

Abstract
The fluctuation of population size has not been well studied in the previous studies of theoretical linkage disequilibrium (LD) expectation. In this study, an improved theoretical prediction of LD decay was derived to account for the effects of changes in effective population sizes. The equation was used to estimate effective population size (Ne) assuming a constant Ne and LD at equilibrium, and these Ne estimates implied the past changes of Ne for a certain number of generations until equilibrium, which differed based on recombination rate. As the influence of recent population history on the Ne estimates is larger than old population history, recent changes in population size can be inferred more accurately than old changes. The theoretical predictions based on this improved expression showed accurate agreement with the simulated values. When applied to human genome data, the detailed recent history of human populations was obtained. The inferred past population history of each population showed good correspondence with historical studies. Specifically, four populations (three African ancestries and one Mexican ancestry) showed population growth that was significantly less than that of other populations, and two populations originated from China showed prominent exponential growth. During the examination of overall LD decay in the human genome, a selection pressure on chromosome 14, the gephyrin gene, was observed in all populations.
Introduction
Linkage disequilibrium (LD) is an important parameter in population genetics. The LD in the human genome has been used to determine the association between variants and traits [1], and efforts to understand selection pressures have been based largely on the LD status of populations [2]–[5], The theoretical basis of expectations for LD was established by the pioneering efforts of theoretical geneticists [6]–[14]. Theoretical studies of LD, which deals with two variables, were more difficult than studying mutations, which dealt with one variable. There are several types of LD, but the most widely used is the squared correlation coefficient, r2. Initial effort on the expectation of LD was based on the diffusion approximations, which indicated that at equilibrium, the ratio of expected values in r2, σd 2, would reach approximately 1/(4Nc) when Nc was larger than 1, where N is the effective population size and c is the recombination rate [7]. Continued efforts enhanced the accuracy of the expectation of r2 based on the ratio of expected values or related improvements [10]–[12], [15]–[18].
Instead of using the ratio of expected values, a recurrence relation was derived for the direct expectation of LD based on a conditional probability of identity by descent at the second locus given identity by descent at the first locus. This approach resulted in the formula 1/(4Nc+1) for small values of c [16], [19], [20]. This formula was derived from the relation where the squared LD (r2) equaled the probability (Q) that two genes at a locus are identical by descent (IBD), given that two genes at a linked locus are IBD [19]. The recurrence relation is indicated below [20]. Including this one, all the previous methods assumed a constant effective population size. As indicated in a previous study [21], the population size of the previous and current generations influences the expected r2 value. Therefore, potential effects of the population size of the previous and current generation should be properly incorporated into a recurrence formula.
Recent advances in genomic technology have enabled the genome-wide observation of LD [22]–[24]. It has been suggested that past population history can be inferred from linkage disequilibrium [25]. Previous studies that estimated Ne based on LD identified recent human population growth [26]–[28]. However, in the method in which the estimation of Ne is based on chromosome segment homozygosity, the assumption of linear Ne with time did not work well with exponential growth or complicated population size changes [27]. In the estimation of Ne based on LD, the method was only applicable to small recombination rates due to approximation [26], [28]. The Ne estimates obtained in these studies did not represent the actual Ne changes at each generation, so it became necessary to simulate results regarding various situations that human populations might have experienced. In addition, the corrections made for sampling errors had significant bias, as noted in a previous study [29].
Despite its importance, there has been no study of the accurate theoretical expectation of squared LD with respect to the fluctuations in effective population sizes at each generation. In this study, a complete theoretical prediction of LD with fluctuating Ne was derived based on the recurrence formula from previous studies [19], [20]. This study investigated the expectation of LD decay involving changes of effective population size under various circumstances, and the results were applied to the human genome using HapMap phase III data to infer the past population history of each human population.
Methods
Theoretical expectation of LD decay in a finite population
If ideal Wright-Fisher population assumptions hold for a given population, variances due to genetic drift and recombination will be the main factors for the decay of linkage disequilibrium. As discussed in the previous study, the simplest random mating system; monoecious with selfing, was considered primarily [12], [21]. In this system, closed populations, discrete generations, and the absence of recurrent mutation were assumed. Let Nn be the population size at the nth generation, and let Nn−1 be the population size at the (n−1)th generation. The population sampling procedure from one generation to the next can be described as follows: 1) sampling of 2Nn individuals from Nn−1 with replacement; 2) generation of gametes from each individual among 2Nn individuals selected; and 3) random pairing of the gametes that were generated. The probability of each gamete differs depending on the recombination rate.
Considering that the probability that genes at both loci are identical by descent equals r2 [19], the influence of effective population size for the previous and current generations was incorporated into the recurrence formula from previous studies [20]. There are two samplings in the population sampling procedure: 1) a sampling of an individual from 2Nn−1 gametes and 2) a sampling of gametes from a gamete pool generated from 1). The sampling of an individual from Nn−1 means a sample of two gametes from the available pool, generated at the previous generation. Here, as in the previous study [19], sampling with replacement was assumed. When both haplotypes of the selected individual are IBD with a probability of 1/(2Nn−1), recombination at the current generation does not change the IBD status. Therefore, an additional departure of LD, c2/(2Nn−1), should be added for the portion of recombination. All the generated gametes are IBD, and a further sampling procedure provides no influence on the IBD status. For the gamete portion with no recombination, the proportion of recombinants in the probability, 1/(2Nn−1), is 2c/(2Nn−1). Therefore, the rest, 1−2c/(2Nn−1), should be applied to the portion without recombination. The random sampling from the generated gamete pool consisted of 2Nn gametes produced the formula described in the Introduction. At this time, there is no further recombination. This approach finally leads to Eq. 1, in which r2 n−1 is the LD of the previous generation, and Nn−1 and Nn represent the effective sizes of the previous and current generations, respectively.
| (1) |
Eq. 1 was verified through simulations using various extreme values for Nn, Nn−1, r2 n−1, and c (Table S1). The detailed simulation procedure is described in the following simulation section. As indicated in the previous study [19], the assumption for sampling with replacement involves a sample size that is not excessively small. For a similar reason, the equation could show expectations deviated from real data for the case of extreme allele frequencies with a small population size.
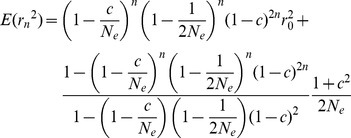 |
(2) |
In Eq. 1, changes in the effective population size were reflected in the linkage disequilibrium. When Ne is constant, the general expression for n generations is Eq. 2. At equilibrium, rn 2 becomes equal to the rn−1 2, which can be expressed as (1+c2)/((1−(1−1/(2Ne))(1−c/Ne)(1−c)2)×2Ne). This expression can be derived from Eq. 2, when the generation (n) approaches infinity. For a constant recombination rate, the expected linkage disequilibrium at equilibrium depended only on the effective population size. If the equilibrium is defined as occurring when the difference between rn 2 and req 2 became less than 1/(2Ne), which is chosen because the r2 values at equilibrium are dependent on population size and the choice makes the equation simpler, the required n generation can be expressed as Eq. 3. As shown in Table 1, the time required to reach equilibrium differed depending on the recombination rate and the effective population size. As the effective population size became large and the recombination rate became small, more time was required to reach equilibrium. However, when the value of r2 at equilibrium is very high (Ne = 100 and c = 0.0001), the time required to reach equilibrium may be expected to decrease.
Table 1. The generation (age) until equilibrium (when the difference between generation becomes less than 1/(2Ne)) depending on Ne and recombination rate (c)).
| Ne | 100 | 500 | 1000 | 5000 | 10000 | |||||
| c | age | E(r2) | age | E(r2) | age | E(r2) | age | E(r2) | age | E(r2) |
| 0.5 | 4 | 0.00831 | 5 | 0.00167 | 11 | 0.00083 | 13 | 0.00017 | 14 | 0.00008 |
| 0.4 | 5 | 0.00902 | 7 | 0.00181 | 15 | 0.00091 | 18 | 0.00018 | 19 | 0.00009 |
| 0.3 | 7 | 0.01060 | 10 | 0.00213 | 21 | 0.00107 | 26 | 0.00021 | 28 | 0.00011 |
| 0.2 | 12 | 0.01427 | 15 | 0.00288 | 34 | 0.00144 | 41 | 0.00029 | 44 | 0.00014 |
| 0.1 | 24 | 0.02592 | 33 | 0.00529 | 72 | 0.00265 | 87 | 0.00053 | 94 | 0.00027 |
| 0.05 | 49 | 0.04892 | 67 | 0.01018 | 147 | 0.00511 | 179 | 0.00103 | 193 | 0.00051 |
| 0.01 | 201 | 0.20084 | 325 | 0.04785 | 717 | 0.02451 | 907 | 0.00500 | 979 | 0.00251 |
| 0.001 | 576 | 0.71440 | 2165 | 0.33345 | 4913 | 0.20008 | 8323 | 0.04764 | 9395 | 0.02440 |
| 0.0001 | 391 | 0.96154 | 4261 | 0.83334 | 10576 | 0.71430 | 44018 | 0.33334 | 64490 | 0.20001 |
| From MAF* | 269 | 1346 | 2692 | 13460 | 26920 | |||||
(*: mean allele age when minor allele frequency (MAF) is 0.4 [53]).
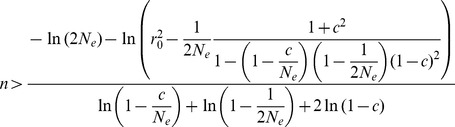 |
(3) |
Data
HapMap Phase III genotype data from the HapMap project, which included the original and expanded HapMap samples, were used for the estimations [22]–[24], [30]. The original HapMap samples were collected from four geographically diverse populations: Yoruba in Ibadan, Nigeria (YRI); Japanese in Tokyo, Japan (JPT); Han Chinese in Beijing, China (CHB); and CEPH (Utah, USA residents with ancestry from northern and western Europe, CEU). Additional samples were collected from seven populations: Maasai in Kinyawa, Kenya (MKK); Luhya in Webuye, Kenya (LWK); Chinese in metropolitan Denver, CO, USA (CHD); Gujarati Indians in Houston, TX, USA (GIH); Tuscans in Italy (TSI); African ancestry in the southwest USA (ASW); and Mexican ancestry in Los Angeles, CA, USA (MEX). The ASW, CEU, MEX, MKK, and YRI were family samples, and only the parents (indicated as ASWp, CEUp, MEXp, MKKp, and YRIp, respectively) were used in this data analysis. A total of 1198 samples were analyzed. For the accuracy of estimates, single nucleotide polymorphisms (SNPs) without missing data were used for analysis of the HapMap data.
Simulation
Simulation was conducted with 1,000 pairs of diallelic loci for various population sizes based on the previous model. Starting from the complete LD (r2 = 1), only two initial haplotypes for each pair at the first generation were generated by a binomial draw based on allele frequencies of 0.5. The LD decay was based on a population sampling procedure similar to that used previously [21]. Let Nep be the population size at the previous generation and Nec be the size at the current generation. First, 2Nec individuals were selected from Nep individuals for mating. Second, one transmitting gamete was generated from each of the 2Nec individuals. When the individual presented haplotypes such that its recombinants were distinguishable from the original haplotypes, whether the gamete would be a recombinant was determined by binomial draw based on the recombination rate. A gamete haplotype was then selected from two haplotypes, either recombinant or not. Finally, random pairings of the gametes generated Nec individuals. This procedure was repeated for N generations. During simulations, frequent allele frequencies were maintained (≥0.1 and ≤0.9), by repeating population sampling with the frequencies below 0.1 or over 0.9.
When the allele frequency is extreme for a given population size, only several limited r2 values are available, which deviates from the expectation of r2. The maintenance of the allele frequencies within a certain range was arranged to avoid such a phenomenon. In reality, these simulation results did not show a distinguishable difference from simulation results with allele frequencies maintained between 0 and 1. All the simulations in this study were executed using the R statistical package with additional C++ coding of the core computation. The generation of every random number was based on the R statistical package.
The simulation results were exactly matched with the equations. As shown in Figure 1, when linkage disequilibrium decayed from the complete linkage disequilibrium value of 1, the simulation results (dots) corresponded well with the expected values (lines) derived from Eqs. 1 and 2. The generations at equilibrium were derived from Eq. 3, and the simulated values showed excellent correspondence with the theoretical equilibrium value, even reflecting the difference of 1/(2Ne), as shown in Figure 1A. To examine LD as the population size changed, the three situations that were most likely, i.e., increment, decrement, and bottleneck of population size, were selected. As shown in Figure 1B, the results indicated excellent agreement between the theoretical and simulated values for all three situations.
Figure 1. Comparisons between theoretical (line) and simulated (dot) r2.

(A) Ne constant: for constant Ne = 100 and 1000, the r2 values at equilibrium decayed from the complete LD are plotted. The 1, 2, and 3 generations indicate r2 values with the decay generation(s) from the complete LD when Ne equaled 1000. (B) Ne changes: After reaching equilibrium with a constant Ne of 1000, three different circumstances of population size were applied for five generations, i.e., increments (1000,1100,1200,1300,1400), decrements (1000 ,500,300,200,100), and bottlenecks (1000,100,100,1000,1000).
Sampling and errors
Sampling induces additional departures to the expected r2 value [12]. When the r2 value of the original population was zero and the haplotype frequencies were estimated using maximum likelihood methods, the sampling variance becomes 1/ns+1/(2Ne), where ns is the sample size and Ne is the original population size [10], [12], [21], [25]. When the expected linkage disequilibrium was not zero and the direct identification of haplotypes was possible, the amount of sampling variance due to ro 2 was reduced by a factor of (1−ro 2), where ro 2 was the original LD before sampling. Eq. 1 simply yielded the expected r2 as (1−1/2ns)r2 0+1/2ns, assuming no recombination. This result is consistent with previous studies [19], [20], where the probability that two genes at both loci are IBD for a selected individual in a sampled population provided the same r2 expectation. For a sampled population, the probability that both genes descend from the same gene is not dependent on the original population size but rather on the sampled population size.
This expectation is applicable when the direct identification of haplotypes is available. The maximum likelihood estimation of haplotype frequencies induces additional departures similar to the case of linkage equilibrium [10], [12], [21], [25]. The sampling variances from the likelihood estimation of haplotype frequencies showed almost the same trends that ro 2 reduced by a factor of (1−ro 2), as the variances obtained from the direct identification of haplotypes, with small mean squared errors based on simulations (Table S2). Increasing the sample size reduced the mean squared error. Therefore, the expectation of linkage disequilibrium due to sampling can be expressed approximately as Eq. 4. Here, rs 2 indicates the linkage disequilibrium of the sampled population. When linkage disequilibrium was not zero and haplotype frequencies were estimated using maximum likelihood methods, an accurate equation was difficult to obtain [10]. Since Eq. 4 fit relatively well, the current study utilized Eq. 4 to obtain r2 values for the original populations.
| (4) |
For simulations, the sampling of ns individuals among Nec individuals with replacement was conducted after the population sampling procedure described above. The allele frequencies of the original population were 0.5, and the r2 0 values were based on fixed haplotype frequencies. The haplotype frequencies for two loci were estimated from the sampled genotypes based on the expectation maximization (EM) algorithm. To examine the difference between the original r2 and the sampled r2, the mean squared errors were obtained from simulations for a population size of 1,000 and various sample sizes (Table S2).
LD and Ne estimation
The LD was measured with distances in each chromosome for each population in the HapMap data, and only autosomes were examined. In samples with an insufficient sample size, SNPs with small minor allele frequencies resulted in r2 values deviated from expectations, due to limited values in the frequencies of haplotypes that consist of minor alleles. To avoid extreme values that impede the correct expectation of r2 at equilibrium, SNPs with minor allele frequencies (MAF) higher than 0.4 were used for estimating r2. In addition, the LD between aged SNPs at equilibrium should be used for Ne estimation. SNPs with high minor allele frequencies would be a reasonable choice for aged SNPs. The haplotype frequencies were derived using the EM algorithm.
In each chromosome, the distances between variants were separated as a unit of 10,000, and the mean of r2 estimates in each unit were used to examine LD decay depending on recombination rates in the human genome. Recombination rates varied depending on genomic region [31], but it was clear that there was an apparent correlation between distance and recombination rate. In addition, the population differences observed in a recent study, in which fine-scale recombination rates were estimated based on the Icelandic genealogy database [32]. Therefore, the recombination rates were derived directly from the distances between variants. The Haldane map was used to convert distances to recombination rates, and 1 Mb was considered to represent approximately 130 cM [33].
Assuming that the effective population size has been constant, Ne estimates with respect to recombination rates were obtained, which enabled inferences regarding past changes of effective population size. For the calculation, the average r2 of all chromosomes except chromosome 14 was used because selection pressures were observed in a large region of chromosome 14 for all populations (see Results section). Since the expression of the sampling variance involved the current effective population size, the current effective population size should be estimated first, based on deviations from the Hardy-Weinberg equilibrium [21]. Relying on Eq. 4, the r2 of the original population (E(r2) in Eq. 5) was obtained and used to find Ne from Eq. 5 by solving a cubic equation derived from Eq. 1, assuming constant Ne except for the current generation. Thus, the r2 at the previous generation became (1+c2)/((1−(1−1/(2Ne))(1−c/Ne)(1−c)2)×2Ne). In Eq. 5, Ne is the constant effective population size, Nc is the current effective population size, and c is the recombination rate. For theoretical predictions for various situations, the same method was applied using the known Nc.
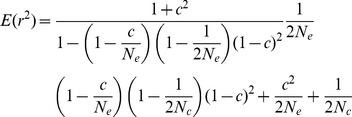 |
(5) |
Descriptions of various population histories for theoretical predictions
To infer past population history from patterns of LD according to recombination rates, three basic changes in population size, increment, decrement, and bottleneck were examined, and simulations were conducted based on possible human population histories. For the human population histories, the overall world population history was first examined and then three important events, increments, decrements and bottlenecks, were further investigated. Because the Ne estimates reflect the recent population history much more than the old population history, the recent population history was examined in priority.
Three basic changes in population size
Because the ancient population histories were not very influential in the Ne estimates for recombination rates (higher than 0.0065), the most recent 100 generations became the focus. For both increment and decrement, three different situations, continuous, terraced, and exponential, were studied (Figure 2). The population size changed from 100 to 1100 or vice versa, and the changes for 100 generations were started when the r2 of the population reached equilibrium for a given initial population size (from Eq. 3). The continuous increment or decrement involved 10 increments or decrements per generation for 100 generations. In the terraced increment, size changes occurred five times for 100 generations, with a rate of 200 per generation. For the exponential changes, the population size increased or decreased exponentially with a base of 1.0366 for 100 generations (Nn = Nn−1+1.0366n; n: 1∼100; the exponential part was rounded). The bottleneck was studied for two different terms of time, duration, and amount of reduction. Therefore, eight different bottlenecks were examined with a population size of 1100: 1) bottleneck for 1∼10 generations with a population size of 100; 2) for 1∼5 generations with a size of 100; 3) for 1∼10 generations with a size of 600; 4) for 1∼5 generations with a size of 600; 5) for 86∼95 generations with a size of 100; 6) for 91∼95 generations with a size of 100; 7) for 86∼95 generations with a size of 600; and 8) for 91∼95 generations with a size of 600.
Figure 2. Three basic changes in population size and the differences in r2 and Ne estimates.

(A) Continuous, terraced, and exponential increment; (B) Continuous, terraced, and exponential decrement; (C) Variable bottlenecks based on time, duration, and amount of reduction.
Real world population history
The global population change from BC 10,000 to AD 2000 [34] was used. Population sizes for the years from 1970 to 2000 were estimates [34]; these estimates fit well with actual population changes and were used in the current study. The average length of human life was less than 36 years before the 18th century but has increased in recent years [35]. Two thousand years ago in Rome, life expectancy was only 22 years. Therefore, differential generation times were applied. Specifically, until AD 500, the generation time was assumed to be 20 years; from AD 500–1400, 22 years; from AD 1400–1900, 25 years; and from AD 1900–2000, 30 years, thus making the total generations for the period from BC 10,000 to AD 2000 result in a sum of up to 4,989 generations (Figure S1). For Ne, the million unit was omitted so that 200 million was considered as 200 for effective population size.
Exponential growth of effective population size
To more closely examine the impact of the recent exponential growth in human population on the estimate of effective population size, three different exponential growths in the most recent generations were modeled. For the first, a constant population size of 100 was sustained for 4969 generations; the population was then exponentially increased with a base of 1.42 for 20 generations, giving a final population size of 3851. The second model featured a constant population size of 100 for 4979 generations; the size of the population was then increased exponentially with a base of 2.4 for 10 generations, giving a final population size of 10,968. In the third model, two different increasing situations were applied. In the first situation, a constant population size of 100 was applied for 4984 generations; the size was then exponentially increased for five generations with a base of 10, producing a final population size of 111,210 (exponential increment 3a in Figure 3). In the second situation, the population size was exponentially increased for the last five generations with a base of 6, yielding a final population size of 9430 (exponential increment 3b in Figure 3).
Figure 3. Past population changes and Ne estimates for various situations.

(A) Various increments of population size; (B) Ne estimates based on (A); (C) A decrement and various bottlenecks in population size; (D) Ne estimates based on (C).
Continuous increment and decrement of effective population size
A continuous increment for the last 89 generations was modeled after a constant population size of 1000. The increment size was 20 per generation. For the model of decreasing effective population size, a constant population size of 3000 was maintained for 4900 generations, and the decrement was continued for 89 generations. The amount of decrement was constant at 20 per generation.
Bottleneck of effective population size
In real human population history, several bottlenecks have occurred. To examine the impact of bottlenecks on the estimates obtained in this work, bottlenecks in three different periods were modeled. One period included generations 4916 to 4925 starting at approximately AD 500 (Bottleneck 1 in Figure 3). Another bottleneck period included generations 4962 to 4966 in the 13th century [36], when the black death raged (Bottleneck 2a in Figure 3). Since more recent bottlenecks would have a greater impact on the Ne estimates, the bottleneck for five recent generations (4970∼4985) was modeled (Bottleneck 3 in Figure 3). In the model, the original population size was 1000, and a 50% reduction was modeled for these bottlenecks. To examine the effects of a greater reduction in population, the second bottleneck was repeated with an 80% reduction (Bottleneck 2b in Figure 3).
Results
LD expectation depending on recombination rates and Ne changes
The past Ne change can be inferred from the changes in Ne estimates according to recombination rates. Table 1 shows the time to equilibrium when constant population size was assumed. Smaller effective population size leads to faster equilibrium. More importantly, as the recombination rate became smaller, the LD between polymorphisms decayed more slowly so that the r2 value represented the changes of effective population sizes for a longer period of time (Table 1). Assuming constant population size, the estimates of Ne reflected past changes of effective population sizes, among which the more recent changes were more highly reflected. Depending on recombination rate, each Ne estimate represented Ne change in a different period of time. The Ne estimates derived from smaller recombination rates represented longer past changes of effective population size. Therefore, approximate inferences regarding past Ne changes were acquired from the estimates depending on the recombination rates.
Figure 2 shows the differences in r2 values and Ne estimates depending on the recombination rates for the various changes in population sizes. The Ne estimates show clear differences for population size changes, even when the r2 values show no big differences. Continuous increments in population size gave almost constant Ne estimates after steep increments at very small recombination rates. The terraced increment showed smaller Ne estimates at large recombination rates than did the continuous increment. Compared to the continuous increment, the exponential increment showed smoother Ne increments, as the recombination rate increased. The same trend was shown for the decrement. Similar to the continuous increment in population size, a continuous decrement in population size gave almost constant Ne estimates after steep decrements at very small recombination rates. The bottlenecks for the 1∼10 generations were not effective for the Ne estimates when the recombination rates were greater than 0.1, but the recent bottlenecks for the 86∼95 generations showed large differences at higher recombination rates. This result indicates that the earlier bottleneck might not be detectable in these Ne estimates and thus we need a detailed examination of Ne estimates in extremely small recombination rates. The size of the change in the bottleneck influenced the amount of reduction in Ne estimates, depending on the recombination rate.
Figure 3 shows Ne estimates for various situations according to human population history. Since the main interest in the current study was the human genome, Ne changes relevant to the history of the human population were examined intensively. Figures 3A and 3B show several exponential increments of population size, including a circumstance based on the real-world human population history [34]. Changes in actual human population size showed a steep increment in Ne estimates depending on recombination rates. As observed in Figure 2, there is a clear exponential increment during recent generations. To explain the Ne estimates based on the HapMap data, three exponential increments that differed in the period of exponential increment, i.e. the last 20, 10, and 5 generations, were examined. As the start point of an exponential increment approached the current generation, the increment in the Ne estimate became steeper at small recombination rates. The magnitude of bases influenced the shape at large recombination rates. As the recombination rate increased, the exponential increment 3a with an exponential base of 10 showed a steeper increment than the exponential increment 3b with an exponential base of 6.
Figures 3C and 3D show that the decrement of Ne decreased the Ne estimates as the recombination rate increased and that bottlenecks caused reductions in the Ne estimates at certain recombination rates. The Ne estimates could be changed depending on the time and amount of the decrement. The impacts of a bottleneck differed depending on the time, duration, and amount of reduction from the original population, as in Figure 2. The time at which the bottleneck began affected the reduction of the Ne estimates at certain recombination rates, and as the duration of the bottleneck increased, its impact on the reduction of the Ne estimate lasted longer. The amount of reduction from the original population directly determined the extent of reduction of the Ne estimates.
LD decay and past population history in the human genome
The theoretical expectation derived in the current study was applied to the LD in the human genome using HapMap data. Figure 4 indicates the LD decay in the human genome based on recombination rates, which can be obtained by converting distances. Each chromosome is indicated by a different color; most of the chromosomes showed similar LD decay overall. The dashed line in Figure 4 indicates the expected r2 at equilibrium when the effective population size is constant and the same as the estimate of current effective population size. It is clear from the figure that all the populations had larger current than past effective population sizes.
Figure 4. LD decay of the human genome depending on recombination rates.

(Dashed lines represent the expected r2 at equilibrium assuming constant Ne with the Ne estimate of the current generation).
Four populations, ASWp, CHB, CHD, and JPT, showed increases in the r2 estimates in chromosome 14. When the positions of variants with high r2 values were examined in the four populations, all the variants in the regions from approximately 65,700 kb to 66,800 kb had high LDs between them, creating a large linkage disequilibrium block (Figure S2). Using variants with all frequencies, the same regions in all the other populations showed a similar LD block. Since the current study only used variants with a minor allele frequency (MAF) higher than 0.4, variants with all frequencies were examined within the specified region. By examining the frequency spectra of the region from 65,500 kb to 67,500 kb, extraordinary spectra were found for all populations; in these spectra the variants with specific frequencies occurred much more often than the variants with other frequencies (Figure S3).
The foregoing phenomenon was expected when selective sweeps occurred in a specific genomic region [2], [5]. This region had not been previously identified as a major region of selective pressure. This specific LD block region in chromosome 14 harbors the complete gephyrin gene (GPHN) (Figure S2). This gene encodes a multifunctional protein that catalyzed the molybdenum cofactor biosynthesis in the liver, kidney, and other non-neuronal organs and that plays a role in the postsynaptic targeting and clustering of glycine and GABAA receptors at inhibitory synapses [37]. The gephyrin gene had a complex intron-exon structure and multiple splice sites. Isoforms of the protein are expressed in a tissue-specific manner and with apparent species-specific differences [37]. The center of the LD block was located within intron 2 of this gene.
As shown in Table 1, the time required to reach equilibrium differed depending on recombination rate and effective population size. Over time, the population sizes had been changed, and the LD, which depends on recombination rate, can indicate past changes in effective population size. The effective population size was estimated depending on the recombination rates. In this estimate of Ne, the current effective population size was not involved. Figure 5 shows that all the populations experienced recent exponential increases in population size, confirming the previous results [27]. From the results shown in Figures 2 and 3, it can be concluded that this exponential increment occurred specifically in most recent generations, probably less than five generations before the current generation; this finding is reasonably acceptable considering the recent explosive growth in the human population.
Figure 5. Ne estimates of human population samples depending on recombination rates.

Two distinctive patterns are illustrated in Figure 5. One is the occurrence of an extreme exponential increment in several very recent generations, shown in CEUp, CHB, CHD, GIH, JPT, TSI, and YRIp; the other is a less extreme exponential increment in the effective population size, shown in ASWp, LWK, MEXp, and MKKp. The former populations showed exponential increments with extremely large magnitudes, whereas the latter populations displayed increments of much smaller magnitudes. The major differences between these two groups result from a very recent, more explosive population growth in one group. When the Ne estimates for small recombination rates (less than 0.3) were examined, all four populations of African descent, ASWp, LWK, MKKp, and YRIp showed slightly larger Ne estimates than those of other populations, suggesting that long ago their population sizes were larger than those of other populations (see the last part of Figure 5 and Figure S4.) This result supports the occurrence of an out-of-Africa expansion.
It was noteworthy that of the populations with milder past growth, one was of African descent, two were African populations, and one was of Mexican ancestry. Among the populations with African ancestry, only YRIp showed a pattern that was similar to other populations. It is clear that suppressive factors impeded population growth in these four populations in comparison to others. As shown in a previous study [21], the current results also indicate that the recent immigrant populations show more similar trends in their countries of origin than in the countries to which they immigrated (CHD, GIH, and MEXp). These results are not surprising because these samples were collected from individuals who identified themselves as having at least three out of four grandparents belonging to their original population. As illustrated in the panel of Figure 5, bottlenecks were observed in many populations, including CEUp and TSI at the approximate time when the black death occurred [36]. However, the effects of these bottlenecks were relatively minor compared to the exponential growth.
Past population history derived from the genome and historical studies
The Ne estimates obtained using HapMap data effectively represent the recent extreme exponential growth in the human population. These results verify previous observations of human population growth [26], [27] and provide further detailed information regarding the individual population histories. Four populations, i.e., ASWp, LWK, MEXp, and MKKp, showed mild exponential growth in effective population size, and CHB and CHD showed prominent exponential growth. Among the four population samples of African descents, YRIp was the only population, showing population growth similar to most other populations. To verify these results, historical reports were examined in this section.
Historically, the slave trade and colonial invasion impeded population growth among Africans in recent centuries [38]. In this region, independence from colonial domination began after World War II ended in 1945 [39]. Both LWK and MKKp are Kenyan and thus might be expected to be relatively less affected by the initial slave trade due to regional effects; however historians have suspected that there was at least a slight population reduction in East Africa during the colonial domination period [38]. The current results partially support this idea and suggest that East Africa did not experience such recent explosive population growth that occurred on most other continents.
Ibadan is located on the West African coast near the Bight of Benin, a central historical port for the slave trade. However, YRIp showed population growth more similar to that of CEUp and TSI than to that of LWK and MKKp. In the early 18th century, Oyo, the Yoruba kingdom located near Ibadan, was an important supplier of slaves, who were mostly acquired in the conquest of surrounding countries [38], [40]. This expanded the money supply and brought trading activities into the region [38], [40], which might have augmented the population growth, making it similar to the growth on other continents.
During the colonial domination period in Africa, most women were married as soon as they reached child-bearing age, and polygamy was relatively common until recently [38]. It is also likely that the average lifetime in Africa during this period was shorter than on other continents. It therefore follows that the time per generation was shorter in Africa than on other continents. On most continents, if a steep population increase occurred during the last 100 years, this increment occurred over three to four generations. However, for Africans, the increment might have occurred over four to five generations, and this should be considered in the interpretations of relevant samples. The relatively early starting points of the exponential growth in four populations of African descent in the last panel of Figure 5 might be explained by this difference.
The ASWp consists of people of African descent who, for the most part, were moved involuntarily to the U.S. due to the slave trade [41]. During their transportation to America, only half the enslaved population survived and remained available for work [38]. Although slavery was abolished in the U.S. in 1865, apparent racial discrimination persisted until 1964 [41], which could have been an influence on the relatively impeded population growth of this population. People in the MEXp sample had Mexican ancestry. Mexicans suffered a recent bottleneck after the colonial invasion, and the population was not restored to half its size at the time of the voyage of Columbus until the late 18th century [42]. Therefore, the relatively small increments observed in both ASWp and MEXp in Figure 5 would be reasonable, but strong bottleneck effects were not observed in these populations compared to other populations.
Polygamy reduces the effective population size [43]; this may partly explain the results with LWK and MKKp, in which the culture persisted up to date. In addition, there might be an unknown effect in admixed populations such as ASWp and MEXp [44]. However, it is obvious that the results obtained with these four population samples indicate that their population growth rate in recent generations has been much smaller than that of other populations, and these results correspond with historical studies.
CHB and CHD were prominent among samples with extreme exponential growth. China has experienced almost continuous population growth from 1400 to the present, except for the period from 1683 to 1700, during which a population decrement occurred [45]. China is currently the world's most populous country. Thus, extreme exponential growth could be expected in the population samples originally from China. Colonial invasion of China began in the late 19th century and lasted for a very short period. In India, colonial invasion began in the late 18th century; however the colonization of India was much more cautious and very different from that of Africa and America [46]. The length of the colonial period in India was approximately 100 years longer than in China; this might explain why the population growth of GIH was not as steep as that of CHB and CHD, even though India is the second most populous country in the world.
Discussion
The current study provides an actual expression of the expectation of LD decay depending on recombination rate and changes in effective population size. The theoretical expectation derived in this study showed an accurate agreement with simulation results. In addition, this study provides an advanced solution for sampling variances in LD estimations. Using the expression, the past population history can be derived from LD data. Naturally, the recent population history has a greater influence on the Ne estimates than does the old population history. Applications to the human genome showed good agreement with the changes in human population that have occurred in relatively recent generations. By enhancing the accuracy of r2 expectation, the current study appears to be useful for studying the patterns of linkage disequilibrium in relation to changes of effective population size, providing a better understanding of past population histories.
However, it should also be noted that the current method provided only an approximate picture what type of population changes the sampled population had experienced, especially with respect to recent generations. For better inferences, a model fitting based on a likelihood could be helpful. Because smaller recombination rates need more time to reach equilibrium, the mean linkage disequilibrium of small recombination rate would involve a wider range of ancient population changes. If the allele ages could be estimated accurately and the inaccuracies due to extreme frequencies and selection pressures were appropriately adjusted, a serial estimation of the effective population size from current to ancient generations might be possible. In addition, the method is based on the assumption of a closed population, which is not true in the real world. Migrations should also be considered for the application to real populations. In that case, a more accurate picture of past population histories could be presented.
The selection pressure on chromosome 14 found in this study was unexpected. Although the LD block was larger than 1 Mb, previous studies that attempted to identify regions particularly affected by selection pressures in the human genome did not detect this region in a selective sweep using the HapMap data [24], [30], [47], [48]. Earlier studies did not list the region because it did not seem significant enough in their result tables representing extensive data analyses. One study, which used HapMap data, listed this region as under strong selection pressure in supplementary information that provided a summary of the strongest regions of selection in Europeans [49]. Another analysis, which used the Perlegen data, did not identify GPHN as a gene under strong selection pressure, but supplementary information indicated that this gene region fell within the significant selective sweep with a p-value of ∼0.0006 in the Chinese sample [4], [50]. In European-American and African-American samples, the p-values of ∼0.10 and ∼0.83, respectively, associated with this region were not significant [50]. A study aimed at identifying positively selected genes by comparing human and chimpanzee genomes showed no evidence for selection pressure on GPHN [51], presumably indicating that the selection pressure on this gene might have appeared relatively recently. Inconsistent results between these methods for finding selection pressures were well indicated in a previous review [52]. Further studies are necessary to elucidate the understanding of the selection pressures in this region and to assess the population differences in more detail.
In the current study, the theoretical expectation for change in effective population size was exactly matched with the simulation results. In the sampling expectation derived from Eq. 4 in the Methods section, the mean squared errors of the expectation after sampling were quite small. However, detailed simulation studies indicated that there was a more complicated relationship between the population LD, ro 2, and the sampling variances than suggested by Eq. 4. When direct acquisition of haplotype frequencies without likelihood estimation was tested, the sampling estimates provided the exact relationship between ro 2 and the variances. Therefore, either the development of an accurate expectation for the sampled genotype data or the direct identification of haplotypes is necessary for solving the problem of residual sampling variances. Since the errors were relatively minor and influenced the data equally, the results of the current estimation might not differ greatly.
Even considering this slight inaccuracy in sampling expectations, the current study provides a much more accurate expectation of LD for sampling than previous studies [26]–[28]. Previously, the reduction of sampling variances was not counted when the original LD was not zero, which may have serious bias into the results. The uncorrected sampling bias inflated Ne estimates at small recombination rates, especially 10,000∼20,000 years ago in their timeframe. The large Ne (up to several thousand individuals) at small recombination rates might result from this bias. In the present study, the bias was largely corrected, and very small Ne values (less than 50) was observed for very small recombination rates. The current results are more reasonable because in the past most people lived in very small communities and available mating was quite limited due to limited transportation systems.
Previously [26]–[28], Ne estimates were considered as the Ne at 1/(2c) generation ago. However, as shown in Table 1, the estimates merely represented the past Ne changes over time until equilibrium. Neither was the value for the generations until equilibrium equivalent to 1/(2c) nor did the estimates indicate the Ne at a certain number of generations in the past. Therefore, this approximate timeframe, 1/(2c), seems not appropriate, and simulations or other methods should be accompanied to infer the past population history properly. In addition, the previous studies were based on approximations that are applicable only to small recombination rates, suggesting that their observations on recent population increments might be biased as well. On the contrary, the current method is accurate for any recombination rate, when the population size is not too small and the frequencies are not extreme.
In this study, variants with MAF higher than 0.4 were used, and most of the variants were of sufficient age to have reached equilibrium, as shown in Table 1. However, for Ne estimates at very small recombination rates, the allele ages might not be sufficient. The smallest recombination rate used in this study was 0.0065, and most of the recombination rates used in this study appear to be safely within the range to result in equilibrium for variants with MAF greater than 0.4. The LD expectation in this study assumed decay from the complete LD value of one between the frequent variants. In most cases, the variants started from single mutation events, and the starting LD (r2) was not one unless they occurred on the same haplotype at the same time. Nevertheless, most aged variants at equilibrium would still provide valid estimates reflecting the Ne changes for different lengths of generations depending on the recombination rates.
In summary, the current study provides the first actual LD expectation regarding changes of effective population sizes. This advanced LD expression enables a more accurate inference of past Ne changes. In addition, this study provides a more accurate method of correcting sample size effect when LD is not zero. The application of the method to human genome data gave good agreement with the actual population changes documented in historical studies. This method provides a simple and accurate picture of the population changes the sampled populations had experienced, which is applicable to other populations of interest. The results of this study confirmed that LD in the genome would be a useful source for obtaining a detailed population history.
Supporting Information
The reconstituted population history based on generation time. (A) from BC 10,000 to AD 2000; (B) an enlargement of (A) from AD 0 to AD 2000.
(PDF)
The LD plot for the region from 65,500 kb to 67,500 kb in chromosome 14 using CHB data (data with missing genotype less than 1% and no monomorphic site).
(PDF)
The histogram of minor allele frequency for the region from 65,500 kb to 67,500 kb in chromosome 14.
(PDF)
The enlargement of the last figure of Figure 4 ; Ne estimates of human population samples depending on recombination rates up to 0.3.
(PDF)
Verification of Eq. 1 by simulations using extreme values.
(XLSX)
Mean squared errors of sampling from fixed r2 for various sampling sizes with 10,000 SNP pairs and a population size of 1000 (r2 o: the original r2 of a population; ss: sample size).
(DOC)
Acknowledgments
The author appreciates the reviewers' comments, which substantially improved the quality of this study.
Funding Statement
This work was supported by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2007-532-C00017) and by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2009-353-C00061). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Weir BS (2008) Linkage disequilibrium and association mapping. Annu Rev Genomics Hum Genet 9: 129–142. [DOI] [PubMed] [Google Scholar]
- 2. Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, et al. (2002) Detecting recent positive selection in the human genome from haplotype structure. Nature 419: 832–837. [DOI] [PubMed] [Google Scholar]
- 4. Nielsen R, Williamson S, Kim Y, Hubisz MJ, Clark AG, et al. (2005) Genomic scans for selective sweeps using SNP data. Genome Res 15: 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim Y, Stephan W (2002) Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160: 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright S (1969) Evolution and the Genetics of Populations: The University of Chicago Press.
- 7.Kimura M, Ohta T (1971) Theoretical Aspects of Population Genetics. Princeton: Princeton University Press.
- 8.Weir BS (1996) Genetic Data Analysis II. Sunderland: Sinauer Associates, Inc.
- 9.Hartl DL, Clark AG (2007) Principles of Population Genetics. Sunderland: Sinauer Associates, Inc.
- 10. Hill WG (1974) Estimation of linkage disequilibrium in randomly mating populations. Heredity 33: 229–239. [DOI] [PubMed] [Google Scholar]
- 11. Hill WG (1975) Linkage disequilibrium among multiple neutral alleles produced by mutation in finite population. Theor Popul Biol 8: 117–126. [DOI] [PubMed] [Google Scholar]
- 12. Weir BS, Hill WG (1980) Effect of mating structure on variation in linkage disequilibrium. Genetics 95: 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ewens WJ (2004) Mathematical Population Genetics; S AS, Marsden JE, Sirovich L, Wiggins S, editors:Springer.
- 14.Feldman MW, editor (1989) Mathematical Evolutionary Theory: Princeton University Press.
- 15. Hill WG, Robertson A (1968) Linkage disequilibrium in Finite Populations. Theor Appl Genet 38: 226–231. [DOI] [PubMed] [Google Scholar]
- 16. Hill WG (1977) Correlation of gene frequencies between neutral linked genes in finite populations. Theor Popul Biol 11: 239–248. [DOI] [PubMed] [Google Scholar]
- 17. Hill WG, Weir BS (1988) Variances and covariances of squared linkage disequilibria in finite populations. Theor Popul Biol 33: 54–78. [DOI] [PubMed] [Google Scholar]
- 18. Song YS, Song JS (2007) Analytic computation of the expectation of the linkage disequilibrium coefficient r2. Theor Popul Biol 71: 49–60. [DOI] [PubMed] [Google Scholar]
- 19. Sved JA (1971) Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor Popul Biol 2: 125–141. [DOI] [PubMed] [Google Scholar]
- 20. Sved JA, Feldman MW (1973) Correlation and probability methods for one and two loci. Theor Popul Biol 4: 129–132. [DOI] [PubMed] [Google Scholar]
- 21. Park L (2011) Effective population size of current human population. Genet Res (Camb) 93: 105–114. [DOI] [PubMed] [Google Scholar]
- 22. The International HapMap Consortium (2003) The International HapMap Project. Nature 426: 789–796. [DOI] [PubMed] [Google Scholar]
- 23. The International HapMap Consortium (2005) A haplotype map of the human genome. Nature 437: 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature 467: 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38: 209–216. [Google Scholar]
- 26. Tenesa A, Navarro P, Hayes BJ, Duffy DL, Clarke GM, et al. (2007) Recent human effective population size estimated from linkage disequilibrium. Genome Res 17: 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayes BJ, Visscher PM, McPartlan HC, Goddard ME (2003) Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res 13: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McEvoy BP, Powell JE, Goddard ME, Visscher PM (2011) Human population dispersal “Out of Africa” estimated from linkage disequilibrium and allele frequencies of SNPs. Genome Res 21: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sved JA, McRae AF, Visscher PM (2008) Divergence between human populations estimated from linkage disequilibrium. Am J Hum Genet 83: 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, et al. (2004) The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584. [DOI] [PubMed] [Google Scholar]
- 32. Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, et al. (2010) Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103. [DOI] [PubMed] [Google Scholar]
- 33. Yu A, Zhao C, Fan Y, Jang W, Mungall AJ, et al. (2001) Comparison of human genetic and sequence-based physical maps. Nature 409: 951–953. [DOI] [PubMed] [Google Scholar]
- 34.McEvedy C, Jones R (1978) Atlas of World Population History: Penguin Books Ltd.
- 35.Pressat R (1970) Population. Baltimore: Penguin Books Inc.
- 36.Scott S, Duncan C (2004) Return of the Black Death: The world's Greatest Serial Killer: John Wiley & Sons, Ltd.
- 37. Fritschy JM, Harvey RJ, Schwarz G (2008) Gephyrin: where do we stand, where do we go? Trends Neurosci 31: 257–264. [DOI] [PubMed] [Google Scholar]
- 38.Iliffe J (1996) Africans: The history of a continent: Cambridge University Press.
- 39.van Dijk L (2005) Die Ceschichte Afrikas. Ahn I, translator: Campus Verlag GmbH.
- 40.Manning P (1990) Slavery and African Life: Cambridge University Press.
- 41.Whitney F, editor (2004) An Outline of American History: Bureau of International Information Programs, U.S. Department of State.
- 42.Villegas DC, Bernal I, Toscano AM, Gonzalez L, Blanquel E (1991) Historia Minima de Mexico. Ko H, translator: Dankook University Press.
- 43. Nomura T (2005) Effective population size under random mating with a finite number of matings. Genetics 171: 1441–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Araki H, Waples RS, Blouin MS (2007) A potential bias in the temporal method for estimating Ne in admixed populations under natural selection. Mol Ecol 16: 2261–2271. [DOI] [PubMed] [Google Scholar]
- 45.Ho P-t (1994) Sutdies on the Population of China, 1368∼1953. Jung C, translator. Seoul: BookWorld.
- 46.Spear P (1993) India, a Modern History. Lee O, translator. Seoul: Shingu Press.
- 47. Grossman SR, Shylakhter I, Karlsson EK, Byrne EH, Morales S, et al. (2010) A composite of multiple signals distinguishes causal variants in regions of positive selection. Science 327: 883–886. [DOI] [PubMed] [Google Scholar]
- 48. Barreiro LB, Laval G, Quach H, Patin E, Quintana-Murci L (2008) Natural selection has driven population differentiation in modern humans. Nat Genet 40: 340–345. [DOI] [PubMed] [Google Scholar]
- 49. Voight BF, Kudaravalli S, Wen X, Pritchard JK (2006) A map of recent positive selection in the human genome. PLoS Biol 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, et al. (2007) Localizing recent adaptive evolution in the human genome. PLoS Genet 3: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nielsen R, Bustamante C, Clark AG, Glanowski S, Sackton TB, et al. (2005) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol 3: e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG (2007) Recent and ongoing selection in the human genome. Nat Rev Genet 8: 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kimura M, Ota T (1973) The age of a neutral mutant persisting in a finite population. Genetics 75: 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The reconstituted population history based on generation time. (A) from BC 10,000 to AD 2000; (B) an enlargement of (A) from AD 0 to AD 2000.
(PDF)
The LD plot for the region from 65,500 kb to 67,500 kb in chromosome 14 using CHB data (data with missing genotype less than 1% and no monomorphic site).
(PDF)
The histogram of minor allele frequency for the region from 65,500 kb to 67,500 kb in chromosome 14.
(PDF)
The enlargement of the last figure of Figure 4 ; Ne estimates of human population samples depending on recombination rates up to 0.3.
(PDF)
Verification of Eq. 1 by simulations using extreme values.
(XLSX)
Mean squared errors of sampling from fixed r2 for various sampling sizes with 10,000 SNP pairs and a population size of 1000 (r2 o: the original r2 of a population; ss: sample size).
(DOC)