

Abstract
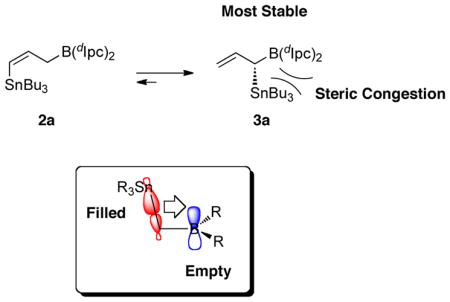
(E)-δ -stannyl homoallylic alcohols are prepared by an allene hydroboration-aldehyde allylboration sequence (Chen et al. J. Am. Chem. Soc. 2010, 132, 7881). Key to this reaction sequence is that the kinetic allene hydroboration product, 2a, is less stable than and isomerizes to the more sterically congested α-stannylallylborane 3a (see abstract figure). An M06-2X density functional analysis shows that the C—Sn to boron σ-π hyperconjugation interaction is sufficiently stabilizing to override the steric congestion in 3a.
The development of new carbonyl allylation reactions remain an important topic in organic synthesis.1 These transformations are widely used in the stereoselective construction of C-C bonds with diverse substitution patterns.1 Our laboratories recently demonstrated that an allenylstannane hydroboration-aldehyde allylation sequence generates (E)-δ-stannyl homoallylic alcohols with high enantioselectivity (Scheme 1).2 This is an important contribution to this area because most allylation reactions generate products with terminal vinyl groups that often require several steps for further functionalizion.3 Our previous experimental studies and computational analysis using the B3LYP density functional approximation (DFA) showed that (dIpc)2BH addition to allene 1 first gives the (Z)-δ-stannylallylborane 2a that then undergoes a rapid and highly diastereoselective 1,3-boratropic shift4 to give 3a.2 Addition of an aldehyde to 3a results in formation of the (E)-δ-stannyl homoallylic alcohol 4. Homoallylic alcohol 5 was not formed in these experiments. That intermediate 3a is thermodynamically more stable than 2a is somewhat surprising given that it places a bulky –SnBu3 group geminal to the very bulky diisopinocampheylborane unit. Here we provide computational evidence that C—Sn σ-bond to boron p-orbital hyperconjugation is sufficiently stabilizing to overcome the steric congestion in 3a.
Scheme 1.

Allenestannyl Hydroboration-Aldehyde Allylation Reaction Sequence. (M06-2X relative energies are in kcal/mol)
B3LYP structures were optimized and verified as minima using Gaussian 03.5 CCSD(T) energy calculations were also carried out in this Gaussian version. M06-2X energy evaluations were carried out in Jaguar 7.76 and Gaussian 09.7 For geometry optimizations the 6-31G(d,p) basis set was used for all atoms except Sn. The Gilbert and Sunderlin basis set was employed for Sn.8 SnBu3 groups were modeled as either SnMe3 or SnH3 groups. For energy evaluations the larger LACV3P++** basis set was used with very dense grids.9 Natural bond orbital (NBO) second-order perturbative delocalization energies (ΔEdeloc) were computed by comparison of the fully relaxed electronic structure (ΔESCF) with a localized Lewis bonding state (ΔElocal) using the keyword NBOdel in the NBO 3.1 program10 attached to Gaussian 09. Although this perturbation technique does not partition the total energy of each molecule it gives qualitative insights into relative electron delocalization energies.
To begin, we have also experimentally carried out the hydroboration reaction of allenylstannane 1 with 9-borabicyclo[3.3.1]nonane (9-BBN, Scheme 1b).11 Similar to the (dIpc)2BH hydroboration of allene 1, allylborane 3b is thermodynamically favored over the (Z)-alkene 2b. Although the steric congestion would not be as severe in 3b compared to 3a, these results again illustrate that the geminal arrangement of C-Sn and C-B bonds is highly favorable.
Scheme 2 gives the computed thermodynamics for the 1,3-boratropic shifts of allylboranes 6 and 8, which were used to model 2 and 3. When the diisopinocampheyl and SnBu3 groups of 2 are replaced with —BH2 and –SnH3 units to give 6 and 7, the equilibrium determined by the M06-2X/LACV3P++** density functional approximation (DFA) shows that 7 is more stable than 6 by 9.6 kcal/mol (3D structures are shown in Scheme 2b). CCSD(T)/6-31G(d,p) theory predicts an almost identical energy difference of 9.5 kcal/mol and validates our use of M06-2X. The B3LYP DFA underestimates this energy difference by ~3 kcal/mol. When methyl groups are used rather than hydrogen atoms on the boron and tin units, the equilibrium between 8 and 9 still favors the geminal species 9 by 8.8 kcal/mol. Comparison of the energy difference between 6 and 7 or 8 and 9 with 2a and 3a shows that there is a ca. 4 kcal/mol decrease in the stability of the geminal species when bulky diisopinocampheyl and SnMe3 groups are in close proximity (ΔE2a-3a = 5.7 kcal/mol). Figure 1 shows that the SnMe3 group is within ~2.3Å of the isopinocampheyl groups.12 The M06-2X energy difference between the hydroboration products of allene 1 and 9-BBN shows that the geminal species 3b is favored by 9.6 kcal/mol over 2b and indicates that the borabicyclo ring structure induces less steric congestion with the SnR3 group.
Scheme 2.

a) M06-2X 1,3-Boratropic Shift Thermodynamics and Transition State Energies for C-B Bond Rotation of 7 and 9. Distances are Reported in Ångstroms.
Figure 1.

Strucutres of 2a and 3a. Hydrogen atoms were removed for clarity. Distances are reported in Ångstroms.
We wondered whether this thermodynamic preference was unique for the SnR3 group. Replacement of this group in 2a and 3a with SiMe3 resulted in structure 3a being favored by only 2.2 kcal/mol while replacement of the SnR3 group with C(CH3)3 showed a 2.8 kcal/mol preference for structure 2a (Scheme 3).
Scheme 3.

M06-2X/LACV3P++** Boratropic Shift Energetics. (B3LYP Energies are Given in Parenthesis)
Because of the underestimation of this hyperconjugation effect, B3LYP predicted that for SiMe3 and C(CH3)3 2a would be more stable than 3a. A similar analysis on the 9-BBN structures, 2b and 3b, revealed a similar trend.
To test whether groups other than BMe2 would also favor the geminal configuration, we computed the thermodynamics for replacement of the dimethylborane group in 8 and 9 with CH3, AlH2, Li, OH, NH2, F, and NH3+ groups (Scheme 4). For nearly all of these units, structure 8 was found to be more stable than 9. The only exception is the cationic ammonium group, which slightly favors the geminal structure. In fact, the AlH2 group, which is isoelectronic with BH2 does not favor structure 9. This indicates that the geminal relationship of SnR3 and BR2 groups provides for a very unique stability that overrides steric congestion between the two groups.
Scheme 4.

M06-2X/LACV3P++** Boratropic Shift Energetics. (B3LYP energies are given in parenthesis)
To understand the novel stability of structure 3a (and model structures 7 and 9), we have explored the possibility of hyperconjugative stabilization between the C-Sn σ-bond and the “empty” boron p-orbital. Figure 2a shows the C-Sn bond delocalization and interaction in a bonding and antibonding fashion with the boron p-oribital. This effect is similar to the well-known β-tin and β-silicon effects that stabilize radicals and cations.13 The bond length differences between structures 2a and 3a (Figure 1) as well as 6 and 7 (Scheme 2b) show that when the C-Sn bond is geminal to the C-B bond it is elongated and the C-B bond is shortened. For example, in 6 the C-Sn bond is 2.131Å and increases to 2.263Å in 7. The C-B bond lengths are 1.565Å and 1.521Å in 6 and 7, respectively. Although the C-Sn bond length elongation can be rationalized by the increased steric congestion, the shorter C-B bond cannot.
Figure 2.

a) Molecular orbitals showing the hyperconjugative interaction of C-Sn and boron p-oribtals. b) Illustration of the diminished hyperconjugation interaction in the C-BR2 bond rotation transition state.
This hyperconjugation hypothesis can also be inspected by computing the barriers for rotation about the C-BR2 bond. Figure 2b illustrates that upon C-B bond rotation the boron p-orbital becomes orthogonal to the donor C-Sn bond. Indeed, in the transition structure for C-B bond rotation of structure 7 (Scheme 2b—right structure) the C-Sn becomes shorter with a bond length of 2.172Å and the C-B bond length increases to 1.578Å, which is longer than the C-B bond length in 6. Most important is that the transition structure for 7 requires an activation energy of 11.4 kcal/mol and for 9 requires 9.4 kcal/mol. These energy values are 1–2 kcal/mol larger than the energy of structures 6 and 8 and indicates that the hyperconjugation interaction that favors 7 and 9 has been severed and the C-Sn bond localized. As a comparison, the rotation barrier for replacement of the SnH3 with a CH3 group in 7 results in a barrier of only 1.3 kcal/mol. The Mulliken atomic charges for the Sn and B atoms also suggest charge delocalization. In 7 the Sn and B charges are +0.51e and +0.14e. In the transition state for bond rotation the charges on Sn and B are +0.55e and +0.24e, respectively. The NBO natural charges in structure 7 for Sn and B atoms are +1.30e and +0.28e and in the transition state are +1.31e and +0.54e. Both of these charge schemes show that the main difference is a significant decrease in charge on the boron center in the transition state strucutre.
As another evaluation of the hyperconjugation interaction in structure 7 we have compared the NBO delocalization energies between 6 and 7 and compared this with the delocalization in the transition structure for bond rotation in 7. NBO delocalization energies (ΔEdeloc) compare a localized bonding state with the fully delocalized state shown in Figure 2a. Although the molecular orbitals show the symmetry of this interaction it does not indicate the energetic relevance of the interaction. The NBOdel keyword localizes the orbitals between two atomic centers and removes all donor-acceptor interactions, which would remove this hyperconjugative interaction. However, it is important to point out that this NBOdel procedure removes all donor-acceptor interactions.
The difference in delocalization energies (ΔΔEdeloc) between 6 and 7 is 50 kcal/mol. This number significantly overestimates the energy difference between 6 and 7 because it is a second-order perturbative analysis and does not partition to total SCF energy. However, this large delocalization energy is in accord with the work of Fernández and Frenking who evaluated the intermolecular interactions in [H2C-CH(SnH3)2]+.14 They found that the p-interactions that are dominated by two C-Sn bond to carbon p-orbital interactions are worth an estimated 60 kcal/mol of stability per interaction. Comparison of this energy to the difference in delocaliztion energies for when the SnH3 group is replaced with CH3 shows that the ΔΔEdeloc value drops to only 10 kcal/mol. The ΔΔEdeloc value for comparison of 7 and the transition structure for 7 shows a slightly larger difference of 60 kcal/mol. This confirms that the filled C-Sn bond to empty boron p-orbital donor-acceptor interaction is in large part responsible for the energy difference between 6 and 7.
In conclusion, we have provided computational evidence that the hyperconjugative interaction between geminal C-Sn and C-B bonds is sufficently stabilizing to overcome steric congestion in structures such as 2a.
Supplementary Material
Acknowledgments
DHE thanks BYU for financial support and the Fulton Supercomputing Center for computational support. WRR thanks the NIH (GM038436) for support of this work at Scripps Florida.
Footnotes
Supporting Information Available. XYZ coordinates, absolute energies, and full Gaussian 03 and 09 references.
References
- 1.(a) Roush WR. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2. Pergamon Press; Oxford, U.K: 1991. p. 1. [Google Scholar]; (b) Yamamoto Y, Asao N. Chem Rev. 1993;93:2207. [Google Scholar]; (c) Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim, Germany: 2000. p. 299. [Google Scholar]; (d) Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Wiley-VCH; Weinheim, Germany: 2000. p. 403. [Google Scholar]; (e) Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]; (f) Lachance H, Hall DG. Org React. 2008;73:1. [Google Scholar]
- 2.Chen M, Ess DH, Roush WR. J Am Chem Soc. 2010;132:7881. doi: 10.1021/ja103041u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For recent examples, see: Amans D, Bareille L, Bellosta V, Cossy J. J Org Chem. 2009;74:7665. doi: 10.1021/jo900945x.Frein JD, Taylor RE, Sackett DL. Org Lett. 2009;11:3186. doi: 10.1021/ol900971r.
- 4.(a) Hancock KG, Kramer JD. J Am Chem Soc. 1973;95:6463. [Google Scholar]; (b) Kramer GW, Brown HC. J Organomet Chem. 1977:132, 9. [Google Scholar]; (c) Hoffmann RW, Zeiss HJ. J Org Chem. 1981;46:1309. [Google Scholar]; (d) Henriksen U, Snyder JP, Halgren TA. J Org Chem. 1981;46:3767. [Google Scholar]; (e) Brown HC, Jadhav PK, Bhat KS. J Am Chem Soc. 1985;107:2564. [Google Scholar]; (f) Wang KK, Gu YG, Liu C. J Am Chem Soc. 1990;112:4424. [Google Scholar]; (g) Gu YG, Wang KK. Tetrahedron Lett. 1991;32:3029. [Google Scholar]; (h) Narla G, Brown HC. Tetrahedron Lett. 1997;38:219. [Google Scholar]; (i) Fang GY, Aggarwal VK. Angew Chem, Int Ed. 2007;46:359. doi: 10.1002/anie.200603659. [DOI] [PubMed] [Google Scholar]; (j) Canales E, González AZ, Soderquist JA. Angew Chem, Int Ed. 2007;46:397. doi: 10.1002/anie.200603467. [DOI] [PubMed] [Google Scholar]; (k) González AZ, Soderquist JA. Org Lett. 2007;9:1081. doi: 10.1021/ol070074g. [DOI] [PubMed] [Google Scholar]
- 5.Frisch MJ, et al. Gaussian 03, Revision D.01. Gaussian, Inc; Wallingford, CT: 2004. See Supporting Information for full reference. [Google Scholar]
- 6.Jaguar, version 7.7. Schrodinger, LLC; New York, NY: 2010. [Google Scholar]
- 7.Frisch MJ, et al. Gaussian 09, Revision A.02. Gaussian, Inc; Wallingford, CT: 2009. See Supporting Information for full reference. [Google Scholar]
- 8.Check CE, Faust TO, Bailey JM, Wright BJ, Gilbert TM, Sunderlin LS. J Phys Chem A. 2001;105:8111. [Google Scholar]
- 9.Keywords gdftmed=-14, gdffine=-14, and gdftgrad=-14 were used to generate a very dense grid.
- 10.Glendening ED, Reed AE, Carpenter JE, Weinhold F. NBO Version 3.1. [Google Scholar]
-
11.Hydroboration of allenylstannane 1 (1 equiv.) with 9-BBN (0.5 equiv.) was carried out at 0 °C for 5 h. The structure of the resulting allylborane was deduced based on the allylboration product of hydrocinnamaldehyde. See Supporting Information for details.

-
12.Diastereoselective rearrangment of (Z)-δ-stannylallylborane 2a can result in either 3a or 3a′. We previously showed (reference 2) that the 1,3-boratropic shift transition state leading to 3a is 2.2 kcal/mol lower than the barrier leading to 3a′. However, 3a′ is slightly more stable thermodynamically than 3a (ca. 2 : 1 experimentally).

- 13.Davis DD, Gray CE. J Org Chem. 1970;35:1303.Jerkunica JM, Traylor TG. J Am Chem Soc. 1971;93:6278.Krusic PJ, Kochi JK. J Am Chem Soc. 1971;93:846.Yamamoto Y, Yatagai H, Maruyama K. J Am Chem Soc. 1981;103:3229.Pereyre M, Quintard JP, Rahm A. Tin in Organic Synthesis. Butterworth; London: 1987. Lambert JB, Wang GT, Teramura DH. J Org Chem. 1988;53:5422.Lambert JB. Tetrahedron. 1990;46:2677.Nguyen KA, Gordon MS, Wang GT, Lambert JB. Organometallics. 1991;10:2798.Lambert JB, Liu X. J Organomet Chem. 1996;521:203.Lambert JB, Zhao Y, Emblidge RW, Salvador LA, Liu X, So JH, Chelius EC. Acc Chem Res. 1999;32:183. (and references therein)Schormann M, Garratt S, Hughes DL, Green JC, Bochmann M. J Am Chem Soc. 2002;124:11266. doi: 10.1021/ja026443f.Müller T, Bauch C, Ostermeier M, Bolte M, Auner N. J Am Chem Soc. 2003;125:2158. doi: 10.1021/ja021234g.
- 14.Fernández I, Frenking G. J Phys Chem A. 2007;111:8028. doi: 10.1021/jp073737k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.