration curve. The protein density on the cells was determined by dividing the number of fluorophores on the cells by the estimated cell surface area. The fluorophore density on the cells was quantified by comparing the total fluorescence intensity on the cells against a calibration curve generated with the FITC standard beads.
Micropipette Measurement of Cell Adhesion Dynamics
Adhesion probability measurements with the micropipette were conducted as described previously (36). Both the C-cadherin-expressing CHO K1 cells (C-CHO) and modified RBCs were incubated in the sample chamber mounted on the microscope stage. The chamber was filled with L-15 medium (Invitrogen) containing 1.26 mM CaCl2, and supplemented with 1% BSA. Cells were aspirated into each of the two micropipettes, which were then used to position the cells adjacent to each other. The contact area was adjusted to ~3 μm2 (~2 μm diameter). One of the micropipette manipulators is interfaced to the computer via a piezoelectric actuator, which controls the cell contact time and changes the micropipette position by moving cells in and out of contact at a speed of 0.1 μm/s. The intercellular contact time is operator-programmed. The RBC deformations during cell contact and retraction are visualized in real time with a CCD camera and TV monitor. An adhesion event is identified from the elongation of the RBC during micropipette retraction. Adhesion is scored as 1, while a non-adhesion event is scored as 0. Each cell pair was subjected to 50–100 contact-retraction cycles, after which we determined the binding probability. The binding probability is defined as the number of binding events detected per total number of cell contact-retraction cycles attempted. For each cell-cell contact time, we measured three to five pairs of cells (n > 150). Data are represented as the mean ± S.D. from the mean.
Cell pairs consisted of (i) a transfected CHO cell and a cadherin-modified RBC or (ii) two cadherin-modified RBCs. The RBCs were modified with either hexahistidine-tagged or Human Fc-tagged soluble ectodomain fragments. The CHO cells expressed either the wild-type C-cadherin or the W2A mutant of the full-length C-cadherin. The three different control measurements consisted of (i) an antibody-coated RBC (no cadherin) interacting with a C-CHO. The second set of control measurements were done with 5 mM EDTA in the chamber medium, and, in the third control, we first incubated the cadherin-coated RBC with 4 μg of anti-C-cadherin, polyclonal blocking antibody for 30 min.
RESULTS
Adhesion between CHO Cells Expressing Wild-type C-Cadherin and RBCs Coated with CEC1–5-His6 Exhibits Biphasic Kinetics
Fig. 2A shows a representative time course of the binding probability obtained with the experimental configuration in Fig. 1. The cadherin densities on the CHO cell and RBC were ~7 and 3/μm2, respectively. Instead of a simple rise to a limiting plateau, as observed with other receptors (36, 38–44), this time course is biphasic, and exhibits two different, consecutive kinetic stages. Within the first 2 s, the background-corrected binding probability increases rapidly to the first plateau at ~0.2; that is, ~20% of cell-cell contacts result in adhesion. This low probability state is followed by a 2–5-s lag phase, and subsequent transition to a high probability binding state and second plateau. At these cadherin densities (Fig. 2A), the upper plateau is at 0.87 ± 0.05. There was no further change in the binding probability after 20 s.
FIGURE 2.
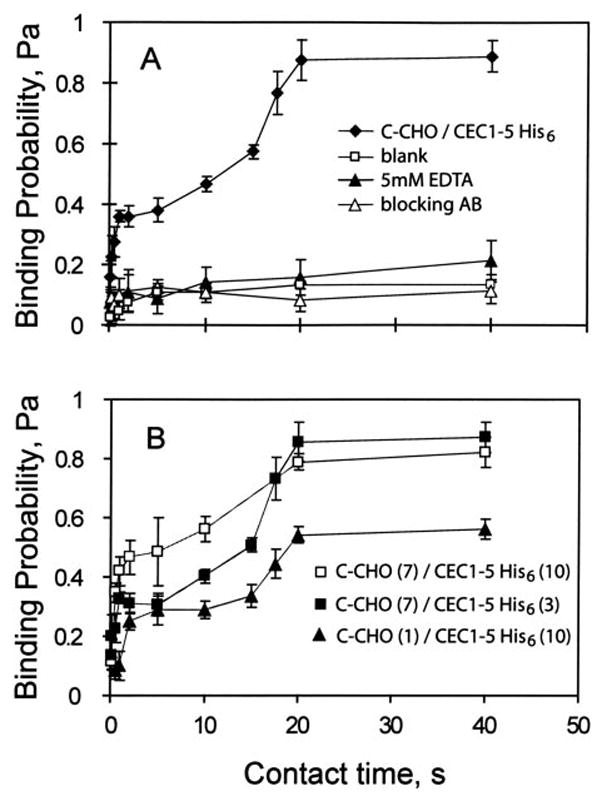
A, binding probability versus contact time between C-CHO and RBCs derivatized with CEC1–5-His6. The black diamonds show the time evolution of the adhesion probability between CHO cells expressing the full-length C-cadherin at 7 cadherin/μm2 and RBCs coated with CEC1–5 His6 at 3 cadherin/μm2. In the blank control (white squares), we measured the adhesion probability between C-CHO and the antibody-coated red blood cells without bound CEC1–5. The black triangles indicate measurements between C-CHO and CEC1–5 His6 RBCs in 5 mM EDTA. The white triangles show the data obtained in the presence of blocking antibody. B, binding probability versus contact time between C-CHO and CEC1–5 His6 RBCs at different cadherin densities. The background-corrected binding time courses were measured between C-CHO (7 cadherin/μm2) and CEC1–5His6 (10 cadherin/μm2) (white squares); C-CHO (7 cadherins/μm2) and CEC1–5His6 (3 cadherins/μm2) (black squares); and C-CHO (1 cadherin/μm2) and CEC1–5His6 (10 cadherin/μm2) (black triangles).
Several control measurements assessed the background adhesion. These included measurements between C-CHO and antibody-modified RBCs without the cadherin ectodomains (blank). A second control was conducted with 5 mM EDTA in the medium. In the third control, measurements were done in the presence of polyclonal anti-C-cadherin antibody, which blocks cadherin adhesion (blocking AB). These controls all reduce the binding probability to ~0.1 (Fig. 2A). The latter is attributed to nonspecific binding. In all subsequent binding time courses reported in this manuscript, the background was subtracted from the data using the following equation (49): Pc = (P − P0)/(1 − P0). Here Pc is the background-corrected binding probability, P is the raw binding probability, and P0 is the binding probability of the control.
The cadherin density does not alter the biphasic kinetic profiles, but it changes them quantitatively (Fig. 2B). Decreasing 2 the CEC1–5 His6 density from ~10 to ~3 cadherin/μm reduced the first plateau from 0.46 ± 0.06 to 0.31 ± 0.03, and may slightly increase the duration of the lag. Decreasing the C-cadherin density on the C-CHO from 7 to 1 cadherin/μm2 decreased the plateau of the high probability state from 0.79 ± 0.03 to 0.56 ± 0.03 (Fig. 2B). This 7-fold reduction in cadherin density also appears to increase the lag slightly.
Biphasic Cadherin Kinetics Do Not Require the Cytoplasmic Domain
To test the impact of the cytoplasmic domain on the two stage (biphasic) kinetics, we measured the binding time course between two RBCs coated with the His6-tagged extra-cellular domain of C-cadherin (CEC1–5-His6), which lacks both the transmembrane and cytoplasmic domains. The resulting binding probability versus time curve (Fig. 3A) is also bipha-sic. Binding increased rapidly in the first 2 s, at both cadherin densities examined. After a subsequent 2–5-s lag, the binding transitioned to the second, high probability state. At both CEC1–5 His6 densities, the binding curve plateaued within 20 s. These profiles are qualitatively similar to those measured between C-CHO and CEC1–5His6 (see Fig. 2), indicating that the ectodomains alone determine the biphasic kinetics within the first 40 s of contact.
FIGURE 3. Effect of cadherin dimerization on biphasic kinetics.

The time-dependent binding probabilities are reported for three different cell configurations. A, background-corrected data show the adhesion probability versus contact time between RBCs modified identically with CEC1–5 His6 at densities of ~4 (black squares) and ~5 (black circles) cadherins/μm2. B, measurements between identically modified CEC1–5 Fc RBCs at 39 cadherins/μm2 (white squares) and 12 cadherins/μm2 (white circles). C, measurements between C-CHO and CEC1–5 Fc RBCs. The cadherin densities on the C-CHO and RBCs were, respectively, 10 and 12 cadherins/μm2 (black squares) and 1 and 38 cadherins/μm2 (black circles). The data also show the blank (white squares) and EDTA control (white circles).
Studies suggest that cadherin dimerization enhances cadherin activity (50). To test the influence of lateral dimerization on the cadherin binding dynamics, we immobilized preformed cadherin dimers on the RBCs. The cadherin ectodomains are dimerized, by fusing them to the human IgG Fc domain, at the C terminus (CEC1–5-Fc). The dimers are then immobilized on the RBCs via anti-human-Fc monoclonal antibodies (CEC1–5 Fc RBC).
The time-dependent binding probability measured between two RBCs modified identically with CEC-Fc dimers is also biphasic (Fig. 3B), but the overall curve shifts slightly to the left, compared with data in Fig. 3A. The first, fast binding stage plateaus within 1s, and is followed by a shorter lag phase and a faster transition to the high probability binding state. Interactions between C-CHO and CEC1–5 Fc RBCs (Fig. 3C) also shift the curve left, relative to the curves in Fig. 2. In this case, the first step plateaus within 2 s, and the second stage plateaus within 15 s, which is intermediate between that measured with the CEC1–5-Fc RBC/CEC1–5-Fc RBC and C-CHO/CEC1–5 His6 RBC cell pairs.
Biphasic Cell Binding Kinetics Require Specific Cadherin Domains
Cadherin adhesion requires the EC1 domain (4, 7, 8, 51). However, increasing evidence shows that the full-length protein and EC3 are required for strong binding (11, 13–15). Here we investigated the impact of removing domains EC3 and EC3–5 on cadherin binding dynamics.
Fig. 4 shows the binding time course measured with the cadherin Fc-tagged domain deletion mutants CEC12-Fc and CEC1245-Fc bound to RBCs via the anti-human-Fc antibody. Removing EC3 substantially alters the kinetic profiles (Fig. 4). The time courses exhibit a single, monophasic rise to a low probability binding state. There is no lag or subsequent transition to a high probability binding state. This is not due to insufficient cadherin surface densities. The densities of the immobilized fragments required to achieve a binding probability of 0.2, as in the first, fast step measured with the full ectodomain (see Fig. 2), were 10 and 158 cadherin/μm2 for CEC1245-Fc and CEC12-Fc, respectively. Even at the highest cadherin density, there was little change in the binding probability after 20 s. Measurements at lower Fc-tagged ectodomain densities also exhibited simple kinetics, and the binding curve plateaued below 0.2 (not shown). The CEC12-Fc binding curve approaches the plateau at a similar rate as the biphasic CEC1–5-Fc binding curves approach the first plateau. This also suggests similar dissociation rates (36). In contrast, the CEC1245-Fc binding curve rises at a slower pace, suggesting slower kinetic rates.
FIGURE 4. Binding probability versus contact time between C-CHO and the domain deletion mutants CEC1245-Fc or CEC12-Fc on RBCs.

Background adhesion was subtracted from all data. The time-dependence of the adhesion probability was measured between C-CHO (7 cadherin/μm2) and CEC1245-Fc RBC (10 cadherin/μm2) (white squares) and between C-CHO (7) and CEC12-Fc RBCs (158 cadherin/μm2) (black circles).
Kinetic Analyses of Monophasic Binding Probability Curves
Binding mechanisms and kinetic rate constants of the receptor-ligand interactions can be determined from the dependence of the binding probability on the cell contact duration. For a simple one-step reaction (Equation 1),
| (Eq. 1) |
the analytical expression for the time-dependence of the adhesion probability between cells with a given contact area AC is in Equation 2 (36).
| (Eq. 2) |
Here mL and mR are the surface density of the receptor and the ligand, respectively. The apparent affinity constant is Ka = kf/kr where kr and kf are the reverse and forward binding rates, respectively. This model predicts a simple monophasic rise in the binding probability with time. With systems that exhibit such binding curves, as in Fig. 4, the parameters Ka and kr can be estimated from nonlinear least squares fits of Equation 2 to the probability time courses. The forward binding rate kf is determined from kf = Kakr.
We thus determined kinetic rates associated with the time courses in Fig. 4, by fitting the data using Equation 2 (solid lines, Fig. 4). The apparent contact area AC of ~3 μm2 was set at the beginning of each series of measurements. The best-fit parameters obtained from the CEC1245-Fc versus C-CHO time course were Ka = (3 ± 1) × 10−3 μM+2 and kr = 0.31 ± 0.06 s−1. The fitted parameters obtained with the CEC12-Fc fragment were Ka = (1.4 ± 0.5) × 10−4 μm−2 and kr = 0.9 ± 0.2 s−1. These results are summarized in Table 1.
TABLE 1.
Summary of thermodynamic and kinetic constants determined from fits of Equation 2 to adhesion time courses in Figs. 4 and 6
| mR | mL | kr | kf | Ka | R2 |
|---|---|---|---|---|---|
|
| |||||
| μm−2 | μm−2 | s−1 | ×10−4 s−1 μm−2 | ×104 μm−2 | |
| CCAD (7) | CEC1245 Fc (10) | 0.31 ± 0.06 | 7.2 ± 0.9 | 30 ± 9 | 0.91 |
| CCAD (7) | CEC12 Fc (155) | 0.9 ± 0.2 | 1.3 ± 0.2 | 1.4 ± 0.5 | 0.78 |
| CCAD (44) | CEC1–5 W2A His6 (556) | 0.08 ± 0.01 | 0.008 ± 0.001 | 0.09 ± 0.02 | 0.91 |
| CCAD W2A (24) | CEC1–5 Fc (452) | 0.10 ± 0.03 | 0.01 ± 0.002 | 0.12 ± 0.05 | 0.78 |
| CCAD W2A (24) | CEC1–5 Fc (328) | 0.07 ± 0.02 | 0.008 ± 0.002 | 0.10 ± 0.05 | 0.81 |
The contact area AC was approximately 3 μm2. The numbers in parentheses are the cadherin densities in #/μm2.
We considered the possibility that cadherin dimers could bind in a two step reaction, if cadherins on the C-CHO also bind as dimers. The reaction scheme in this case is Equation 3.
| (Eq. 3) |
We tested this mechanism by fitting the data (not shown) to the analytic expression for the time-dependent binding probability for this dimer-binding mechanism.5 This model did not improve the fits. An F-test indicated that the more complicated reaction scheme was not statistically justified. Calculated F values were 0.37 and 0.04 for fits to the CEC1245-Fc and CEC12-Fc data, respectively.
The mass action effects on the kinetics can be investigated, by rearranging Equation 2. We applied a log transformation to the plateau adhesion probabilities Pa(∞), and plotted −ln[1 − Pa(8)] versus the product of the cadherin densities expressed or coated on the two cells, mL × mR (Fig. 5) (36). In this case, −ln[1 − Pa(∞)] is proportional to mL × mR. The triangles in Fig. 5 are the thus normalized data measured between identically modified CEC1245 Fc RBCs. The CEC12-Fc data fall much below this line (not shown), suggesting a much lower apparent affinity.
FIGURE 5. Semilog plot of the probability of total adhesion (−Ln(1 − Pi)) versus the product of surface densities of cadherin on the two cells (mR × mL).

The black squares show the data from Fig. 2B normalized relative to the first plateau (P1) and the white squares are data normalized relative to the second plateau (P2). The black triangles show the data obtained with C-CHO and CEC1245-Fc RBCs normalized relative to the limiting plateau in Fig. 4.
We similarly normalized the data obtained with the full ectodomains by ln(1 − P1) and by ln(1 − P2), where P1 and P2 are the respective magnitudes of the first and second plateaus. These normalized data are also shown in Fig. 5, for comparison. It is important to note that the low probability binding data fall along a straight line (black squares), which is collinear with the CEC1245 data. The linear dependence on the cadherin density product suggests that the formation 5C. Zhu, unpublished manuscript. of the first binding state is proportional to the molecular densities on the respective cell surfaces. The slope of the line also represents the product of the affinity and contact area, ACKa. Based on the colinearity of these two normalized data sets (Fig. 5, black symbols), one could postulate (i) that the underlying kinetic mechanisms are the same and (ii) that the CEC1245-Fc interaction has an apparent affinity equal to that of the first, fast binding step of the wild-type protein.
The normalized data for the high probability state (white triangles) do not exhibit an obvious linear dependence. This may be due to the greater error in the measured adhesion probabilities above ~0.8, relative to the mid-ranged values.
W2A Mutants Exhibit Simple Binding Time Courses with Slow Kinetics
The second amino acid from the N terminus in the mature cadherin, Trp2 is reportedly critical for robust cell adhesion. These studies investigated the impact of the W2A mutation on cadherin kinetics. Binding probability curves were measured (i) between C-CHO and W2A CEC1–5-His6 RBCs and (ii) between CEC1–5-His6 RBCs and W2A C-CHO at two different expression levels (Fig. 6). In all three cases, the cells adhere, and the binding time course exhibits simple, monopha-sic kinetics. The binding probability increases with contact time to a limiting plateau, similar to previous studies (see Fig. 4). The profiles are similar qualitatively regardless of whether the mutation is in the soluble ectodomain fragment or in the full-length cadherin on the C-CHO.
FIGURE 6. Adhesion time courses between cadherin W2A mutants and wild-type cadherin.

Adhesion probabilities measured between C-CHO (44 cadherin/μm2) and CEC1–5-His6 W2A on the RBC (556 cadherin/μm2) (black triangles); C-CHO W2A (24 cadherin/μm2) and CEC1–5-Fc RBC (328 cadherin/ μm2) (black circles); and C-CHO W2A (24 cadherins/μm2) and CEC1–5-Fc RBC (453 cadherin/μm2) (white squares). The background binding probability was subtracted from all data. The solid lines through the data are the weighted nonlinear least squares fits to Equation 2. The best-fit parameters are summarized in Table 1.
The W2A mutant densities required to achieve comparable binding frequencies were substantially higher than required for CEC12-Fc and CEC1245-Fc fragments (Fig. 6 and Table 1). Nonlinear least squares fits of the data using Equation 2 (Fig. 6, solid lines) show that this can be explained by a lower apparent affinity than either the CEC12-Fc or the CEC1245-Fc fragments. The apparent affinities Ka of the W2A mutant for wild-type ectodomains, at ~1 × 10−5 μm2 are more than two orders of magnitude lower than that of CEC1245-Fc, and the dissociation rates are nearly 10-fold slower than that of CEC12-Fc. Again, the simple kinetic model (Equation 2) describes the data, and fitting to the dimer-binding model was not justified statistically. In all three data sets (Fig. 6), the best-fit parameters (Table 1) were, however, statistically similar to each other, regardless of whether the W2A mutation was on the soluble fragment or on the full-length protein on the CHO cell. The independence of the fitted parameters on the protein densities also confirms that equation 1 describes the binding mechanism.
DISCUSSION
The significant new findings of this study are that the initial, cadherin-mediated cell contact formation occurs by a complex kinetic process that requires the full ectodomain, and that this kinetic behavior is independent of the cytoplasmic domain. The cadherin binding curves exhibit a fast, low probability binding state and a second, high probability binding state, which forms more slowly and requires the full extracellular segment. The two kinetic states are separated by a lag, the duration of which depends in part on the cadherin surface density and on the state of cadherin oligomerization. Measurements between RBCs coated identically with the truncated ectodomains demonstrate that, within the first 40 s, the cell adhesion kinetics are independent of the cytoplasmic domain.
The cytoplasmic domain requirement for the formation of robust intercellular junctions is well established (52). Importantly, these micropipette assays do not contradict the previous studies because they do not quantify the adhesion strength. The probability time courses reflect the mechanism of cadherin binding and the associated kinetic rates. Our results show that the cytoplasmic domain does not alter the biphasic binding kinetics during the first steps of cell-cell contact formation. Over longer time scales (>40 s) the cytoplasmic domain is likely involved in adhesion strengthening via mechanisms such as cadherin clustering (2, 53).
These findings also raise interesting questions concerning the mechanisms and impact of inside out signaling on cadherin activity. There is increasing evidence that cadherin function is regulated by several different mechanisms, which include clustering on the cell surface, internalization, and association with cell surface proteins such as growth factor receptors (1, 28, 50, 53–55). The activation of small GTPases and Src regulates cadherin activity (31, 56, 57), and growth factors such as EGF alter cadherin activity, independent of changes in cell surface expression (1, 28, 54, 55). Any or all of these mechanisms could involve the cytoplasmic domain in some (as yet undefined) way. Our findings allow for possible perturbations, such as described above. Such perturbations could alter the binding kinetics and consequently the intrinsic cadherin activity. In this study, we did not perturb the CHO cells in ways that could alter the intrinsic cadherin activity. However, these micropipette assays demonstrate new possibilities for interrogating quantitatively the impact of such perturbations on cadherin function.
The qualitative features of the biphasic time courses require specific ectodomain segments. The first, low probability state requires EC12. Domain deletion mutants containing EC12 bind wild-type cadherin with rapid kinetics comparable to the kinetics of the first, fast forming low probability state measured between full-length ectodomains. The normalized data (Fig. 5) further support the equivalence of these two bonds. This conclusion agrees with surface force measurements, which similarly mapped the CEC1245-Fc bond to the EC1 domain (11). Together these findings provide compelling evidence that EC1 mediates the first, fast step in cadherin-dependent cell contact formation. This agrees with single bond rupture studies, which also linked the fast, weak bonds between full ectodomains to EC12 (14, 15).
A second direct parallel with previous force measurements is that the slower forming, high probability state requires EC3. In force measurements, weak EC12 bonds form rapidly, but the additional strong bonds, which require the full ectodomain and EC3, form more slowly (14, 15). Consistent with our micropipette data, increasing the protein contact time from 0.1 s to 3 s increased the population of strong E-cadherin bonds at the expense of the weak EC12 bonds (15). Surface force measurements also demonstrated that the strongest cadherin bond requires EC3 (11). The distance dependence of the adhesion by the extracellular fragments EC345 (ΔEC12); EC1245 (ΔEC3), and EC1–5 showed that removing EC3 eliminates the strongest cadherin bond. The requirement of domains other than EC1 for strong adhesion is supported by single bond rupture measurements (14, 15) and by cell adhesion assays (13).
Based on the numerous parallels between these cell adhesion results and the earlier biophysical studies, one can not help but speculate that the high probability binding state and the strong adhesive bonds detected in prior molecular force measurements are directly linked, and possibly the same. Importantly, these micropipette measurements demonstrate that C-cadherin binding at the molecular level (10, 11, 14) translates to intercellular adhesion.
A much higher CEC12-Fc density (158 cadherins/μm2) achieved a slightly lower limiting plateau (~0.48) than CEC1245-Fc (~0.55) at (10 cadherins/μm2). The lower CEC12-Fc activity also agrees with prior biophysical measurements (11, 13). Here, we show quantitatively that this is due to a lower apparent affinity (Table 1), which is likely due to structural perturbations due to the protein truncation. Recent reports show that subtle structural changes can significantly alter cadherin function (7, 16, 17). An alternative explanation is that its shorter molecular length may reduce the apparent two-dimensional affinity of CEC12-Fc relative to the longer CEC1245-Fc (58).
The W2A mutant binds to wild type ectodomains, although this mutation reportedly abrogates cell adhesion (7, 8, 17, 59). Nevertheless, our results agree with reports that both canine E-cadherin W2A EEC1–5 Fc and Xenopus C-cadherin W2A CEC1–5-Fc aggregate beads (16, 17). The apparent discrepancy between these micropipette results and other cell adhesion assays is attributed to the lower (single bond) detection threshold of the micropipette measurements. The much lower affinity of the W2A mutant (Table 1) accounts for its impact on cell adhesion, bead aggregation, and force measurements. The estimated affinity of the W2A mutant for wild-type cadherin is an order of magnitude lower than that of CEC12-Fc fragment and more than two orders of magnitude lower than CEC1245-Fc (Table 1). The substantial affinity reduction caused by this substitution is consistent with the critical role of Trp2 in cadherin adhesion.
Although the residual W2A binding might be attributed to H-bond formation between Glu89 and the N terminus of an opposed β-strand (7), this explanation is not supported by these and other findings. First, loss of Trp2 docking would weaken the EC1 bond, but this would not affect the biphasic probability curve. This technique measures binding probability, and not adhesion. Merely enhancing or weakening adhesion at the same site would not alter biphasic kinetics. Second, force measurements showed that the W2A mutant of E-cadherin forms a single weak bond to wild-type cadherin and to a second W2A mutant, in contrast to the wild-type protein, which forms two bonds (16). The mutant also adhered at the identical membrane separation as binding between opposed EC345 mutants (ΔEC12), which lack EC1 entirely (11).
Structure-based models postulate that cadherin-mediated cell adhesion only requires the strand exchange between Trp2 residues and hydrophobic pockets on opposite EC1 domains. This is equivalent to the receptor-ligand binding reaction described by Equation 1, and would only result in simple binding kinetics, as shown in Figs. 4 and 6. Although we have not yet determined the detailed reaction mechanism and kinetic rates for all steps in the biphasic time course, the simple strand exchange mechanism would not account for either the two stage binding time course or its dependence on the presence of either EC3 or EC3–5 domains.
In summary, these kinetic measurements of single cell interactions show that cadherin-mediated binding occurs in a two stage process that is independent of the cytoplasmic domain. The biphasic kinetics requires multiple extracellular domains: namely, the first, fast forming state requires EC12, but the slower, high probability binding state requires EC3. By contrast, W2A, CEC12, and CEC1245 all exhibit monophasic binding time courses characteristic of a simple binding mechanism. Furthermore, kinetic analyses also show that only the formation of a single, mutual bond between full-length ectodomains would not generate the biphasic reaction time course observed in these measurements.
Acknowledgments
We thank B. M. Gumbiner for the C-cadherin constructs used in this study.
The abbreviations used are
- EC
extracellular
- BSA
bovine serum albumin
- FACS
fluorescent-activated cell sorting
- FITC
fluorescein isothiocyanate
- RBC
red blood cell
Footnotes
This work was supported in part by National Institutes of Health Grants R01 GM51338 (to D. E. L.) and by R01 AI38282 and R01 AI44902 (to C. Z.).
References
- 1.Gumbiner BM. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 2.Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Curr Biol. 1997;7:308–315. doi: 10.1016/s0960-9822(06)00154-0. [DOI] [PubMed] [Google Scholar]
- 3.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 4.Nose A, Tsuji K, Takeichi M. Cell. 1990;61:147–155. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]
- 5.Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- 6.He W, Cowin P, Stokes DL. Science. 2003;302:109–113. doi: 10.1126/science.1086957. [DOI] [PubMed] [Google Scholar]
- 7.Harrison OJ, Corps EM, Berge T, Kilshaw PJ. J Cell Sci. 2005;118:711–721. doi: 10.1242/jcs.01665. [DOI] [PubMed] [Google Scholar]
- 8.Shan WS, Tanaka H, Phillips GR, Arndt K, Yoshida M, Colman DR, Shapiro L. J Cell Biol. 2000;148:579–590. doi: 10.1083/jcb.148.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sivasankar S, Brieher W, Lavrik N, Gumbiner B, Leckband D. Proc Natl Acad Sci U S A. 1999;96:11820–11824. doi: 10.1073/pnas.96.21.11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivasankar S, Gumbiner B, Leckband D. Biophys J. 2001;80:1758–1768. doi: 10.1016/S0006-3495(01)76146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu B, Chappuis-Flament S, Wong E, Jensen IE, Gumbiner BM, Leckband D. Biophys J. 2003;84:4033–4042. doi: 10.1016/S0006-3495(03)75129-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prakasam AK, Maruthamuthu V, Leckband DE. Proc Natl Acad Sci U S A. 2006;103:15434–15439. doi: 10.1073/pnas.0606701103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chappuis-Flament S, Wong E, Hicks LD, Kay CM, Gumbiner BM. J Cell Biol. 2001;154:231–243. doi: 10.1083/jcb.200103143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bayas MV, Leung A, Evans E, Leckband D. Biophys J. 2006;90:1385–1395. doi: 10.1529/biophysj.105.069583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perret E, Leung A, Feracci H, Evans E. Proc Natl Acad Sci U S A. 2004;101:16472–16477. doi: 10.1073/pnas.0402085101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prakasam A, Chien YH, Maruthamuthu V, Leckband DE. Biochemistry. 2006;45:6930–6939. doi: 10.1021/bi060213m. [DOI] [PubMed] [Google Scholar]
- 17.Tsuiji H, Xu L, Schwartz K, Gumbiner BM. J Biol Chem. 2007;282:12871–12882. doi: 10.1074/jbc.M611725200. [DOI] [PubMed] [Google Scholar]
- 18.Ahrens T, Pertz O, Haussinger D, Fauser C, Schulthess T, Engel J. J Biol Chem. 2002;277:19455–19460. doi: 10.1074/jbc.M200606200. [DOI] [PubMed] [Google Scholar]
- 19.Koch AW, Pokutta S, Lustig A, Engel J. Biochemistry. 1997;36:7697–7705. doi: 10.1021/bi9705624. [DOI] [PubMed] [Google Scholar]
- 20.Pertz O, Bozic D, Koch AW, Fauser C, Brancaccio A, Engel J. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pokutta S, Herrenknecht K, Kemler R, Engel J. Eur J Biochem. 1994;223:1019–1026. doi: 10.1111/j.1432-1033.1994.tb19080.x. [DOI] [PubMed] [Google Scholar]
- 22.Tomschy A, Fauser C, Landwehr R, Engel J. EMBO J. 1996;15:3507–3514. [PMC free article] [PubMed] [Google Scholar]
- 23.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 24.Luo BH, Carman CV, Takagi J, Springer TA. Proc Natl Acad Sci U S A. 2005;102:3679–3684. doi: 10.1073/pnas.0409440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vorup-Jensen T, Ostermeier C, Shimaoka M, Hommel U, Springer TA. Proc Natl Acad Sci U S A. 2003;100:1873–1878. doi: 10.1073/pnas.0237387100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Proc Natl Acad Sci U S A. 2001;98:2393–2398. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo BH, Takagi J, Springer TA. J Biol Chem. 2004;279:10215–10221. doi: 10.1074/jbc.M312732200. [DOI] [PubMed] [Google Scholar]
- 28.Brieher WM, Gumbiner BM. J Cell Biol. 1994;126:519–527. doi: 10.1083/jcb.126.2.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhong Y, Brieher WM, Gumbiner BM. J Cell Biol. 1999;144:351–359. doi: 10.1083/jcb.144.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG, Frame MC. Nat Cell Biol. 2002;4:632–638. doi: 10.1038/ncb829. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 32.Panorchan P, Thompson MS, Davis KJ, Tseng Y, Konstantopoulos K, Wirtz D. J Cell Sci. 2006;119:66–74. doi: 10.1242/jcs.02719. [DOI] [PubMed] [Google Scholar]
- 33.Kaplanski G, Farnarier C, Tissot O, Pierres A, Benoliel AM, Alessi MC, Kaplanski S, Bongrand P. Biophys J. 1993;64:1922–1933. doi: 10.1016/S0006-3495(93)81563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierres A, Benoliel AM, Bongrand P. J Biol Chem. 1995;270:26586–26592. doi: 10.1074/jbc.270.44.26586. [DOI] [PubMed] [Google Scholar]
- 35.Pierres A, Benoliel AM, Bongrand P. J Immunol Methods. 1996;196:105–120. doi: 10.1016/0022-1759(96)00103-2. [DOI] [PubMed] [Google Scholar]
- 36.Chesla SE, Selvaraj P, Zhu C. Biophys J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans E, Berk D, Leung A. Biophys J. 1991;59:838–848. doi: 10.1016/S0006-3495(91)82296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, Marcus WD, Goyal NH, Selvaraj P, Springer TA, Zhu C. J Biol Chem. 2005;280:42207–42218. doi: 10.1074/jbc.M510407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang J, Edwards LJ, Evavold BD, Zhu C. J Immunol. 2007;179:7653–7662. doi: 10.4049/jimmunol.179.11.7653. [DOI] [PubMed] [Google Scholar]
- 40.Chesla SE, Li P, Nagarajan S, Selvaraj P, Zhu C. J Biol Chem. 2000;275:10235–10246. doi: 10.1074/jbc.275.14.10235. [DOI] [PubMed] [Google Scholar]
- 41.Long M, Zhao H, Huang KS, Zhu C. Ann Biomed Eng. 2001;29:935–946. doi: 10.1114/1.1415529. [DOI] [PubMed] [Google Scholar]
- 42.Williams TE, Nagarajan S, Selvaraj P, Zhu C. Biophys J. 2000;79:1867–1875. doi: 10.1016/S0006-3495(00)76436-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams TE, Selvaraj P, Zhu C. Biophys J. 2000;79:1858–1866. doi: 10.1016/S0006-3495(00)76435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams TE, Nagarajan S, Selvaraj P, Zhu C. J Biol Chem. 2001;276:13283–13288. doi: 10.1074/jbc.M010427200. [DOI] [PubMed] [Google Scholar]
- 45.Nose A, Nagafuchi A, Takeichi M. Cell. 1988;54:993–1001. doi: 10.1016/0092-8674(88)90114-6. [DOI] [PubMed] [Google Scholar]
- 46.Gold ER, Fudenberg HH. J Immunol. 1967;99:859–866. [PubMed] [Google Scholar]
- 47.Kofler R, Wick G. J Immunol Methods. 1977;16:201–209. doi: 10.1016/0022-1759(77)90198-3. [DOI] [PubMed] [Google Scholar]
- 48.Dumaswala UJ, Wilson MJ, Jose T, Daleke DL. Blood. 1996;88:697–704. [PubMed] [Google Scholar]
- 49.Zhu C, Williams TE. Biophys J. 2000;79:1850–1857. doi: 10.1016/S0006-3495(00)76434-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brieher WM, Yap AS, Gumbiner BM. J Cell Biol. 1996;135:487–496. doi: 10.1083/jcb.135.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patel SD, Chen CP, Bahna F, Honig B, Shapiro L. Curr Opin Struct Biol. 2003;13:690–698. doi: 10.1016/j.sbi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 52.Yap AS, Brieher WM, Gumbiner BM. Annu Rev Cell Dev Biol. 1997;13:119–146. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 53.Yap AS, Niessen CM, Gumbiner BM. J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gumbiner BM. J Cell Biol. 2000;148:399–404. doi: 10.1083/jcb.148.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weidner KM, Behrens J, Vandekerckhove J, Birchmeier W. J Cell Biol. 1990;111:2097–2108. doi: 10.1083/jcb.111.5.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braga VM, Yap AS. Curr Opin Cell Biol. 2005;17:466–474. doi: 10.1016/j.ceb.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 57.Kaibuchi K, Kuroda S, Fukata M, Nakagawa M. Curr Opin Cell Biol. 1999;11:591–596. doi: 10.1016/s0955-0674(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 58.Huang J, Chen J, Chesla SE, Yago T, Mehta P, McEver RP, Zhu C, Long M. J Biol Chem. 2004;279:44915–44923. doi: 10.1074/jbc.M407039200. [DOI] [PubMed] [Google Scholar]
- 59.Ozawa M. J Biol Chem. 2002;277:19600–19608. doi: 10.1074/jbc.M202029200. [DOI] [PubMed] [Google Scholar]