

Abstract
Background
Mercury is a well-known neurotoxin implicated in a wide range of neurological or psychiatric disorders including autism spectrum disorders, Alzheimer’s disease, Parkinson’s disease, epilepsy, depression, mood disorders and tremor. Mercury-induced neuronal degeneration is thought to invoke glutamate-mediated excitotoxicity, however, the underlying mechanisms remain poorly understood. Here, we examine the effects of various mercury concentrations (including pathological levels present in human plasma or cerebrospinal fluid) on cultured, rat cortical neurons.
Results
We found that inorganic mercuric chloride (HgCl2 –at 0.025 to 25 μM) not only caused neuronal degeneration but also perturbed neuronal excitability. Whole-cell patch-clamp recordings of pyramidal neurons revealed that HgCl2 not only enhanced the amplitude and frequency of synaptic, inward currents, but also increased spontaneous synaptic potentials followed by sustained membrane depolarization. HgCl2 also triggered sustained, 2–5 fold rises in intracellular calcium concentration ([Ca2+]i). The observed increases in neuronal activity and [Ca2+]i were substantially reduced by the application of MK 801, a non-competitive antagonist of N-Methyl-D-Aspartate (NMDA) receptors. Importantly, our study further shows that a pre incubation or co-application of MK 801 prevents HgCl2-induced reduction of cell viability and a disruption of β-tubulin.
Conclusions
Collectively, our data show that HgCl2-induced toxic effects on central neurons are triggered by an over-activation of NMDA receptors, leading to cytoskeleton instability.
Keywords: Mercury Chloride, Rat cortical neurons, Toxicity, MK 801, NMDA receptor, Excitotoxicity, Cytoskeleton
Background
Mercury is a potent environmental contaminant that exerts toxic effect on a variety of vital organs in the human body. It exists in three predominant forms: elemental (Hg0), organic (such as methylmercury -MeHg), and inorganic (mercuric chloride - HgCl2) mercury [1]. The major sources of mercury load in humans are food contamination, drug and vaccine preservatives, dental amalgams, or occupational exposure [2]. Both acute and chronic exposure to mercury is also known to cause a variety of neurological or psychiatric disorders [3-6]. The central nervous system (CNS) is one of the most vulnerable organs affected by mercury toxicity. Within the CNS, two of the most often affected areas are the cerebral cortex [7,8] and the cerebellum [9,10]. While the mechanisms underlying mercury-induced toxicity remain largely unknown, several studies have shown toxic effects of organic MeHg on the CNS as it can easily cross the blood–brain barrier and accumulate in the brain at high concentrations [1,11]. MeHg has been reported to interact with a wide range of cellular targets and affect multiple cellular functions. These toxic effects include, but are not limited to, the inhibition of neuronal ion channels [12-14], disruption of presynaptic transmitter release and postsynaptic receptor function [15-17], damage to neuronal cytoskeleton components and DNA structures [8,18,19], and alteration of Na+/K+ ATPase and mitochondrial function [20,21] (also see review [22]). Moreover, a large body of recent evidence has also drawn the link between mercury and glutamate-mediated excitotoxicity. For instance, mercury has been shown to affect several aspects of glutamatergic signaling, including the inhibition of glutamate reuptake in astrocyte [23], inhibition of glutamine synthetase activity [24], and an enhancement of spontaneous glutamate release from neurons [25]. These effects result in an increase in glutamate concentration at the synaptic cleft. MeHg has also been found to impact postsynaptic N-Methyl-D-Aspartate (NMDA) receptors. For instance, Basu et al. (2007) have demonstrated that mercury decreases the number of NMDA receptors in several brain regions including the basal ganglia, cerebellum, brainstem, and occipital cortex of mink [26]. In contrast, Ndountse and Chan (2008) have reported that MeHg increased NMDA receptor expression, which in turn may be responsible for neurotoxicity [27]. Another study by Miyamoto et al. (2001) suggested that the enhanced sensitivity of NMDA receptors might contribute to the vulnerability of developing cortical neurons to MeHg neurotoxicity [28]. Although the mercury-induced toxic effects on the nervous tissue have been well documented, the findings are often conflicting and the underlying mechanisms remain poorly defined.
In contrast to MeHg, much less is known about the toxic effect of inorganic HgCl2, despite the fact that it can easily cross the blood–brain barrier and accumulate in the brain at much higher concentrations [29,30]. Moreover, HgCl2 is a more reactive form of mercury as it interacts with many macromolecules and exhibits a long latency of neurotoxicity [24,31]. For instance, HgCl2 is known to be five times as potent as MeHg in blocking L-type Ca2+ channels in hippocampal neurons [32]. It is also a more potent inhibitor of glutamate reuptake and glutamine synthetase activity than MeHg [24]. However, little is known about its actions on the NMDA receptors. In the present study, we first assessed the impact of inorganic HgCl2 on neuronal viability and network integrity in both developing and matured rat cortical neurons in cell culture. Next, we investigated the cellular mechanisms underlying HgCl2-induced toxic effect by studying the involvement of glutamatergic NMDA receptor activity. Our results show that HgCl2 disturbs neuronal network structure and induces cell apoptosis in cortical neurons. These actions involve a degradation of β-tubulin, an important component of the neuronal cytoskeleton, and these effects were evoked by NMDA-receptor function. Inhibition of NMDA-receptor activity by MK 801 not only inhibited HgCl2-induced increase in synaptic currents and intracellular Ca2+, but also prevented HgCl2-induced disassembling of cytoskeleton protein and disruption of neuronal network integrity.
Results
HgCl2 inhibits neuronal outgrowth and induces degeneration of both developing and mature cortical neurons in culture
To examine how mercury may impact neuronal outgrowth and network formation between central neurons, we first investigated the effects of HgCl2 on neurite ini-tiation and network formation during early outgrowth. To this end, rat neonatal cortical neurons were cultured either in the absence (control) or presence of HgCl2 at the concentrations of 25 nM, 100 nM, and 25 μM for 3 days. Neuronal viability, neurite outgrowth and network formation were evaluated 3 days later. The choices of these experimental doses were based on the studies showing that in the Minamata Bay exposure in Japan, the mercury concentration in the brains of patients was found to be around 1.7 μM to 26 μM, whereas in non-exposed controls its levels averaged around 30 nM [33,34]. Additionally, several studies have revealed that mercury levels in brain tissues are 2–10 fold higher in patients with dental amalgam fillings, Alzheimer’s disease, autism spectrum disorders, epilepsy or hydrocephalus [35-38]. Figure 1A shows a typical culture dish grown under control conditions (no mercury) yielding a healthy population of both neurons and glial cells (indicated by a circle) with extensive neurite outgrowth and network formation (indicated by an arrow). Although cells developed extensive neurites and neuronal network in the presence of both 25 nM and 100 nM HgCl2 (Figure 1B & C, indicated by arrows), it is interesting to note that HgCl2, at 100 nM, appeared to cause cell body damage, abnormal neurite outgrowth, or degeneration of some neurites (Figure 1C, indicated by an asterisk and an arrow head). Cells cultured in the presence of 25 μM of HgCl2 however failed to develop neurite processes and hence were devoid of network connectivity (Figure 1D, indicated by an arrow head and asterisks). To quantify the effect of HgCl2 on cdead cell assay /dead cell assay (LIVEDEAD® ViabilityCytotoxicity Kit, Invitrogen, see Materials and Methods). Specifically, cells were maintained in culture for 3 days either in the absence or presence of HgCl2 at concentrations as described above. On day 3, neurons were incubated and stained with green-fluorescent calcein-AM for 30 mins to indicate intracellular esterase activity (live cells), and red-fluorescent ethidium homodimer-1 to determine the damage to plasma membrane integrity (dead cells) (Figure 1E & F). Fluorescent labeling of live and dead cells was visualized using a confocal microscope (LSM-510) and images were acquired under the same confocal parameter settings using a 10x or 20x objective. The number of live (green) and dead (red) cells in the above culture conditions was counted using imageJ software. Specifically, neurons were counted based on a randomized selection of three areas of 1 mm2 under each culture condition. The mean value of cell viability reflected by the percentage of live cells was calculated and compared. Figure 2A showed that in four experiments cell viability under control conditions was 89.2 ± 3.5% (n = 4). There was however no significant difference between 25 nM (90.7 ± 3.7%, n = 4, P = 0.352) and 100 nM (89.6 ± 3.0%, n = 4, P = 0.914) of HgCl2. HgCl2 used at the concentration of 25 μM did, however, significantly (11.4 ± 1.5%, n = 4, P < 0.0001) compromised cell viability compared to control (Figure 2A).
Figure 1.

HgCl2 exposure perturbs neurite outgrowth and compromises neuronal viability in cultured cortical neurons. Rat cortical neurons were grown either in the absence (A, control) or presence of HgCl2 at 25 nM (B), 100 nM (C), and 25 μM (D) for 3 days. Phase contrast images were taken on day 3. A. Cortical culture under control conditions exhibited healthy population of neurons and glial cells (indicated by a circle), exhibiting extensive neurite outgrowth and network formulation (indicated by an arrow). B. Cells cultured in the presence of 25 nM HgCl2 also developed extensive neurite processes and formed extensive network. C. Neuronal populations cultured in the presence of 100 nM of HgCl2 underwent apoptotic cell death (indicated by an asterisk) and exhibited collapse or retraction of neurites (indicated by an arrow head). D. HgCl2 at 25 μM induced cell death (indicated by asterisks) and inhibited neurite extension and network formation (indicated by an arrow). E &F shows representative confocal images of live/dead assay of cortical neurons cultured either in the absence (E) or presence (F) of 25 μM of HgCl2 for 3 days. Live cell bodies and their neurites were stained with calcein-AM (green), whereas dead cells with compromised membranes were stained with ethidium homodimer-1 (red). Scale bars, 20 μm.
Figure 2.
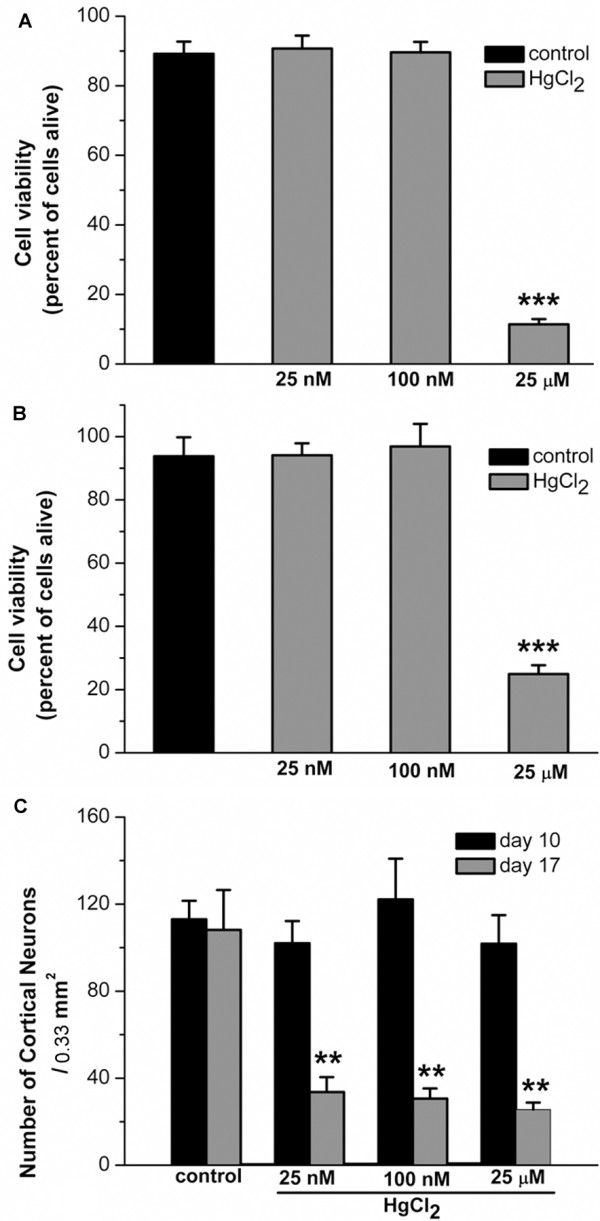
Statistical data showing the acute and chronic effects of HgCl2 on cell viability of developing and mature cortical network. A. To evaluate the effect of HgCl2 on neurite initiation and network formation during the early stage of neuronal development, rat cortical cells were cultured in the absence (control) or presence of HgCl2, at 25 nM, 100 nM, and 25 μM, for 3 days. Cell viability/cytotoxicology assay were performed on day 3. The number of live and dead cells in randomly chosen areas of 1 mm2 was counted. The percentage of live cells is presented in the bar graph. These data show that cultures in the presence of HgCl2 at 25 μM, but not 25 or 100 nM, exhibited significant reduction of cell viability. B. To evaluate the acute effect (< 24 hrs) of HgCl2 exposure on the newly developed network, 4 day old cortical cultures were exposed to HgCl2, at 25 nM, 100 nM, and 25 μM, for 16 to 24 hrs. Subsequent live/dead cell assay showed that acute exposure to higher concentration of HgCl2 at 25 μM significantly reduced the cell viability. C. To evaluate the chronic effect (7 days) of HgCl2 exposure on the neuronal network, cells were grown for 10 days and were maintained for another 7 days in the absence or presence of HgCl2. Phase contrast images of neurons in the same area of 0.33 mm2 were taken on day 10 and day 17. The statistic data show that the numbers of neurons were significantly reduced by exposure to HgCl2 for 7 days at all concentrations examined. Statistical significance was determined using ANOVA (A &B) and Student’s paired t-test (C). Post hoc analysis was conducted using Tukey’s test. ** P < 0.01. *** P < 0.001. Error bars indicate SEM for all figures.
We next sought to determine whether the established cortical neuronal network at early stages of cell culture could be affected by shorter exposure (< 24 hrs) to HgCl2 at different concentrations. To do this, cells were maintained in culture for 4 days and allowed to exhibit neurite outgrowth and network connectivity. Neurons were then cultured in medium containing either no HgCl2 (as control) or HgCl2 at 25 nM, 100 nM or 25 μM for 16–24 hrs. To evaluate the effects of mercury on neuronal network and cell viability, neurons were again stained with calcein-AM (live) and ethidium homodimer-1 (dead) after treatment. The protocol for live/dead cell counting and cell viability calculation was as described in Figure 2A. As shown in Figure 2B, HgCl2 at lower doses such as 25 nM (93.8 ± 6%, n = 4, P = 0.072) and 100 nM (94.1 ± 3.8%, n = 4, P = 0.254) did not affect the established network and cell survival as compared with control (96.9 ± 7.1%, n = 4). However, a shorter (16–24 hrs) exposure of cells to 25 μM of HgCl2 not only remarkably disrupted the neuronal physical connectivity, but also significantly reduced the cell viability (24.9 ± 2.8%, n = 4, P < 0.001) (Figure 2B).
Finally, we sought to determine whether longer-term exposure (>7 days) of cells to HgCl2 could affect fully developed neurons and their network. To test this, cortical cells were first allowed to grow in culture for 10 days and establish outgrowth and extensive networks. Cells were subsequently treated with HgCl2 at 25 nM, 100 nM and 25 μM and maintained in culture for another 7 days. Neuronal viability and network formation were examined at day 17. We found that at this stage, the cortical neurons exhibited extremely dynamic neuronal outgrowth pattern, and the active growth of glial cells formed enormous layers in the culture dish. The extensive background labeling of glial cell layers made the survival assay, particularly the counting of live cells, very difficult. We therefore opted to evaluate the effect of HgCl2 on neuronal viability using phase contrast images. Specifically, cortical neurons were cultured on glass cover slips with a grid (Bellco Glass, aid for counting). Phase contrast images were taken before exposure to HgCl2 on day 10 and after an addition 7-day exposure to HgCl2 (i.e., on day 17). The average numbers of neurons were calculated based on a randomized selection of five different areas of 0.33 mm2 in each dish. With the aid of the grid on cover slips, we were able to take images of the same five areas in each dish on day 10 and day 17. The mean numbers of survived neurons were compared between day 10 and day 17. Neurons were identified based on their size, shape, arrangement and number of processes. Our results showed that 7-day exposure to HgCl2, at all concentrations examined, caused a significant reduction in the rate of neuronal survival. Specifically, control cultures had on average 113 ± 8.5 neurons/0.33 mm2 on day 10 and it did not change after 7 days in culture (108.2 ± 18.3, n = 5, P = 0.842). Cultures exposed to 25 nM HgCl2 for 7 days had a substantial reduction in the number of neurons (33.6 ± 6.9, n = 5, P = 0.005) as compared with that before HgCl2 exposure on day 10 (102 ± 10.2, n = 5). Similarly, HgCl2 at 100 nM (from 122.2 ± 18.7 to 30.6 ± 4.7, n = 5, P = 0.003) and 25 μM (from 101.8 ± 13.1 to 25.6 ± 3.2, n = 5, P = 0.002) caused significant decrease in cell viability (Figure 2C). These data indicated that chronic exposure to HgCl2 at concentrations, as low as 25 nM to 100 nM, caused detrimental effects on central neurons even at a more matured developing stage. It is, however, important to note that because the mercury-induced effects are not neuron-type specific, the significant reduction in the number of neurons at all concentrations examined might also involve a secondary toxic effect involving the glial cells. Specifically, mercury toxicity to glial cells may prevent astrocytes from forming a firm “bottom layer” which plays an essential role for neuronal outgrowth and survival under our culture condition. The toxic impact of mercury on glial cells may have thus exasperated neuronal loss in our culture condition. However, our experiments together with previous studies warn against the potential effects of chronic low-dose mercury exposure [39,40].
Based on the fact that HgCl2 used at 25 μM consistently caused neuronal damage to both developing and more mature neurons during short-term and long-term exposure, we next investigated the mechanisms underlying the toxic effect of HgCl2 on cortical neurons using the concentration of 25 μM in all subsequent experiments, unless stated otherwise.
HgCl2-induced cell damage is associated with loss of cytoskeleton structures
The architecture and survival of the central neurons rely significantly upon their cytoskeleton components, specifically the actin filament (F-actin) and microtubules such as β-tubulin. Any disturbance of these dynamic structures or their polymerization/depolymerization status due to environmental or genetic factors could compromise cell survival [41-43]. Based on our observations that HgCl2 induces cell damage and retraction of neuronal processes, we hypothesized that HgCl2 toxicity may involve disruption of neuronal cytoskeleton proteins such as the F-actin and β-tubulin. To test this hypothesis, cortical neurons were first cultured for four to ten days, which allowed for the development of neurite outgrowth and network connectivity. Cells were then exposed to culture medium containing either no HgCl2 (as control) or HgCl2 at 25 μM for 24 hrs. The preparations were then fixed with 4% paraformaldehyde and stained with anti β-tubulin antibody and rhodamine-phalloidin (to label F-actin). As shown in Figure 3, the pyramidal cortical neurons in control condition (A) exhibited bigger cell bodies with multiple healthy processes and extensive network. In contrast, neurons treated with HgCl2 (B) appeared to be much smaller in size with degraded neurite and compromised network. Accordingly, a strong fluorescent labeling of cell bodies and neurites with both β-tubulin (A) and F-actin (C) antibodies were observed in control neurons. The cells exposed to HgCl2 at 25 μM, however, remarkably lost the fluorescent intensity of β-tubulin (B) although there was no apparent change in the intensity of F-actin staining (D). These results clearly demonstrated that the β –tubulin protein is most likely affected by HgCl2-induced toxicity in the CNS. The reduction of overall intensity of β –tubulin is likely due to HgCl2-induced degeneration of neurons rather than cell death as the level of F-actin intensity remained unaffected by HgCl2. One would need to be cautious, however, that these experiments may not preclude the possibility that mercury exposure might also change the conformation of β –tubulin altering access of the primary antibody to the epitope. Future investigation and analysis using western blot techniques are still required for further validation of these data.
Figure 3.

Exposure of HgCl2 compromised cytoskeleton components, mainly the β-tubulin, in cortical neurons. Cells were first cultured for 4 to 10 days and allowed to develop neurite and networks and were then exposed to 25 μM of HgCl2 for up to 24 hrs. Cultures were subsequently fixed and stained with antibodies against β-tubulin and rhodamine-phalloidin (A-D). E &F are the merged confocal images of β-tubulin and F-actin staining. The fluorescence intensity of β-tubulin, and not F-actin was reduced in neurons that were treated with 25 μM of HgCl2. Scale bar, 25 μm.
HgCl2 triggers sustained intracellular Ca2+ rise in cortical neurons, which is blocked by NMDA receptor antagonists
Recent studies have linked the degeneration of cytoskeleton structures to glutamatergic excitoxicity. Specifically, over stimulation of NMDA receptors and the subsequent overloading with Ca2+ have been shown to cause rapid disruption of established cytoskeleton components or inhibition of the assembly of new cytoskeleton elements such as microtubules, F-actin and neurofilament proteins in a variety of preparations [44-46]. Moreover, NMDA receptors have been shown to play a pivotal role in mercury-induced neurotoxicity [19,28]. We next sought to determine whether HgCl2 affects cortical Ca2+ homeostasis through NMDA receptors. To this end, Ca2+ ratiometric experiments using Fura-2 AM were performed on cortical neurons. Neurons were maintained in culture for about five to ten days and subsequently loaded with 5 μM of Fura-2 AM for 30 mins at 37°C. Cells were then washed with normal saline containing free Mg2+ and 3 μM of glycine in order to activate NMDA receptors at the resting level. Ca2+ images and the ratio of Fura-2 fluorescence intensity at 340 nm and 380 nm (reflection of intracellular Ca2+ levels) were acquired prior to and post exposure to HgCl2 at 25 μM. Representative traces of four different cells are shown in Figure 4A (red, green, black and blue traces). As shown in Figure 4A, intracellular Ca2+ transients were recorded before HgCl2 (indicated by an arrow). After local application of HgCl2, the amplitude and frequency of calcium transients were augmented. After 5 to 10 mins of HgCl2 exposure, sustained rises in intracellular Ca2+ were observed in almost all cells examined (Figure 4A). The ratio of Fura-2 fluorescence intensity increased by 2.26 ± 0.2 times (n = 8) after 10 mins of HgCl2 application and 4.55 ± 1.44 (n = 11) times compared to control levels after 15 mins of HgCl2 application (Figure 4C, two columns on the left panel). Next, we examined the involvement of NMDA receptor activity in HgCl2-induced rise in intracellular Ca2+. To do this, the basal Ca2+ levels were first monitored for 5 mins and again intracellular Ca2+ transients were recorded in control conditions (Figure 4B, indicated by an arrow). The cells were then exposed to 10 μM of the NMDA receptor antagonist MK 801 for 5 to 10 mins. Note that upon application of MK 801, the intracellular Ca2+ transients were almost completely abolished indicating an involvement of tonic NMDA receptor activity in normal Ca2+ oscillations. It is also important to note that subsequent application of HgCl2 (25 μM) in the continued presence of MK 801, failed to induce rises in Ca2+. In MK 801 treated neurons, the fluorescence ratio in the continued presence of MK 801 was 1.09 ± 0.13 (n = 11) times after 10 mins of HgCl2 exposure and 1.37 ± 0.33 times (n = 8) after 15 mins of HgCl2 exposure (Figure 4C, two columns on the right panel). The rise in intracellular Ca2+ in the presence of MK 801 was significantly smaller than that of MK 801 (Figure 4C). These results indicate that NMDA receptor activity may play a crucial role in HgCl2-induced alteration of intracellular Ca2+ homeostasis, which might contribute to HgCl2-induced disturbance of cytoskeleton network.
Figure 4.
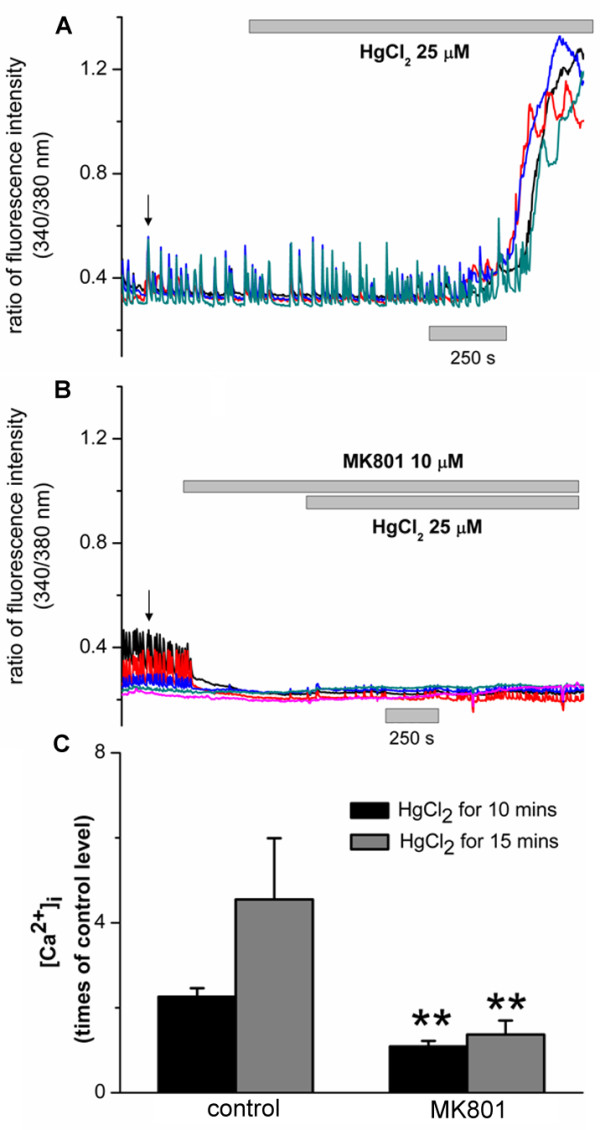
HgCl2 induced sustained elevation of intracellular Ca2+ concentration ([Ca2+]i) in cortical neurons and this rise in [Ca2+]i was inhibited by the NMDA receptor antagonist MK 801. A. Representative traces of changes in fura-2 fluorescence ratio before and after the application of HgCl2 (25 μM) in four different cortical neurons (shown by red, green, blue and black traces). Cells were loaded with fura-2 AM and the ratio of fluorescence intensity, at the 340 and 380 nm excitation wavelengths, was measured B. Representative traces of fura-2 fluorescence ratio recorded before and after application of HgCl2 (25 μM) in four different cortical neurons (shown by red, green, blue and black traces) that were previously exposed to MK 801 (10 μM), an antagonist of NMDA receptors. C. Fold- rises in fura-2 fluorescence ratio induced by HgCl2, after 10 and 15 mins of exposure time, in the absence or presence of MK 801. Note that, HgCl2-induced rise in [Ca2+]i in MK 801-treated neurons was significantly smaller than that in control cells in the period of 10 mins and 15 mins. Arrows indicate the presence of spontaneous Ca2+ transients in cortical neurons under resting conditions.
It is important to note that HgCl2-induced sustained elevations of [Ca2+]i occurred simultaneously in groups of cells that were measured as shown in Figure 4A. This led us to propose that HgCl2 may initially affect the synaptic activity of cortical neurons by acting through their NMDA receptors. These changes in neuronal excitability may in turn alter the status of dynamic or balanced excitability within a network or their neighboring cells.
HgCl2 increased synaptic inward currents and cause sustained membrane depolarization in pyramidal cortical neurons through NMDA receptors
NMDA receptor-mediated synaptic activity plays an important role in early neuronal development and also in the later stages of synaptic maturation [47-50]. To further determine the role of NMDA receptors in HgCl2-induced toxicity, we next explored whether HgCl2 acts to alter NMDA receptor-mediated synaptic activity such as synaptic currents or membrane discharges. We first tested whether HgCl2 affected spontaneous synaptic currents in pyramidal neurons. To test this, whole cell voltage-clamp experiments were performed on pyramidal neurons in culture for 7 to 15 days and spontaneous inward currents were recorded by holding the cells at −70 mV. As shown in Figure 5A, spontaneous inward currents were recorded in pyramidal cortical neurons under control condition as shown in the insert (Ai). Upon local application of HgCl2 (25 μM), large amplitude inward currents were elicited. It is important to note that HgCl2-elicited inward currents were often followed by high frequency, spontaneous postsynaptic events, which lasted for a long period (see insert Aii, indicated by an arrow). To test the involvement of NMDA receptors in HgCl2-induced increase in synaptic currents, the inward currents were recorded from cells that were first exposed to MK 801 to block the NMDA-receptors, and then to both MK 801 and HgCl2. Figure 4B shows that spontaneous synaptic currents were again recorded under control conditions. Application of MK 801 eliminated events with bigger amplitudes indicating the involvement of NMDA-currents in these events. The remaining synaptic events in the presence of MK 801 are likely the non-NMDA currents. Interestingly, in the continued presence of MK 801, HgCl2 failed to further enhance synaptic currents that were observed in Figure 4A, while the frequency and amplitude of non-NMDA currents remained unchanged by HgCl2. These experiments suggest that HgCl2 mainly increased the NMDA-currents in pyramidal cortical neurons.
Figure 5.

HgCl2 enhanced spontaneous inward currents and caused sustained membrane depolarization in pyramidal cortical cells. A. A representative trace of spontaneous synaptic currents recorded before and after the application of HgCl2 (25 μM) from a pyramidal neuron. The cell was held at the potential of −70 mV. Spontaneous inward currents were recorded before (Ai) and during the application of HgCl2. Both the amplitude and frequency of inward current were substantially increased. Robust small, spontaneous events followed the big amplitude of currents as shown in the insert (Aii, indicated by an arrow). B. A representative trace of synaptic currents recorded before and after HgCl2 exposure in the presence of 10 μM of MK 801. Note that application of MK 801 prevented HgCl2 from inducing increase of synaptic currents indicating that NMDA currents was involved in HgCl2 action on cortical neurons (n = 7). C. Membrane potential of a pyramidal cell recorded under current-clamp mode before and after the application of HgCl2 at 25 μM. HgCl2 increased the spontaneous electrical activity and triggered sustained rise in membrane potentials. Subsequent application of MK 801 (10 μM) abolished HgCl2-induced firing of action potentials but did not affect the sustained membrane depolarization. D. A representative trace of membrane potential recorded from a pyramidal cell pretreated with MK 801 for 10 mins before and after HgCl2 exposure,. Pretreatment of cells with MK 801 greatly reduced HgCl2 - induced increase in action potential firing and sustained membrane depolarization. E. Statistical data shows that the increased amplitude of sustained membrane depolarization by HgCl2 (25 μM) in MK 801-treated cells is significantly smaller than that in control cells.
The above data suggest that HgCl2-induced increase in synaptic inputs in pyramidal cortical neurons may in turn change the membrane excitability in these neurons. To test this possibility, we next monitored the membrane potential changes by performing whole-cell current-clamp experiments on pyramidal cortical neurons. Figure 5C shows a representative membrane potential trace recorded from a pyramidal neuron before and after the application of 25 μM of HgCl2. In this particular cell, the resting membrane potential of the cell was about −56 mV. Application of HgCl2 initially enhanced the spontaneous synaptic discharge and triggered firing of action potentials followed by sustained membrane depolarization to a potential of around −42 mV. Interestingly, subsequent application of MK 801 (10 μM) did abolish the firing of action potentials, but did not affect the sustained membrane depolarization indicating the involvement of secondary-mechanism in the sustained depolarizing components. It is interesting to note that pre-incubation of cells with MK 801 at 10 μM for about 10 mins almost completely prevented HgCl2 from triggering action potentials and eliciting large sustained membrane depolarization as shown in Figure 5D (n = 7). Specifically, we found that HgCl2 failed to induce an increase in synaptic potentials and firing of action potentials after 10 to 15 mins of exposure time, although a small elevation of membrane potential was still detectable in some cells. The mean amplitudes of sustained membrane depolarization either in the absence (control) or presence of MK 801 is shown in Figure 5E. These data reveal that the mean increase in membrane potentials of sustained components after 10 mins of HgCl2 exposure in control neurons was 20.4 ± 2.8 mV (n = 7). The increased amplitude of membrane potential was significantly reduced to 6.86 ± 1.3 mV (n = 5) in the presence of MK 801. In summary, HgCl2 increased synaptic current inputs and caused enhancement of membrane discharges in pyramidal cortical neurons, and this increase in synaptic activity could be substantially attenuated by NMDA receptor block.
Inhibition of NMDA receptor activity prevents mercury-induced impairment of network integrity and disruption of cytoskeleton component in cortical neurons
Perturbation of synaptic activity or Ca2+ homeostasis, especially excessive loading of cells with Ca2+, activates a series of downstream cascades. These steps include proteinases, reactive oxygen species, and mitochondrial failure, which are lethal to cell cytoskeleton components or DNA structures [51-53]. Based on our results showing that MK 801 could significantly inhibit HgCl2-induced increases in synaptic activity and [Ca2+i, we predicted that MK 801 may exert a protective action on cortical neurons during exposure to HgCl2. To examine this, cells cultured for 10–12 days were exposed to the following four conditions: (1) control; (2) 25 μM of HgCl2; (3) co-application of HgCl2 at 25 μM and MK 801 at 5 or 10 μM, or following 30 mins of pretreatment with MK 801; (4) MK 801 alone for 16–24 hrs. Phase contrast pictures were taken the next day. We found that pretreatment or co application of MK 801, at both 5 and 10 μM, prevented HgCl2 - induced disruption of neuronal network. Specifically, Figure 6 shows that control neurons exhibit healthy cell bodies with well-established networks (A). HgCl2 alone severely disrupted neuronal network formation and triggered cell apoptosis (B). However, cells exposed to both MK 801 and HgCl2 still exhibited healthy morphologies with extensive neurite outgrowth and integrated network (C). MK 801 alone did not cause damage to neurons and their networks (D).
Figure 6.

Inhibition of NMDA receptor by MK 801 prevents HgCl2-induced disruption of neuronal network and degradation of cytoskeleton components. To evaluate the protective effect of MK 801 on HgCl2-induced neurotoxicity, cells were grown for about 10 days and allowed to extend neurites and form network. Cultures were then maintained in the following conditions: control (A), 25 μM of HgCl2 (B), 25 μM of HgCl2 and 5–10 μM of MK 801 (C), and MK 801 alone (D). Phase contrast images were taken 16–24 hr later. Cells were subsequently fixed and stained with antibodies against β-tubulin and F-actin (E-H). Neurons exposed to HgCl2 exhibited retraction of neurite processes (B) and lower fluorescence intensity of β-tubulin (F) as compared with control cultures (A &E). HgCl2 in the presence of MK 801 failed to induce disturbance of network integrity and cytoskeleton staining (C &G). MK 801 alone did not affect neuronal network and fluorescence intensity of cytoskeleton staining (D &H). Scale bars, 25 μm.
To further evaluate the protective action of MK 801 on HgCl2-induced neurotoxicity, we investigated whether MK 801 could also prevent HgCl2-induced damage to cytoskeleton components. To test this, cortical neurons were exposed to four conditions overnight as listed above, and cells were then fixed and stained with antibodies against β-tubulin and F-actin the next day. Confocal images showed that the fluorescence intensity of β-tubulin but not F-actin was substantially reduced in cells treated with HgCl2 alone (Figure 6F) as compared with control (Figure 6E). This result is consistent with what we observed previously (see Figure 3). Cells in dishes con-taining MK 801 at 5 μM and HgCl2 at 25 μM exhibited intense fluorescence for both β-tubulin and F-actin (Figure 6G). MK 801 alone did not affect staining in both instances (Figure 6H). In fact, the fluorescence intensity of F-actin in cells exposed to HgCl2 was even stronger in some cases as shown in Figure 5F & G.
In summary, our data indicate that an inhibition of NMDA-receptor activity indeed attenuates the degenerative effects of HgCl2 on cultured cortical neurons. These data further support the notion that NMDA synaptic transmission or receptor function plays an essential role in initiating HgCl2-induced toxicity in the CNS.
Discussion
This study is the first to investigate HgCl2 neurotoxicity on rat cortical neurons from a variety of aspects – ranging from neuronal morphology to excitability. Specifically, our data demonstrate that HgCl2 elicits detrimental effects on young, developing cortical neurons – ranging from reduced cell survival, impaired neurite outgrowth, compromised network assembly to the degene-ration of mature networks. Importantly, our data caution against the possible detrimental effects of mercury at low nanomolar levels (sub-lethal doses). An apparent lack of short-term (3 days) or acute (< 24 hrs) effect of HgCl2, at 25 and 100 nM, on cortical cell via-bility does not necessarily rule out cellular toxicity, as the detrimental changes may either be subtle or require longer exposures. In fact, HgCl2 at 100 nM has been demonstrated to cause remarkable disruption of neurite membrane integrity, and induce robust retraction and collapse of growth cones in Lymnaea Stagnalis neurons [39]. Low nanomolar concentrations of mercury have also been shown to affect the production of pathological hall-marks of Alzheimers’ diseases in cultured neurons [40,54] as well as affect cardiac function [55,56]. Therefore, a detailed evaluation of the effect of HgCl2 at these concentrations may still be required. Specially, the effect of HgCl2 on the motile structure of growth cones, the length of neurite elongation, and cell viability would need to be monitored using proper neuronal model types in which these parameters can be easily measured as shown in our previous study [57].
Mercury neurotoxicity has been implicated to involve myriad mechanisms and cellular targets. These include perturbation of Ca2+ homeostasis, dysfunction of mitochondria, glutamatergic excitability, disruption of cytoskeleton structures, reactive oxygen species (ROS), and many others [22,28,58]. However, the steps that initiate, mercury-induced neuronal degeneration and the underlying mechanisms remain largely unknown. In this study, we first explored the potential involvement of Ca2+ in mercury toxicity because Ca2+ is an important integrator of neuronal viability and excitability. The proper regulation of intracellular Ca2+ level plays an important role in regulation of growth cone motility, neurite initiation and outgrowth [59-61]. Intracellular Ca2+ levels either fall below or rise significantly above an optimal range have been shown to inhibit both of the growth cone motility and neurite outgrowth [59,62-64]. The data presented in this study show that HgCl2 triggered a sustained rise in [Ca2+i in all cortical neurons examined and this effect on Ca2+ is not reversible (data not shown). The sustained elevation of [Ca2+i in cortical neurons by HgCl2 may thus last for a long term and reach a level which would be detrimental for growth cone motility, neurite outgrowth and hence the network assembly during early neuronal development. Our data further demonstrated that HgCl2 also induced degradation of mature neurite and network connectivity (Figure 3 &6) suggesting that neuronal ultrastructure components such as cytoskeleton proteins may also be affected by HgCl2 –induced disturbance in Ca2+ homeostasis. In support of this hypothesis, our study demonstrates that HgCl2 indeed induced disassembly of cytoskeleton, primarily the β-tubulin proteins in cortical neurons. Our findings are consistent with previous studies showing that mercury induce disintegration of β -tubulin protein in several other species [39,65-67]. Neuronal cytoskeletal proteins have been shown to be extremely sensitive to intracellular Ca2+ levels and their assembly and disassembly can be influenced by Ca2+ either directly or indirectly via regulation of cytoskeleton associated proteins such as tau, a tubulin binding protein [68,69]. For instance, studies have shown that experimentally-induced, sustained elevation of [Ca2+i either by Ca2+ ionophores, or depolarizing agents causes hyperphosphorylation of tau resulting in microtubule depolymerization and neuronal degeneration in cultured human cortical neurons [69]. As the microtubule cytoskeleton forms the basis for not only the structural integrity, but also for functional communications between neurons, the damage to the microtubules may result in abnormal physiological functions of the brain and hence aberrant animal behaviors.
Because Ca2+ is also an important regulator of cell excitability and gene expression, its entry (via NMDA receptors)-mediated synaptic activity has been shown to play crucial roles in neuronal development, synaptic plasticity, cell survival, and synaptic circuitry refinement [47,48,50]. Disturbance of neuronal activity even within one element of the network has been found to perturb the development of the entire circuitry and its physiological functions [47,50]. Considering the predominant effects of HgCl2 on NMDA receptor-mediated synaptic current inputs and membrane discharges in pyramidal cells, we postulated that Ca2+ entry via NMDA receptors may alter the membrane excitability and cellular signaling in pyramidal neurons resulting in a deficit in overall network activity and/or Ca2+ homeostasis. This is supported by our observations that HgCl2-induced rise in Ca2+ occurred almost simultaneously in a group of neurons within a network. The disturbance of Ca2+ homeostatic or other cellular signaling pathways resulting from an over stimulation of NMDA receptors might ultimately lead to, cytoskeleton disruption and cell death which are hallmarks of HgCl2-induced glutamatergic excitoxicity. Future studies are however required to decipher the precise involvement of NMDA receptors by HgCl2. In regard to the possibility of mercury directly modulating NMDA receptor function, the sulfhydral group in cysteine residues of NMDA receptors has been found to be the potential targets for regulation of NMDA receptor function by redox or alkylating agents. Oxidizing agents are known to decrease while sulfhydryl reducing agents potentiate NMDA receptor activity [70,71]. For instance, Tang and Aizenman (1993) demonstrated that alkylation of the NMDA redox site by the sulfhydryl alkylating agent N-ethylmaleimide (MEM) enhanced NMDA receptor response in cortical neurons [72]. Based on the fact that HgCl2 is a small molecular as MEM and has high affinity to sulfhydryl group, it is possible that HgCl2 may potentiate NMDA receptor function by directly interacting with its cysteine residue sulfhydryl groups, rendering the NMDA receptor dysfunctional. These postulates however need to be determined experimentally.
Conclusions
Taken together, our data demonstrate that pre exposure of cells to MK 801 could remarkably inhibit HgCl2-induced increase in neuronal excitability, Ca2+ rise, and importantly prevent the damage to cytoskeleton components and network integrity. Our data suggest that over activation of NMDA receptors is likely the initial trigger for the downstream of cascades involved in HgCl2-induced toxicity. Inhibition of NMDA receptor activity is a potential and effective way to block the initiation of injurious effect of mercury in central neurons.
Methods
Cortical neuron cultures
All animal procedures were approved by the University of Calgary institutional animal use and care committee. Conditions were met with the standards established by the Canadian Council on Animal Care. The culture of cortical neuron was made using Sprague–Dawley rat pups on the day they were born. Dissociated cortical neurons were plated onto cover slips coated with poly-D-lysine (30 μg/ml, Sigma P6407) and laminin (2 μg/ml, Sigma L2020). Cortical neurons were cultured in neurobasal medium (Invitrogen, no. 21103–049) supplemented with 2% B27 (Invitrogen, no. 17504–044), L-Glutamine (200 mM) (Invitrogen, no. 25030–081), 4% FBS (Invitrogen, no. 12483–020), and penicillin-streptomycin (Invitrogen, no. 15140–122). Approximately 80% of the culture media was replaced every 3–4 days. Cultures were maintained at 37°C in an incubator circulated with air and 5% carbon dioxide. For counting studies, cortical neurons were plated onto cover slips with grids (Bellco Glass, no. 1916–91818).
To study the effect of Mercury (II) Chloride (HgCl2) on neuronal process initiation, neurite outgrowth and network formation, freshly dissociated cortical neurons were cultured either in the absence (control) or presence of different concentrations of HgCl2 (25 nM, 100 nM and 25 μM) for 3 days. Neurite outgrowth, cell viability and network formation were evaluated on day 3. To examine the acute effect (up to 24 hrs) of HgCl2 on newly established neuronal processes and network, cells were first cultured for 4 days to allow for the establishment of neurite outgrowth and network. Neurons were then exposed to HgCl2 at different concentrations overnight, and the effects of HgCl2 were examined the next day. To study the chronic effect (7 days exposure) of HgCl2 on well-established mature network, 10 day-old cells cultured on cover slips with grid were exposed to different HgCl2 concentrations and the effect was evaluated on day 17.
Live/dead cell viability assay for cortical neurons
To determine and quantify the impact of HgCl2 on cell viability, cortical cultures that were maintained in control or drug-treated conditions were subsequently loaded with the LIVEDEAD® ViabilityCytotoxicity Kit (Molecular probes, L-3224) for 30 mins at room temperature (21-22°C). This two-color assay was developed based on the fact that intracellular esterase activity and an intact plasma membrane are unique characteristics of live cells. The LIVEDEAD® ViabilityCytotoxicity Kit discriminates live from dead cells by simultaneously staining with green-fluorescent calcein-AM to indicate intracellular esterase activity and red-fluorescent ethidium homodimer-1 to indicate loss of plasma membrane integrity. Preparations were visualized using confocal microscopy (LSM 510 Meta, Zeiss, Germany) under a 10x or 20x objective at 488 nm excitation (green) and 548 nm (red) wavelength and images were collected using a band pass filter (560–615 nm). The number of cells labeled with both colors was subsequently counted using imageJ software.
Whole-cell patch-clamp recordings in cortical neurons
Whole-cell recordings of synaptic inward currents and membrane potentials were performed on pyramidal cortical neurons after 7 to 15 days in culture at room temperature (21-22°C) using a Multiclamp 700B amplifier (Axon Instruments; Sunnyvale, CA, USA) connected to an analog-to-digital interface Digidata 1322 (Axon Instruments). Signals were acquired and stored through pClamp 9.2 software (Axon Instruments). Synaptic currents were recorded under voltage-clamp mode with the holding potential of −70 mV throughout. Membrane potentials of pyramidal cells were recorded under current-clamp mode. The external solution contained (in mM) NaCl, 135; CaCl2, 3; KCl, 5; MgCl2, 2; HEPES, 10; D-Glucose, 10; pH adjusted to 7.3 with NaOH. The internal pipette solution was composed of (in mM) K+-Gluconate, 130; NaCl, 7; MgCl2, 0.3; HEPES, 10; EGTA, 0.1, ATP-Mg, 3; GTP-Na, 0.6; pH adjusted to 7.3 with KOH. The osmolarity for internal solution was approximately 300 mOsm (295–305) and for external solution approximately 330 mOsm (320–340). Borosilicate patch pipettes (A-M Systems, Inc; Sequim, WA, USA) were pulled using a horizontal micropipette puller (Model P-97, Sutter instrument Co., USA) and had a tip resistance ranging from 3–5 MΩ after filling with internal solutions. Only cells with series resistances less than ~15 MΩ and leaks less than ~100 pA were selected for the analysis. Data were analyzed using Clampfit 9.0 software (Axon Instruments), and traces were plotted using OriginPro 8.0 SRO (OriginLab Corporation; Northampton, MA, USA).
Ca2+ Imaging
Fura-2 acetoxymethyl ester (AM) ratiometric Ca2+ imaging experiments were performed to study the action of HgCl2 and/or N-methyl-D-Aspatate (NMDA) receptor activity on intracellular Ca2+ level in cortical neurons. Specifically, neurons that were cultured for 5 to 10 days were loaded with the membrane permeable and ratiometric Ca2+ sensor Fura-2 AM (Molecular Probes, Carlsbad, CA) at 5 μM for 30 mins at 37°C. Cells were subsequently washed three times with normal saline solution containing 0 Mg2+ and 3 μM glycine to activate NMDA receptor at resting level. The composition of normal saline included (in mM): NaCl, 135; CaCl2, 3; KCl, 5; HEPES, 10; D-Glucose, 10; pH adjusted to 7.3 with NaOH. Culture dishes were then mounted on an inverted microscope and neurons were exposed to excitation wavelengths of 340 and 380 nm delivered using a LAMBDA DG4 high-speed wavelength switcher (Sutter Instrument, Novato, CA) through a 40x oil objective. The emitted fluorescence signal was collected at 510 nm by a Regina Exi camera. Images were acquired with the Northern Eclipse software ionwave program (Empix Imaging, Canada). The change in intracellular Ca2+ level was reflected by the ratio of fluorescence intensity obtained at 340 nm and 380 nm.
Immunochemistry and confocal microscopy
Cultured cells were fixed for 1 h with pre-warmed 4% paraformaldehyde and subsequently washed four times with 1x PBS. Cell were permeabilized for 1 h with incubation media (IM) (0.5% Triton in 1x PBS with 10% goat serum) and then incubated overnight with monoclonal anti-β-tubulin antibody produced in mouse (1:500) (Sigma, T0198). Cells were rinsed two times with 1x PBS the next day. Cells were then incubated with AlexaFluor 488 goat anti-mouse Ig6 secondary antibody (1:100) (Invitrogen, A-11001) for 1 h at room temperature (21-22°C) under dark conditions. Cultures were subsequently rinsed two times with 1x PBS and incubated for 30 minutes with rhodamine phalloidin (1:20) (Invitrogen, no. R415). Following two time wash with 1 x PBS and one quick rinse with double distilled H2O cover slips were mounted using MOWIOL mounting media with 4’6-diamidino-2-phenylindole dihydrochloride (Sigma-Aldrich). Samples were viewed using confocal microscopy (LSM 510 Meta, Zeiss, Germany) under a 20x dry or 40x water objective at 488 nm excitation (green, β-tubulin) and 548 nm (red, F-actin) wavelength. Images were collected using a band pass filter (560–615 nm). To assess the level of ß-tubulin and F-actin staining, image acquisition parameters for control and drug-treated neurons were kept the same.
Chemicals
All chemicals including Mercury (II) chloride (HgCl2, M1136) and MK 801 (M107) were purchased from Sigma-Aldrich (Oakville, Ontario, Canada). Fura-2 acetoxymethyl ester (AM) was purchased from (Molecular Probes, Carlsbad, CA). HgCl2 was dissolved in deionized water (for electrophysiological and Ca2+ imaging experiments), or in culture medium (for toxic study) to a stock concentration of 10 mM or 10 μM. The applied solutions (0.025-25 μM) were diluted with normal external solution or culture medium.
Statistical analysis
Data were analyzed statistically using Student’s t test or one-way analysis of variance (ANOVA) as appropriate. Post hoc analysis was conducted using Tukey’s test. Values were considered statistically significant at the level of P < 0.05. The data are presented as mean ± S. E.M. Each experiment was replicated a minimum of four times; the actual number of replicates for each experiment is listed in the corresponding figure legend or in the text.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FX, SF, SS, FXZ and LC conducted experiments and data analyses. Specifically, FX, SF, SS, LC performed the toxicity and cell viability study. FX and FXZ conducted the calcium imaging and electrophysiological experiments. FX, NIS and GWZ drafted and edited the manuscript. All authors read and approved the final manuscript.
Contributor Information
Fenglian Xu, Email: fxu@ucalgary.ca.
Svetlana Farkas, Email: sfarkasc@ucalgary.ca.
Simone Kortbeek, Email: skortbeek@me.com.
Fang-Xiong Zhang, Email: fxzhang@ucalgary.ca.
Lina Chen, Email: linchen@ucalgary.ca.
Gerald W Zamponi, Email: zamponi@ucalgary.ca.
Naweed I Syed, Email: nisyed@ucalgary.ca.
Acknowledgements
This work was supported by Natural Sciences and Engineering Research Council (NSERC) grant to NIS. GWZ is supported by a grant from the Canadian Institutes of Health Research (CIHR). He is a Canada research Chair and AI-HS Scientist. FXZ is supported by the Hotchkiss Brain Institute Fong fellowship and Alberta Innovate- Health Solutions fellowship.
References
- Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol. 2006;36:609–662. doi: 10.1080/10408440600845619. [DOI] [PubMed] [Google Scholar]
- Clarkson TW. The Three Modern Faces of Mercury. Environ Health Perspect. 2002;110(Suppl 1):11–23. doi: 10.1289/ehp.02110s111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BH. Kim R C and Peckham N H: Hydrocephalus Following Prenatal Methylmercury Poisoning. Acta Neuropathologica. 1988;75:325–330. doi: 10.1007/BF00687784. [DOI] [PubMed] [Google Scholar]
- Clarkson TW, Magos L, Myers GJ. The Toxicology of Mercury – Current Exposures and Clinical Manifestations. N Engl J Med. 2003;349:1731–1737. doi: 10.1056/NEJMra022471. [DOI] [PubMed] [Google Scholar]
- Meyer-Baron M, Schaeper M, Seeber A. A Meta-Analysis for Neurobehavioural Results Due to Occupational Mercury Exposure. Arch Toxicol. 2002;76:127–136. doi: 10.1007/s00204-002-0327-9. [DOI] [PubMed] [Google Scholar]
- Mutter J, Naumann J, Schneider R, Walach H, Haley B. Mercury and Autism: Accelerating Evidence? Neuroendocrinol Lett. 2005;26:439–446. [PubMed] [Google Scholar]
- Ferraro L, Tomasini MC, Tanganelli S, Mazza R, Coluccia A, Carratu MR, Gaetani S, Cuomo V, Antonelli T. Developmental exposure to Methylmercury elicits early cell death in the cerebral cortex and long-term memory deficits in the rat. Int J Dev Neurosci. 2009;27:165–174. doi: 10.1016/j.ijdevneu.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Eto K, Yasutake A, Kuwana T, Korogi Y, Akima M, Shimozeki T, Tokunaqa H, Kaneko Y. Methylmercury poisoning in common marmosets–a study of selective vulnerability within the cerebral cortex. Toxicol Pathol. 2001;29(5):565–573. doi: 10.1080/019262301317226375. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Vilaro MT, Babot Z, Rodriguez-Farre E, Sunol C. Mercury Compounds Disrupt Neuronal Glutamate Transport in Cultured Mouse Cerebellar Granule Cells. J Neurosci Res. 2005;79:545–553. doi: 10.1002/jnr.20375. [DOI] [PubMed] [Google Scholar]
- Korogi Y, Takahashi M, Shinzato J, Okajima T, Farina MR. et al. Findings in 7 Patients With Organic Mercury-Poisoning (Minamata-Disease) Am J Neuroradiol. 2011;1994(15):1575–1578. [PMC free article] [PubMed] [Google Scholar]
- Farina M, Rocha JBT, Aschner M. Mechanisms of Methylmercury- Induced Neurotoxicity: Evidence From Experimental Studies. Life Sci. 2011;89:555–563. doi: 10.1016/j.lfs.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekel M, Platt B, Busselberg D. Mercury (Hg2+) Decreases Voltage-Gated Calcium-Channel Currents in Rat Drg and Aplysia Neurons. Brain Research. 1993;632:121–126. doi: 10.1016/0006-8993(93)91146-J. [DOI] [PubMed] [Google Scholar]
- Peng SQ, Hajela RK, Atchison WD. Effects of Methylmercury on Human Neuronal L-Type Calcium Channels Transiently Expressed in Human Embryonic Kidney Cells (HEK-293) J Pharmacol Exp Ther. 2002;302:424–432. doi: 10.1124/jpet.102.032748. [DOI] [PubMed] [Google Scholar]
- Sirois JE, Atchison WD. Effects of Mercurials on Ligand- and Voltage-Gated Ion Channels: A Review. Neuro toxicology. 1996;17:63–84. [PubMed] [Google Scholar]
- Castoldi AF, Coccini T, Ceccatelli S, Manzo L. Neurotoxicity and Molecular Effects of Methylmercury. Brain Res Bull. 2001;55:197–203. doi: 10.1016/S0361-9230(01)00458-0. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Atchison WD. Methylmercury Differentially Affects GABAA Receptor-Mediated Spontaneous IPSCs in Purkinje and Granule Cells of Rat Cerebellar Slices. Journal of Physiology-London. 2003;550:191–204. doi: 10.1113/jphysiol.2003.040543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan YK, Atchison WD. Methylmercury Induces a Spontaneous, Transient Slow Inward Chloride Current in Purkinje Cells of Rat Cerebellar Slices. J Pharmacol Exp Ther. 2005;313:751–764. doi: 10.1124/jpet.104.080721. [DOI] [PubMed] [Google Scholar]
- Castoldi AF, Barni S, Turin I, Gandini C, Manzo L. Early Acute Necrosis, Delayed Apoptosis and Cytoskeletal Breakdown in Cultured Cerebellar Granule Neurons Exposed to Methylmercury. J Neurosci Res. 2000;59:775–787. doi: 10.1002/(SICI)1097-4547(20000315)59:6<775::AID-JNR10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Juarez BI, Portillo-Salazar H, Gonzalez-Amaro R, Mandeville P, Aguirre JR, Jimenez ME. Participation of N-Methyl-D-Aspartate Receptors on Methylmercury-Induced DNA Damage in Rat Frontal Cortex. Toxicology. 2005;207:223–229. doi: 10.1016/j.tox.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Insug O, Datar S, Koch CJ, Shapiro IM, Shenker BJ. Mercuric Compounds Inhibit Human Monocyte Function by Inducing Apoptosis: Evidence for Formation of Reactive Oxygen Species, Development of Mitochondrial Membrane Permeability Transition and Loss of Reductive Reserve. Toxicology. 1997;124:211–224. doi: 10.1016/S0300-483X(97)00153-4. [DOI] [PubMed] [Google Scholar]
- Limke TL, Atchison WD. Acute Exposure to Methylmercury Opens the Mitochondrial Permeability Transition Pore in Rat Cerebellar Granule Cells. Toxicol Appl Pharmacol. 2002;178:52–61. doi: 10.1006/taap.2001.9327. [DOI] [PubMed] [Google Scholar]
- Atchison WD, Hare MF. Mechanisms of Methylmercury-Induced Neurotoxicity. FASEB J. 1994;8:622–629. doi: 10.1096/fasebj.8.9.7516300. [DOI] [PubMed] [Google Scholar]
- Aschner M, Yao CP, Allen JW, Tan KH. Methylmercury Alters Glutamate Transport in Astrocytes. Neurochem Int. 2000;37:199–206. doi: 10.1016/S0197-0186(00)00023-1. [DOI] [PubMed] [Google Scholar]
- Allen JW, Mutkus LA, Aschner M. Mercuric Chloride, but Not Methylmercury, Inhibits Glutamine Synthetase Activity in Primary Cultures of Cortical Astrocytes. Brain Research. 2001;891:148–157. doi: 10.1016/S0006-8993(00)03185-1. [DOI] [PubMed] [Google Scholar]
- Brookes N. Invitro Evidence for the Role of Glutamate in the Cns Toxicity of Mercury. Toxicology. 1992;76:245–256. doi: 10.1016/0300-483X(92)90193-I. [DOI] [PubMed] [Google Scholar]
- Basu N, Scheuhammer AM, Rouvinen-Watt K, Grochowina N, Evans RD, O'Brien M, Chan HM. Decreased N-Methyl-D-Aspartic Acid (NMDA) Receptor Levels Are Associated With Mercury Exposure in Wild and Captive Mink. Neuro toxicology. 2007;28:587–593. doi: 10.1016/j.neuro.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Ndountse LT, Chan HM. Methylmercury Increases N-Methyl-D-Aspartate Receptors on Human SH-5Y5Y Neuroblastoma Cells Leading to Neurotoxicity. Toxicology. 2008;249:251–255. doi: 10.1016/j.tox.2008.05.011. [DOI] [PubMed] [Google Scholar]
- Miyamoto K, Nakanishi H, Moriguchi S, Fukuyama N, Eto K, Wakamiya J, Murao K, Arimura K, Osame M. Involvement of Enhanced Sensitivity of N-Methyl-D- Aspartate Receptors in Vulnerability of Developing Cortical Neurons to Methylmercury Neurotoxicity. Brain Research. 2001;901:252–258. doi: 10.1016/S0006-8993(01)02281-8. [DOI] [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Mercury Neurotoxicity - Mechanisms of Blood–brain- Barrier Transport. Neuroscience and Bio behavioral Reviews. 1990;14:169–176. doi: 10.1016/S0149-7634(05)80217-9. [DOI] [PubMed] [Google Scholar]
- Charleston JS, Body RL, Bolender RP, Mottet NK, Vahter ME, Burbacher TM. Changes in the Number of Astrocytes and Microglia in the Thalamus of the Monkey Macaca Fascicularis Following Long-Term Subclinical Methylmercury Exposure. Neuro toxicology. 1996;17:127–138. [PubMed] [Google Scholar]
- Castoldi AF, Coccini T, Ceccatelli S, Manzo L. Neurotoxicity and Molecular Effects of Methylmercury. Brain Res Bull. 2001;55:197–203. doi: 10.1016/S0361-9230(01)00458-0. [DOI] [PubMed] [Google Scholar]
- Sziics A, Angiello C, Salanki J, Carpenter DO. Effects of Inorganic Mercury and Methylmercury on the Ionic Currents of Cultured Rat Hippocampal Neurons. Cell Mol Neurobiol. 1997;17:273–288. doi: 10.1023/A:1026338217097. [DOI] [PubMed] [Google Scholar]
- Friberg L, Mottet NK. Accumulation of Methylmercury and Inorganic Mercury in the Brain. Biological Trace Element Research. 1989;21:201–206. doi: 10.1007/BF02917253. [DOI] [PubMed] [Google Scholar]
- Gopal KV. Neurotoxic Effects of Mercury on Auditory Cortex Networks Growing on Microelectrode Arrays: a Preliminary Analysis. Neuro toxicology and Teratology. 2003;25:365–369. doi: 10.1016/S0892-0362(03)00037-0. [DOI] [PubMed] [Google Scholar]
- Hock C, Drasch G, Golombowski S, Muller-Spahn F, Willershausen-Zonnchen B, Schwarz P, Hock U, Growdon JH, Nitsch RM. Increased Bleed Mercury Levels in Patients With Alzheimer's Disease. Journal of Neural Transmission. 1998;105:59–68. doi: 10.1007/s007020050038. [DOI] [PubMed] [Google Scholar]
- Mutter J, Naumann J, Schneider R, Walach H. Mercury and Alzheimer's Disease. Fortschritte der Neurologie Psychiatrie. 2007;75:528–540. doi: 10.1055/s-2007-959237. [DOI] [PubMed] [Google Scholar]
- Mutter J, Naumann J, Schneider R, Walach H, Haley B. Mercury and Autism: Accelerating Evidence? Neuroendocrinol Lett. 2005;26:439–446. [PubMed] [Google Scholar]
- Sieger FAS, Silva GAD, Ardila GP, Garcia RG. Mercury Chronic Toxicity Might Be Associated to Some Cases of Hydrocephalus in Adult Humans? Medical Hypotheses. 2012;79:13–16. doi: 10.1016/j.mehy.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Leong CCW, Syed NI, Lorscheider FL. Retrograde Degeneration of Neurite Membrane Structural Integrity of Nerve Growth Cones Following in Vitro Exposure to Mercury. Neuro report. 2001;12:733–737. doi: 10.1097/00001756-200103260-00024. [DOI] [PubMed] [Google Scholar]
- Olivieri G, Brack C, Muller-Spahn F, Stahelin HB, Herrmann M, Renard P, Brockhaus M, Hock C. Mercury Induces Cell Cytotoxicity and Oxidative Stress and Increases Beta-Amyloid Secretion and Tau Phosphorylation in SHSY5Y Neuroblastoma Cells. J Neurochem. 2000;74:231–236. doi: 10.1046/j.1471-4159.2000.0740231.x. [DOI] [PubMed] [Google Scholar]
- Poulain FE, Sobel A. The Microtubule Network and Neuronal Morphogenesis: Dynamic and Coordinated Orchestration Through Multiple Players. Mol Cell Neurosci. 2010;43:15–32. doi: 10.1016/j.mcn.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Feng J. Microtubule: A Common Target for Parkin and Parkinson's Disease Toxins. Neuroscientist. 2006;12:469–476. doi: 10.1177/1073858406293853. [DOI] [PubMed] [Google Scholar]
- Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S. Neuro behavioural and Molecular Changes Induced by Methylmercury Exposure During Development. Neurotox Res. 2007;11:241–260. doi: 10.1007/BF03033570. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and Mitochondrial Function in Glutamate-Induced Neuronal Cell Death. FEBS Lett. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- Chung RS, McCormack GH, King AE, West AK, Vickers JC. Glutamate Induces Rapid Loss of Axonal Neuro filament Proteins From Cortical Neurons in Vitro. Exp Neurol. 2005;193:481–488. doi: 10.1016/j.expneurol.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Slemmer JE, De Zeeuw CI, Weber JT. Don't Get Too Excited: Mechanisms of Glutamate-Mediated Purkinje Cell Death. Creating Coordination in the Cerebellum. 2005;148:367–390. doi: 10.1016/S0079-6123(04)48029-7. [DOI] [PubMed] [Google Scholar]
- Blankenship AG, Feller MB. Mechanisms Underlying Spontaneous Patterned Activity in Developing Neural Circuits. Nat Rev Neurosci. 2010;11:18––29. doi: 10.1038/nrn2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM. Activity Regulates the Synaptic Localization of the NMDA Receptor in Hippocampal Neurons. Neuron. 1997;19:801–812. doi: 10.1016/S0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- Shu YS, Hasenstaub A, McCormick DA. Turning on and Off Recurrent Balanced Cortical Activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Thivierge JP. How Does Non-Random Spontaneous Activity Contribute to Brain Development? Neural Networks. 2009;22:901–912. doi: 10.1016/j.neunet.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic Calcium Overload in a Subpopulation of Mitochondria Triggers Delayed Death in Hippocampal Neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of Cell Death: The Calcium-Apoptosis Link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan WZ. "Apoptotic" Biochemical Cascades in Synaptic Compartments: Roles in Adaptive Plasticity and Neurodegenerative Disorders. J Neurosci Res. 1999;58:152–166. doi: 10.1002/(SICI)1097-4547(19991001)58:1<152::AID-JNR15>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Pendergrass JC, Haley BE. Inhibition of Brain Tubulin-Guanosine 5'-Triphosphate Interactions by Mercury: Similarity to Observations in Alzheimer's Diseased Brain. Metal Ions in Biological Systems. 1997;34:461–478. [PubMed] [Google Scholar]
- de Assis GPS, Silva CEC, Stefanon I, Vassallo DV. Effects of Small Concentrations of Mercury on the Contractile Activity of the Rat Ventricular Myocardium. Comparative Biochemistry and Physiology C-Toxicology & Pharmacology. 2003;134:375–383. doi: 10.1016/S1532-0456(03)00005-X. [DOI] [PubMed] [Google Scholar]
- Wiggers GA, Stefanon I, Padilha AS, Pecanha FM, Vassallo DV, Oliveira EM. Low Nanomolar Concentration of Mercury Chloride Increases Vascular Reactivity to Phenylephrine and Local Angiotensin Production in Rats. Comparative Biochemistry and Physiology C-Toxicology & Pharmacology. 2008;147:252–260. doi: 10.1016/j.cbpc.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Xu F, Luk C, Richard MP, Zaidi W, Farkas S, Getz A, Lee A, van MJ, Syed NI. Antidepressant Fluoxetine Suppresses Neuronal Growth From Both Vertebrate and Invertebrate Neurons and Perturbs Synapse Formation Between Lymnaea Neurons. Eur J Neurosci. 2018;31:994–1005. doi: 10.1111/j.1460-9568.2010.07129.x. [DOI] [PubMed] [Google Scholar]
- Farina M, Rocha JBT, Aschner M. Mechanisms of Methylmercury- Induced Neurotoxicity: Evidence From Experimental Studies. Life Sci. 2011;89:555–563. doi: 10.1016/j.lfs.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kater SB, Mills LR. Regulation of Growth Cone Behavior by Calcium. J Neurosci. 1991;11:891–899. doi: 10.1523/JNEUROSCI.11-04-00891.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike T. Nerve Growth Factor-Induced Neurite Outgrowth of Rat Pheochromocytoma PC-12 Cells - Dependence on Extracellular Mg2+ and Ca2+ Brain Research. 1983;289:293–303. doi: 10.1016/0006-8993(83)90030-6. [DOI] [PubMed] [Google Scholar]
- Schubert D, Lacorbiere M, Whitlock C, Stallcup W. Alterations in the surface properties of cells responsive to nerve growth factor. Nature. 1978;273:718–721. doi: 10.1038/273718a0. [DOI] [PubMed] [Google Scholar]
- Almohanna FA, Cave J, Bolsover SR. A Narrow Window of Intracellular Calcium-Concentration Is Optimal for Neurite Outgrowth in Rat Sensory Neurons. Developmental Brain Research. 1992;70:287–290. doi: 10.1016/0165-3806(92)90209-F. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium As Sculptor and Destroyer of Neural Circuitry. Exp Gerontol. 1992;27:29–49. doi: 10.1016/0531-5565(92)90027-W. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Kater SB. Calcium Regulation of Neurite Elongation and Growth Cone Motility. J Neurosci. 1987;7:4034–4043. doi: 10.1523/JNEUROSCI.07-12-04034.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura K, Inokawa M, Imura N. Effects of Methylmercury and Some Metal-Ions on Microtubule Networks in Mouse Glioma-Cells and Invitro Tubulin Polymerization. Toxicol Appl Pharmacol. 1984;73:218–231. doi: 10.1016/0041-008X(84)90327-2. [DOI] [PubMed] [Google Scholar]
- Stoiber T, Degen GH, Bolt HM, Unger E. Interaction of Mercury(II) With the Microtubule Cytoskeleton in IMR-32 Neuroblastoma Cells. Toxicol Lett. 2004;151:99–104. doi: 10.1016/j.toxlet.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Cadrin M, Wasteneys GO, Jonesvilleneuve EMV, Brown DL, Reuhl KR. Effects of Methylmercury on Retinoic Acid-Induced Neuroectodermal Derivatives of Embryonal Carcinoma-Cells. Cell Biology and Toxicology. 1988;4:61–80. doi: 10.1007/BF00141287. [DOI] [PubMed] [Google Scholar]
- Weisenbe RC. Microtubule Formation In-Vitro in Solutions Containing LowCalcium Concentrations. Science. 1972;177:1104. doi: 10.1126/science.177.4054.1104. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Engle MG, Rychlik B. Effects of Elevated Intracellular Calcium Levels on the Cytoskeleton and Tau in Cultured Human Cortical- Neurons. Mol Chem Neuropathol. 1991;15:117–142. doi: 10.1007/BF03159951. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Lipton SA, Loring RH. Selective Modulation of Nmda Responses by Reduction and Oxidation. Neuron. 1989;2:1257–1263. doi: 10.1016/0896-6273(89)90310-3. [DOI] [PubMed] [Google Scholar]
- Lazarewicz JW, Wroblewski JT, Palmer ME, Costa E. Reduction of Disulfide Bonds Activates Nmda-Sensitive Glutamate Receptors in Primary Cultures of Cerebellar Granule Cells. Neurosci Res Commun. 1989;4:91–97. [Google Scholar]
- Tang LH, Aizenman E. The Modulation of N-Methyl-D-Aspartate Receptors by Redox and Alkylating Reagents in Rat Cortical-Neurons In vitro. Journal of Physiology-London. 1993;465:303–323. doi: 10.1113/jphysiol.1993.sp019678. [DOI] [PMC free article] [PubMed] [Google Scholar]