

Abstract
Background
The FabAB pathway is one of the unsaturated fatty acid (UFA) synthesis pathways for Pseudomonas aeruginosa. It was previously noted that this operon was upregulated in biofilms and repressed by exogenous UFAs. Deletion of a 30 nt fabA upstream sequence, which is conserved in P. aeruginosa, P. putida, and P. syringae, led to a significant decrease in fabA transcription, suggesting positive regulation by an unknown positive regulatory mechanism.
Methods/Principal Findings
Here, genetic and biochemical approaches were employed to identify a potential fabAB activator. Deletion of candidate genes such as PA1611 or PA1627 was performed to determine if any of these gene products act as a fabAB activator. However, none of these genes were involved in the regulation of fabAB transcription. Use of mariner-based random mutagenesis to screen for fabA activator(s) showed that several genes encoding unknown functions, rpoN and DesA may be involved in fabA regulation, but probably via indirect mechanisms. Biochemical attempts performed did fail to isolate an activator of fabAB operon.
Conclusion/Significance
The data suggest that fabA expression might not be regulated by protein-binding, but by a distinct mechanism such as a regulatory RNA-based mechanism.
Introduction
The common opportunistic human pathogen, Pseudomonas aeruginosa, causes serious infections in immunocompromised or cystic fibrosis patients. Fatty acid synthesis is essential for cellular function by providing metabolic precursors for synthesis of many cellular components. The fatty acid biosynthetic enzymes are therefore potential targets for new antibacterials. It was previously established that the biosynthesis of fatty acids in P. aeruginosa closely resembles the pathway established in Escherichia coli, and consists of two phases, initiation and elongation (reviewed in [1]). Besides exhibiting dehydratase activity, FabA also catalyzes the isomerization of trans fatty acids produced during de novo synthesis of fatty acids containing a cis-double bond. These UFAs are then condensed with malony-ACP by FabB, thus bypassing the FabI (enoyl-ACP reductase)-catalyzed step and maintaining the double bond. In subsequent rounds of elongation, full-length UFAs are formed [2]. This pathway is the main route for producing the 16:1Δ9- and 18:1Δ11-ACP thioesters that are incorporated into phospholipids by the glycerol-phosphate and acyl-glycerol-phosphate acyltransferases (PlsB and PlsC) [3].
Since UFAs are required for maintaining the fluidity of bacterial membranes, numerous studies on UFAs have been performed in many prokaryotes, especially E. coli. However, the pathways of UFA synthesis remained mostly uncharacterized in P. aeruginosa.
In a recent study, it was demonstrated that UFA biosynthesis in P. aeruginosa can occur via three different pathways depending on the availability of oxygen: 1) the aerobic pathways encoded by the fatty acid desaturase genes desA and desBC [3], and 2) anaerobic pathway encoded by the fabAB operon [2]. It was shown that a ΔfabA mutant required UFA supplementation during anaerobic growth, but not during aerobic growth, demonstrating that although the aerobic pathway supports UFA formation, the fabAB operon is indispensable for the UFA synthesis under anaerobic conditions [3].
In E. coli, two distinct transcriptional regulators regulate expression of the fabA and fabB genes involved in UFA synthesis. FadR, which negatively regulates expression of the fad genes involved in fatty acid transport and β-oxidation [4], also acts as a positive transcriptional regulator of fabA and fabB gene expression [5], [6]. It was shown that an E. coli fadR mutant synthesized less UFAs compared to wild-type strains and that fabA(Ts) fadR double mutants required UFA supplementation for growth, suggesting that FadR also functions as an activator of UFA synthesis [7]. In addition, fabA activation by FadR is repressed by exogenous long acyl-CoAs by binding to the FadR protein [6], [8]. Furthermore, fabB was also shown to be positively controlled by FadR because fadR mutants exhibit cerulenin hypersensitivity, and show conditional lethal phenotypes in fadR fabB double mutants [5]. In contrast, FabR negatively controls UFA synthesis in E. coli by binding to similar sequences in the fabA and fabB upstream regions [9]. Although a FabR homologue PA4890 showing 50% similarity to the E. coli protein was found in P. aeruginosa, it seems to play only a minor role, if any, in fabA regulation [3].
Previous searches failed to identify a P. aeruginosa FadR homolog responsible for activation of fabAB transcription in P. aeruginosa [10] and thus, the regulation of UFA synthesis in P. aeruginosa seems distinct from E. coli. Even though the activity of FabA and FabB from P. aeruginosa is very similar to that of the same enzymes from E. coli, there is one obvious distinction: whereas the E. coli fabA and fabB genes are located in two different regions of the chromosome, the P. aeruginosa fabAB genes form an operon [1], [2]. The different organization of these genes may imply the existence of distinct regulatory pathways of UFA biosynthesis in P. aeruginosa.
Here, we demonstrate that P. aeruginosa fabAB operon expression seems to be controlled by a complex regulatory network composed of a combination of directly acting factors and indirectly acting transcriptional or translational regulators and other metabolic factors, rather than transcriptional regulators directly acting in the fabAB regulatory region.
Materials and Methods
Bacterial strains, plasmids, media and culture conditions
The bacterial strains and plasmids in this study are listed in Table 1. E. coli and P. aeruginosa strains were maintained on Luria-Bertani medium (LB; 10 g per liter tryptone, 5 g per liter yeast extract, 10 g per liter NaCl; Becton, Dickinson & Co., Sparks, MD). Since P. aeruginosa cells are able to utilize citrate as a sole carbon source and energy source but not E. coli, citrate-based VBMM medium (Vogel-Bonner Minimal Medium; 3.0 g Na3Citrate [citric acid Na3 salt], 2.0 g Citric acid [free acid], 10.0 g K2HPO4, 3.5 g NaNH4PO4×4 H2O, 1 mM MgSO4×7 H2O, and 0.1 mM CaCl2) was used for counter-selection against E. coli after biparental matings between E. coli and P. aeruginosa. For plasmid maintenance in E. coli, the media were supplemented with 100 µg/ml ampicillin, 25 µg/ml chloramphenicol, 35 µg/ml kanamycin or 15 µg/ml gentamycin. For marker selection in P. aeruginosa, 200 µg/ml carbenicillin, 30 µg/ml of gentamycin and 150 µg/ml of spectinomycin was used, as appropriate. If necessary, the monounsaturated fatty acid oleic acid (OA) was added together with 0.05% Brij-58 to solubilize the fatty acid at a final concentration of 0.05%.
Table 1. Bacterial strains and plasmids used in this study.
| Strains or plasmids | Relevant properties | Reference or source | |
| E. coli | |||
| SM10lacIq | thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu Km lacIq | T. Hoang | |
| SM10 (λpir) | thi thr leu tonA lacY supE recA::RP4-2-Tc::Mu Km λpir | [15] | |
| DL291 | F- araD139 Δ(argF-lac)U169 rpsL150 deoC1 relA1 rbsR ptsF25 flbB5301 glpR2 gyrA Δ(glpT-glpA)593 recA1 | [18] | |
| DL291-Tn7 b | Gmr; DL291 derivative containing mini-Tn7T-fabA′-′lacZY gene fusion | This study | |
| P. aeruginosa | |||
| PAO1 | Prototroph | [36] | |
| PAO434 | PAO1 with mini-Tn7T-lacZ | This study | |
| PAO435 | PAO1 with mini-Tn7T-pfabA′-lacZ a resulted from integration of pPS1460 | This study | |
| PAO459 | PAO1 with mini-Tn7T-pfabA′-lacZ resulted from integration of pPS1491 | This study | |
| PAO460 | PAO1 with mini-Tn7T-pfabAΔ30′-lacZ | This study | |
| PAO483 | PAO434 with ΔPA4890::FRT a | This study | |
| PAO484 | PAO435 with ΔPA4890::FRT | This study | |
| PAO491 | PAO434 with ΔPA1539::FRT | This study | |
| PAO492 | PAO435 with ΔPA1539::FRT | This study | |
| PAO495 | PAO460 with ΔPA4890::FRT | This study | |
| PAO497 | PAO460 with ΔPA1539::FRT | This study | |
| PAO517 | PAO435 with ΔPA4890::FRT ΔPA1539::FRT | This study | |
| PAO194 | PAO1 with temperature-sensitive fabA(Ts) | This study | |
| PAO1010 | PAO435 with Δanr::FRT | This study | |
| PAO1151 | Gmra; PAO1 with mini-Tn7T-pPA1612′-lacZ | This study | |
| Plasmid | |||
| pPS752 | Apr; fabA+ fabB + (4.8 kb chromosomal BamHI-EcoRI fragment cloned between the same sites of pUC18) | [2] | |
| pPS790 | Apr; fabA+ fabB + (2,039 bp PCR amplified BamHI-KpnI fragment from pPS752 cloned between the same sites of pUC19) | [2] | |
| pPS1450 | Apr, Gmr; mini-Tn7 delivery vector | [37] | |
| pPS1453 | Apr, Gmr; mini-Tn7 delivery vector with a promoter-less lacZ gene | [37] | |
| pPS1460 | Apr, Gmr; ligation of ∼300 bp SalI-SmaI fragment from pPS790 to XhoI-NruI fragment of pPS1453 | This study | |
| pPS1482 | Apr; pCR2.1 with PCR-amplified PA1611-fabA intergenic region from PAO1 | This study | |
| pPS1488b | Apr; pCR-Blunt II-TOPO with PCR-amplified PA1611-fabA intergenic containing a 30 bp deletion | This study | |
| pPS1491 | Apr, Gmr; pUC18-mini-Tn7T-pfabA′-lacZ delivery vector obtained by ligation of ∼230 bp KpnI-SmaI fragment of pPS1482 into the same sites of pPS1453 | This study | |
| pPS1492 | Apr, Gmr; pUC18-mini-Tn7T-pfabAΔ30::lacZ delivery vector obtained by ligation of ∼230 bp KpnI-SmaI fragment of pPS1488 into the same sites of pPS1453 | This study | |
| pDONR221 | Kmr; Gateway donor vector | Invitrogen | |
| pEX18Ap | Apr; gene replacement vector with MCS from pUC18 | [13] | |
| pEX18ApGW | Apr; Gateway destination vector | [14] | |
| pPS1669 | Apr, Gmr; pUC18-mini-Tn7T-Gm-lacZ-GW | [14] | |
| pFLP2 | Apr; source for Flp recombinase | [13] | |
| pPS856 | Apr, Gmr; Gm-FRT cassette | [13] | |
| pBT20 | Apr, Gmr; mariner transposon delivery vector | S. Lory | |
| pPS1472 | Apr, Kmr; pCR2.1 with amplified PA1611 from PAO1 | This study | |
| pPS1476 | Apr; pEX18Ap-PA1611 (ligation of ∼2.0 kb blunt-ended EcoRI fragment of pPS1472 to PstI+EcoRI cleaved and blunt-ended pEX18Ap) | This study | |
| pPS1479 | Apr, Gmr; pEX18Ap-ΔPA1611::Gm (ligation of blunt-ended KpnI-NcoI fragment of pPS1476 to ∼1.1 kb SmaI Gmr::FRT fragment of pPS856) | This study | |
| pPS1503b | Apr, Gmr; pEX18Ap-ΔPA1627::Gm (ligation of the StuI-XhoI fragment of PCR-amplified ΔPA1627::Gm from PAO1 into the SmaI-SalI sites of pEX18Ap) | This study | |
| pPS1506b | Apr, Gmr; pEX18Ap-ΔPA1539::Gm (ligation of the MscI-XhoI fragment of PCR-amplified ΔPA1539::Gm into the SalI-SmaI sites of pEX18Ap) | This study | |
| pPS1505b | Apr, Gmr; pEX18Ap-ΔPA4890::Gm (ligation of the XhoI-SmaI fragment of PCR amplified ΔPA4890::Gm into SalI-SmaI sites of pEX18Ap | This study | |
| pPS1634 | Apr, Gmr; pUC18R6K-mini-Tn7T-Gm-pfabA::lacZY (ligation of three fragments, BamHI-XhoI fragment of pPS1633, ∼250 bp BamHI-EcoRV fragment of pPS790 and ∼6.2 kb SmaI-SalI fragment of pMC1403 into pPS1633) | This study | |
| pPS1633 | Apr, Gmr; pUC18R6K-mini-Tn7T-Gm | [37] | |
| pMC1403 | Apr; lacZY fusion cloning vector for construction of translational fusions | [17] | |
| pPS1733 | Apr, Kmr ; anr + (ligation of 0.8 kb PCR fragment amplified with Anr-UP-KpnI and Anr-DN-BamHI to pCR2.1 vector) | This study | |
| pPS1682 | Spra; anr + (ligation of 0.8 kb KpnI-BamHI fragment of pPS1733 into KpnI-BamHI cleaved pVLT35) | This study | |
| pPS1684 | Apr; anr + (ligation of 0.8 kb KpnI-BamHI fragment of pPS1733 into KpnI-BamHI cleaved pUCP20) | This study | |
| pPS1991b | Apr, Gmr; pPS1669 with attL1-pPA1612-attL2 to promote lacZ expression | This study | |
Abbreviations: Ap, ampicillin; att, λ attachment site (s); FRT, Flp recombinase target site; Gm, gentamycin; Km, kanamycin; MCS, multiple cloning site; p, promoters; Sp, spectinomycin
see text for plasmid or strain construction details.
DNA manipulations and vector constructions
Routine procedures were employed for manipulation of DNA [11]. Plasmid DNA was isolated using the QIAprep Mini-spin kit (Qiagen, Valencia, CA) and P. aeruginosa chromosomal DNA was isolated using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). DNA fragments were purified from agarose gels utilizing the QIAquick gel extraction kit (Qiagen, Valencia, CA).
The lacZ fusion plasmid containing a 30 bp deletion within the fabA upstream region was created in several steps. The fabA upstream region was PCR-amplified from PAO1 genomic DNA using primers IR187-UP and IR187-DN (sequences of primers used in this study are listed in Table 2.). These primers introduced SmaI and SalI restriction enzyme cleavage sites at each end of the PCR fragment. The PCR fragment was purified and ligated into the TA cloning vector pCR2.1 (Invitrogen), resulting in pPS1482. This plasmid was used as a template in a two-step PCR reaction to generate pPS1488 containing a 30 bp deletion in the fabA upstream region. In the first PCR reaction, two flanking DNA segments harboring overlapping sequences were amplified using two sets of primers (PCRSOE-A+IR187SOE-B) and (IR187SOE-C+PCRSOE-D). In the second PCR, each purified 1st round PCR fragment was used to perform splicing by overlap extension (SOE) PCR using primers PCRSOE-A and PCRSOE-D. The resulting PCR product was cloned into pCR-Blunt II-TOPO vector (Invitrogen), resulting in a plasmid containing the fabA upstream sequences with a 30 bp deletion. Finally, pPS1491 and pPS1492 were constructed by ligating the KpnI-SmaI fragments of pPS1482 and pPS1488, respectively, into pPS1453 cleaved with the same enzymes.
Table 2. Primers used in this study.
| Primer Name | Sequence (5′ → 3′) |
| fabA upstream primers | |
| pfabA0 | cgggaatgaacgattacctg |
| pfabA-attB1a | GGGGACAAGTTTGTACAAAAAAGCAGGCTcatgaccggatcgccttcgaa |
| pfabA1-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTtctacgacaagggcggcaagc |
| pfabA2-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTggacgcgggaataaagtgaac |
| pfabA3-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTatctgttcgccggacactgtg |
| pfabA4-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTactttcaccgcaacgcaacag |
| pfabA4a-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTctgtgactttcaccgcaacg |
| pfabA5-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTtctatgactaggctgccgctg |
| pfabA6-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTcgacgccgatacaataacccg |
| pfabA7-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTgcgcgacggccgctggacgaa |
| pfabA8-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTccgccacaaccctgcagttca |
| pfabA9-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTgggatttttgaggagctcgc |
| pfabA10-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTatgaccaaacaacacgccttc |
| pfabA11-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTatcagcgatgtcggcggcaag |
| pPA1612-UP-attB2 | GGGGACCACTTTGTACAAGAAAGCTGGGTcgccacctgctctacttca |
| fabA Δ30 | |
| PCRSOE-A | AGGTATCCGGTAAGCGGCAG |
| IR187SOE-B | TGTCCGGCGAACAGATGTTC |
| IR187SOE-C | GAACATCTGTTCGCCGGACATGACTAGGCTGCCGCTGCGA |
| PCRSOE-D | CGGTTCCTTTAGCAGCCCTT |
| PA1627 deletion | |
| 1627SOE-A | gtgatcagttgcagcatcaccg |
| 1627SOE-B | TCAGAGCGCTTTTGAAGCTAATTCGacccgggacagacccatgact |
| 1627SOE-C | AGGAACTTCAAGATCCCCAATTCGggaagaaacatccaatcatcggat |
| 1627SOE-D | accagcagtactaccaggaacc |
| PA4890 deletion | |
| 4890SOE-A | tgaaggattccgtctgcaagcc |
| 4890SOE-B | TCAGAGCGCTTTTGAAGCTAATTCGgaggacatacggcttcctttgg |
| 4890SOE-C | AGGAACTTCAAGATCCCCAATTCGgctgcgtttcatcatgatcggc |
| 4890SOE-D | tagttgaactccgcctcgccat |
| PA1539 deletion | |
| 1539SOE-A | gcagggtagtagttgtgcgaca |
| 1539SOE-B | TCAGAGCGCTTTTGAAGCTAATTCGtcgctcatgttccacctggttg |
| 1539SOE-C | AGGAACTTCAAGATCCCCAATTCGcgagtgacagctcgatgtcctt |
| 1539SOE-D | cgactaccgtttctgaatccgc |
| anr deletion | |
| anr-UpF-GWL | TACAAAAAAGCAGGCTttgacagggtgcgacaggta |
| anr-UpR-Gm | TCAGAGCGCTTTTGAAGCTAATTCGaatccttgcagtgtgcttgg |
| anr-DwnF-Gm | AGGAACTTCAAGATCCCCAATTCGggaagtgcacatcctcgact |
| anr-DwnR-GWR | TACAAGAAAGCTGGGTacgaagctgtccacggtcat |
| Nested PCR primers | |
| Rnd1-TnM | TATAATGTGTGGAATTGTGAGCGG |
| Rnd1-Pa1 | GGCCACGCGTCGACTAGTACNNNNNNNNNNGATAT |
| Rnd2-TnM | ACAGGAAACAGGACTCTAGAGG |
| Rnd2-Pa | GGCCACGCGTCGACTAGTAC |
| TnMSeq | CACCCAGCTTTCTTGTACAC |
| TnMRev | TGCACCGTGCAGTCGATGATAA |
| Other primers | |
| IR187-UP | GAGGGGAcCCGGgATGATCTACGACAA |
| IR187-DN | AGGTCTTCTCGGGTaccGGCGTGTTGT |
| Gm-UP | TGGAGCAGCAACGATGTTAC |
| Gm-DN | TGTTAGGTGGCGGTACTTGG |
| FabU | GGGATCCGGAATGATCTACGACAAGG |
| FabD | GGGTACCAAGTTTAGCCCGTTCATGC |
| PA1611-UP | AAATCCCTGAACTGCAGGGTTGTG |
| PA1611-DN-in | ACCTGCGCCAAGAATTCACCCATA |
| GmFRT-UP | CGAATTAGCTTCAAAAGCGCTCTGA |
| GmFRT-DN | CGAATTGGGGATCTTGAAGTTCCT |
| Anr-UP-KpnI | ATCGCGGCTGCGGTACCCTT |
| Anr-DN-BamHI | ATACAACGGATCCGCGCTGAG |
| EcglmS-DN | TGCAGCTGCTGGCTTACCATG |
| Tn7R | CACAGCATAACTGGACTGATTTC |
| DIG-fabA | DigN-ATGCGATCGATCATCAGCATGTTG |
| Bio-fadR-for | Biotin-GGACACTGTGACTTTCACCGCAACGCAACAGTCTATGACT |
| fadR-Rev | AGTCATAGACTGTTGCGTTGCGGTGAAAGTCACAGTGTCC |
| fadR-UP | GAACCGGACACTGTGACTTTCACCGCAACGCA-ACAGTCTATGACTA |
| fadR-DN | TAGTCATAGACTGTTGCGTTGCGG-TGAAAGTCACAGTGTCCG |
| 30-concatamer-for | (TAGACTGTTGCGTTGCGGTGAAAGTCACAG)3 |
| 30-biotin-rev | Biotin-CTGTGACTTTCACCGCAACGC |
Sequences in capital letters are common for all genes amplified in a particular experiment and overlap with the sequences of the other genes that they were spliced to, e.g. Gm or attB primer sequences. Lower-case letters indicate gene-specific sequences.
For promoter localization experiments, 11 different portions of the fabA intergenic sequence were fused to a promoter-less lacZ contained on a mini-Tn7 delivery vector utilizing Gateway universal cloning technology (Invitrogen) as described in Choi et al., 2006. The fabA upstream region was amplified using 11 different primer pairs consisting of combinations of pfabA-attB1 and pfabA1-attB2 to pfabA11-attB2, giving PCR segments containing progressively shorter lengths of the fabA upstream region at 20∼21 bp intervals. To construct a lacZ fusion with the PA1612 promoter, a fragment containing the PA1612 promoter, the PA1612-PA1611 operon, and the entire PA1611-fabA intergenic region was also amplified by PCR using the primer pair pfabA-attB1 and pPA1612-UP-attB2. The individual PCR fragments thus obtained contain attB1 and attB2 sites which facilitate high-throughput cloning into the universal donor vector pDONR221 (Invitrogen) containing attP1 and attP2 sites by a site-specific recombination between attB and attP sites, resulting in plasmids harboring different inserts of fabA upstream sequences with flanking hybrid att sites, attL1 and attL2. This was followed by a second site-specific recombination between the attL and attR sites of the resulting plasmids and the destination vector pPS1669, respectively, resulting in mini-Tn7 elements containing transcriptional lacZ fusions with different lengths of fabA upstream sequences.
Random mariner-based insertional mutagenesis
mariner-based random mutagenesis of PAO1 containing a temperature-sensitive fabA allele (PAO1 fabA(Ts)) [2] was performed by electroporating pBT20 [12] containing a mariner transposon with a Gm-resistance marker. It was reasoned that knock-out of potential fabAB activators would create unconditional mutants requiring OA supplementation at all temperatures. Identification of OA auxotrophs was performed by patching Gmr transformants on LB containing 30 µg/ml gentamycin with and without OA. Chromosomal fragments from OA auxotrophs were transferred into the PAO1 fabA(Ts) parental strain containing a chromosomally integrated fabA′-lacZ fusion to assess whether the mutation generated by mariner transposon insertion directly affected fabAB expression. β-galactosidase assays were performed in cells grown in the presence of OA.
mariner-based random mutagenesis was also performed in PAO1-fabA(Ts) strain containing a chromosomally integrated fabA′-lacZ fusion. A knock-out of a potential activator would create an unconditional mutant requiring OA supplementation at all temperatures and result in lighter blue colonies. Lighter blue colonies were tested for OA auxotrophy by patching Gmr transposon mutants on LB containing 30 µg/ml gentamycin with or without OA supplementation. In addition, β-galactosidase assays were performed in cells grown in the presence of OA. The transposon insertion points in the mutants harboring an OA auxotrophy were first determined by PCR amplification using Gm-UP and Gm-DN primers to verify mariner transposon insertions in the PAO1 chromosome. Secondly, the absence of insertions in the fabAB operon was confirmed by PCR amplification using fabAB specific primers FabU and FabD. The insertion sites of transposons located outside of the fabAB genes were determined by two-step random PCR amplifications using mariner specific primers (Rnd1-TnM and Rnd2-TnM), random PAO1 chromosomal primers (Rnd1-Pa1), and its nested primer Rnd2-Pa. The resulting PCR fragments were sequenced by using primer TnMSeq to determine chromosomal mariner transposon insertion sites (S. Lory, personal communication). An alternative method employed to map mariner insertion sites was as follows. Chromosomal DNAs from OA auxotrophs were digested with BamHI which cuts the mariner transposon once, followed by self-ligation of chromosomal DNA fragments self-ligated and PCR amplification with primers Rnd2-TnM and TnMRev. The resulting PCR fragments were sequenced as above.
Gene deletion
For construction of a gene replacement vector containing a deletion allele of the PA1611 gene, this gene was amplified by using primers PA1611-UP and PA1611-DN-in. The PCR fragment was cloned into pCR2.1 to yield pPS1472 followed by the ligation of the ∼2.0 kb blunt-ended EcoRI fragment of pPS1472 to PstI+EcoRI cleaved and blunt-ended pEX18Ap vector. The PA1611 coding sequence was deleted by replacement of a blunt-ended KpnI-NcoI fragment of pPS1476 with a 1.1 kb SmaI Gmr::FRT fragment of pPS856 [13], resulting in pEX18Ap-ΔPA1611::Gm.
The plasmids pPS1503, pPS1505, pPS1506 containing ΔPA1627::Gm, ΔPA4890::Gm and ΔPA1539::Gm, respectively, were constructed by SOEing PCR as described in the following procedure. Five ng of pPS856 plasmid was used for amplification of Gm-FRT cassette using primers GmFRT-UP and GmFRT-DN Flanking gene-specific DNA fragments were amplified using the following primer pairs: 1627SOE-A+1627SOE-B and 1627SOE-C+1627SOE-D; 4890SOE-A+4890SOE-B and 4980SOE-C+4890SOE-D; or 1539SOE-A+1539SOE-B and 1539SOE-C+1539SOE-D for PA1627, PA4890, or PA1539, respectively. Since each primer-B and primer-C contains sequences that overlap with the Gm-FRT fragment, the resulting three PCR fragments - i.e., the two flanking DNA fragments and the Gm-FRT segment - were annealed together and subjected to second round PCR amplification using each primer-A and primer-D. PCR amplification resulted in the generation of a deletion allele of Gene::Gm, which was subcloned into the gene replacement vector pEX18Ap [13]. These gene replacement vectors with ΔPA1611::Gm, ΔPA1627::Gm, ΔPA4890::Gm and ΔPA1539::Gm were transformed into E. coli SM10lacIq cells and then conjugally transferred to PAO1 and PAO1 harboring mini-Tn7T-lacZ or the mini-Tn7T-fabA′lacZ chromosomal fusion by biparental mating for 5 h, followed by plating the recombinants on VBMM plates containing 30 µg/ml of gentamycin. Merodiploid recombinants were purified on the same medium and were resolved by streaking individual Gmr colonies on a LB plate containing 5% sucrose. Mutant constructions were verified by colony PCR, followed by Flp-mediated excision of Gmr marker, which was performed by the conjugal transfer of the Flp-producing pFLP2 into the recombinants [13].
For creation of an anr deletion mutant, a deletion allele of Δanr::Gm was created by PCR amplification using two primer pairs of anr-UpF-GWL+anr-UpR-Gm and anr-DwnF-Gm+anr-DwnR-GWR and Gateway cloning technology was utilized as described in Choi et al. [14]. For complementation experiments, anr was amplified by using primers Anr-UP-KpnI and Anr-DN-BamHI, the PCR fragment was ligated into pVLT35 [15] or pUCP20 [16], resulting in pPS1682 or pPS1684, respectively. The plasmids were individually transformed into the wild-type and Δanr mutant strain containing a chromosomal pfabA′-lacZ transcriptional fusion by using the rapid electroporation method described in Choi et al. [10]. Transformants were selected on LB plates containing 150 µg/ml of spectinomycin or 200 µg/ml of carbenicillin for pVLT35 or pUCP20, respectively.
Construction of an E. coli host strain and a genomic library of P. aeruginosa
For identification of an activator of fabA expression in a heterologous host, gene fusion technology was employed (Fig. 1). First, the pfabA′-′lacZY translational gene fusion-containing Tn7 delivery plasmid pPS1634 was constructed by ligation of three fragments: a BamHI-XhoI fragment of pPS1633, an ∼250 bp BamHI-EcoRV fragment of pPS790 and an ∼6.2 kb SmaI-SalI fragment of pMC1403 [17], resulting in an in-frame fusion of a portion of the fabA coding sequence plus fabA-PA1611 intergenic region to lacZY (Fig. 1A). Second, an E. coli strain containing a chromosomal insertion of the fabA′-′lacZY fusion construct was created by co-electroporating pPS1634 and pTNS1 into the Δlac E. coli strain DL291 [18], resulting in the site-specific integration of fabA′-lacZY fusion at attTn7 downstream of glmS in the E. coli chromosome. The integration into attTn7 was verified by PCR amplification using primer pair EcglmS-DN and Tn7R (Fig. 1B). Third, a PAO1 library was created by ligation of EcoRI+PstI partially-digested chromosomal DNA fragments ranging from 0.75 kb to 5.0 kb into PstI+EcoRI-digested pUC18 (Fig. 1C). After transforming the library into E. coli containing the fabA′-′lacZY gene fusion, putative activator-encoding plasmids were selected as dark blue colonies on LB-ampicillin plates containing 40 µg/ml of X-Gal and 25 µM of IPTG (Fig. 1D). Plasmids isolated from blue colonies were retransformed into endA − E. coli cells to facilitate recovery of high quality plasmid DNA. Plasmids from isolates showing dark blue color were mapped by digestion with restriction enzymes PstI and EcoRI, BamHI, HindIII or SphI. For the identification of the putative fabA-activating gene encoded by these plasmids, vector-chromosomal DNA junction sequences were determined using the M13 forward and reverse sequencing primers.
Figure 1. Construction of an E. coli host strain and a genomic library of P. aeruginosa.

A) A fabA′-′lacZY translational fusion was assembled on a mini-Tn7 suicide delivery vector. B) The mini-Tn7 vector was co-electroporated with the Tn7 transposase expressing helper plasmid pTNS1 into an E. coli Δlac strain. Since the suicide delivery vector cannot replicate in E. coli due to the presence of the conditional protein-dependent oriR6K, gentamycin-resistant (Gmr) transformants will result from site- and orientation-specific integration at the chromosomal attTn7 site which is located immediately downstream of the glmS gene in the glmS - pstS intergenic region. C) A PstI-EcoRI P. aeruginosa chromosomal DNA library was constructed by ligation of partially digested PstI-EcoRI fragments into pUC18. D) The library was used to transform a P. aeruginosa strain harboring a chromosomally integrated fabA′-′lacZY fusion. Since fabA′-′lacZY is only expressed at low levels, the host strain will only form light blue colonies on X-Gal-containing indicator medium. Transformants expressing putative activating proteins indicated by “+” will appear as darker blue colonies. Abbreviations: Apr, ampicillin resistance; FRT, Flp-recombinase target; Tn7L and Tn7R, left and right end of Tn7, respectively.
β-galactosidase assays
Cells were grown at 37°C with shaking to exponential phase (optical density at 540 nm ∼0.4–0.8) in LB medium with 0.05% Brij 58+/−0.05% oleic acid. β-galactosidase (βGal) activity was measured in chloroform/sodium dodecyl sulfate-permeabilized cells and its activity calculated as previously described [19]. The experiment was performed in triplicate.
RT-PCR analysis
Total RNA was extracted from PAO1 cells grown at 37°C and 16°C using the hot phenol method [20]. Using 1 µg of RNA, cDNA was synthesized using primer DIG-fabA following the procedure described in the Invitrogen manual with minor modifications. The cDNA was then used as a template for PCR amplification using different primer sets consisting of DIG-fabA, and primers annealing at various positions in the intergenic region, which included pfabA1-attB2 through pfabA9-attB2 or the primer pfabA0 within the PA1611 coding region. PCR fragments were analyzed by agarose gel electrophoresis.
Purification of fabA activator
First, streptavidin beads were used to purify a FadR-like fabA activator A biotinylated, double-stranded DNA oligonucleotide (Bio-fadR-for and fadR-Rev) composed of the 30 bp putative regulatory region and 5 bp flanking sequences was prepared as follows: 1 µg of both oligonucleotides (Bio-fadR-for and fadR-Rev) were denatured by incubation for 5 min at 95°C and then re-annealed either by slow cooling to room temperature or reducing the temperature to 25°C in 2.5°C increments in a thermal cycler. For preparation of crude cell extracts, three liters of P. aeruginosa PAO1 cells were grown at 37°C until they reached an optical density (540 nm) of 0.8. Cells were harvested by centrifugation, then passed through a French press three times in 27 ml of buffer (20 mM Tris-HCl [pH 7.9], 0.5 mM NaCl, 10% glycerol and 1 mM PMSF). Cell debris was removed by centrifugation at 12,000×g for 10 min followed by ultracentrifugation at 34,000×g for one hour. The supernatant was concentrated by addition of ammonium sulfate to 60% saturation. The protein precipitate was collected by centrifugation and resuspended in 3 ml of 10 mM Tris-HCl (pH 7.5), 10% glycerol and then dialyzed at 4°C overnight against 3 liters of the same buffer. Three µg of the resulting protein extract was added to a DNA binding reaction composed of 1 µg of poly-dI-dC, 1 µg of dsDNA sequence, and 5× binding buffer (100 mM Hepes [pH 7.6], 5 mM EDTA, 50 mM (NH4)2SO4, 50 mM DTT, 1% Tween 20 and 150 mM KCl). After incubation at room temperature for 15 min, 100 µl of μMACS streptavidin microbeads were added and the mixture was applied to a column which was equilibrated with 100 µl of protein equilibration buffer provided in the kit and rinsed twice with 100 µl of binding buffer in the magnetic separation unit. After sample application, the column was washed six times with binding buffer before DNA-bound proteins were eluted by the addition of binding buffer containing 0.2–1.0 M NaCl. The protein contents of eluted samples were analyzed by 10% SDS-PAGE.
Second, CNBr-activated Sepharose beads were used to purify fabA activator according to the procedure described by other research group with minor modification [21]. A duplex of two oligonucleotides (fadR-UP and fadR-DN) was created same as above. Similar to the method described above, dialyzed sample was prepared. The dialyzed mixture was applied to a column packed with CNBr-activated Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ). Unbound proteins were removed by washing with three volumes of column buffer. DNA-bound protein(s) were eluted by the addition of TE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA) containing 0.2–0.8 M NaCl. The protein contents in eluted samples were analyzed by electrophoresis on a 10% SDS-PAGE gel. All fractions showing distinct bands were collected and proteins were concentrated with microcon centrifugal filter units (Millipore). Protein bands were cut from the SDS-PAGE gel, subjected to in-gel trypsin digestion, and their identities confirmed using peptide mass fingerprinting.
Third, affinity chromatography was performed using a 30 bp concatamer generated by PCR amplification using 30-concatamer-for and its complementary 30-biotin-rev primer. Then DIG-labeled concatamer was prepared as the manufacturer's manual (Roche Applied Science, Indianapolis, IN). Cell lysates from PAO1 cells grown overnight in 200 ml of LB medium were prepared by cell lysis in a high salt buffer (20 mM Tris-HCl [pH 8.0], 0.5 M NaCl, 1 mM PMSF, 1 mM EDTA, 0.1% Triton X-100 and 10 µg/ml lysozyme) using freeze-thaw cycles. The cell lysates were subjected to ultracentrifugation at 34,000×g for 1 h (Beckman, SWTi28 rotor) followed by dialysis against 4.5 liters of buffer (10 mM Tris-HCl [pH 7.5], 10% glycerol). DNA binding reactions contained 1 µg of poly-L-lysine, 1 µg of poly-dI-dC, binding buffer (20 mM Hepes [pH 7.6], 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM DTT, 0.2% Tween 20 and 30 mM KCl), 10–40 ng of concatamer DNA and 0.2–2 µg of protein extract, and the mixtures were incubated at room temperature for 30 min. The reaction mixtures were loaded onto a 6% native polyacrylamide gel prepared and electrophoresed in 0.5× TBE buffer. DNA fragments were transferred to neutral or positively charged nylon membranes for biotin or DIG detection, respectively. The detection of biotin- or DIG-labeled DNA was performed following the protocols of the NEBlot Phototope detection kit (New England Biolabs, Beverly, MA) or the DIG gel shift kit (Roche Applied Science, Indianapolis, IN), respectively.
DINAMelt analysis
This was performed on “http://www.bioinfo.rpi.edu/applications/hybrid/hybrid2.php” using the sequences in fabA 5′ untranslated region (UTR).
Results
Characterization of fabA regulatory region
The 188 bp PA1611-fabA intergenic region contains several recognizable features. It contains a putative −10 region (TACAAT), but no corresponding −35 region, suggesting a promoter requiring an activator for RNA polymerase binding to initiate fabAB transcription (Fig. S1). It also contains a 30 bp sequence which is conserved in all Pseudomonas species sequenced to date (Fig. S2). This conservation includes PA1611 which encodes a probable sensor kinase/response regulator hybrid protein. This strong sequence conservation suggests that the 30 bp sequence may be an important fabAB regulatory region. A highly conserved fatty acid biosynthesis repressor (FabR, or DesT in P. aeruginosa) binding site was also found upstream of the 30 bp sequence in P. aeruginosa, which may be involved in negative regulation of fabA expression. The DesT binding site is highly conserved in fabA upstream regions of several other bacteria (Fig. S3). We hypothesized that fabA regulation may be complex and mediated by yet unknown regulatory protein(s) which act at conserved upstream regions. However, the organization of the fabA upstream regions of E. coli and P. aeruginosa is different. Whereas the E. coli FabR repressor binds at a site overlapping the −10 region and the FadR activator binds at a site overlapping the −40 region, P. aeruginosa contains a putative DesT repressor-binding site upstream of the 30 bp putative activator-binding site (Fig. S4). To assess whether the 30 bp sequence plays a role in fabA regulation, it was deleted and the corresponding region was fused to lacZ. After single copy insertion into the PAO1 chromosome, βGal activities were measured in the absence or presence of OA and compared to that expressed in the wild-type fabA′-lacZ fusion strain. As shown in Fig. 2, OA repressed fabAB expression in the wild-type strain harboring a chromosomal fabA′-lacZ fusion, whereas fabAΔ30′-lacZ expression was significantly decreased compared to wild-type fabA′-lacZ. In addition, fabAB expression in the fabAΔ30 deletion construct was no longer repressible in the presence of OA. These results suggested that the 30 bp sequence is indeed involved in regulation of fabA expression and may be a binding site for an unknown activator or other effector molecule.
Figure 2. lacZ expression in PAO1 containing chromosomally integrated lacZ, fabA′-lacZ or fabAΔ30′-lacZ transcriptional fusions.
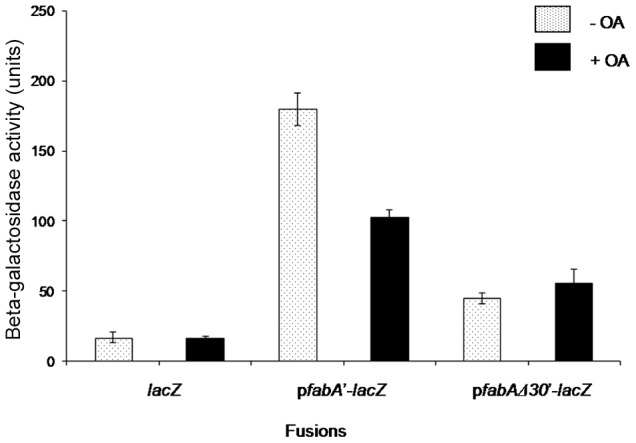
Strains were grown to mid-log phase in LB medium with or without oleate (OA) supplementation and β-galactosidase activities were measured. Activities are expressed in Miller Units.
This was confirmed by analyses using lacZ transcriptional fusions containing progressively shorter fabA upstream fragments (Fig. 3A). The data shown in Fig. 3B demonstrate that deletion of the 30 bp sequence and beyond led to loss of lacZ activity and OA responsiveness. The 5 bp addition in primer 4a, which restores a complete 30 bp sequence, recovered lacZ expression indicating that this sequence is important for fabA transcription (Fig. 3B).
Figure 3. Characterization of fabA regulatory region.

A) Positions of primer-binding sites in the fabA-PA1611 intergenic region. Each primer is symbolized as follows: P0, fabA0; P1-P11, fabA1-attB2 through fabA11-attB2; R, fabA-attB1. Primer P11 is placed at the 124th–144th nucleotide from the first nucleotide of the fabA coding region. The sequence shaded and boxed in the gray box indicates the putative 30 bp regulatory element. Vertical arrow heads indicate the end points of sequences present in the lacZ fusion constructs analyzed in Fig. 3B. B) The 30 bp sequence is important for fabAB expression. PAO1 contained fabA′-lacZ vectors with fabA upstream regions amplified with primers 1 through 11. The 5 bp addition in primer 4a, which restores a complete 30 bp sequence, recovered lacZ expression indicating that it is important for fabA transcription. Cells were grown and β-galactosidase activities were measured as described in the legend to Fig. 2 with and without oleate (OA) supplementation. C) Characterization of the promoter region of fabA using RT-PCR analysis of fabA expression. RNA was extracted from PAO1 grown at 37°C using the hot phenol extraction method. cDNA was synthesized using the Superscript III First-strand kit (Invitrogen) and primer R. Resulting cDNAs were used as templates for PCR amplification utilizing primer R and the indicated primers (see Fig. 3A for location of primer-binding sites). D) β-galactosidase activities in PAO1 containing lacZ fusions with various fabA upstream fragments. The upstream fragments were amplified with primers pPA1612, and primers p1 and p2 (see Fig. 3A for primer-binding sites).
In order to further define the promoter region of fabA, RT- PCR was performed. RNA extracted from wild-type PAO1 cells grown at 37°C was reverse-transcribed to cDNAs, which were then used as templates for PCR amplification utilizing primer R and the primers indicated in Fig. 3A. PCR products were apparent in all reactions containing either primer 0 which primes within the PA1611 coding region, or primers 1 through 6 which prime in the fabA intergenic region (Fig. 3C; see Fig. 3A for the location of fabA primer binding sites). To compare fabA transcription levels from promoters located in far upstream or PA1611-fabA intergenic sequences, βGal activities were measured in lacZ fusion strains harboring the putative PA1612-PA1611 operon promoter, the entire PA1612-PA1611 operon and the PA1611-fabA intergenic sequence (PA1612′-lacZ) or only the PA1611-fabA intergenic region (p1′-lacZ or p2′-lacZ) (Fig. 3D). It is evident that βGal levels expressed from PA1612′-lacZ were two times higher than those expressed from p1′-lacZ or p2′-lacZ (Fig. 3D). Also, predictions of transcription terminators within PA1613 through fabB using TransTerm software (http://cbcb.umd.edu/software/transterm/ttgenbank50/Pseudomonas aeruginosa.tt) indicated that there are no putative transcriptional terminators in the PA1612 through fabB interval, but only downstream of fabB. In summary, both RT-PCR and gene fusion analyses suggested that fabA may be co-transcribed with PA1612-PA1611 or another promoter located outside of the PA1611-fabA intergenic region. This probably explains why identification of transcription start sites using primers with binding sites in the fabA 5′ region failed (data not shown). Similarly, other research group determined that the fabA mRNA transcribed by another promoter located outside of the PA1611-fabA intergenic region was identified and seemed to be the most abundant compared to the shorter transcript using 5′-RACE and Northern blot analysis [22].
Negative regulators of fabAB expression
Besides FadR, an additional regulator of fabA and fabB gene expression in E. coli was originally suggested by a computational footprinting method. An actual binding of this “virtual” regulator was subsequently confirmed using DNA affinity chromatography [9], [21]. Later, FabR (YijC) was found to function as a repressor of fabB gene expression [9]. P. aeruginosa has two FabR homologs, PA4890 and PA1539, which have about 50% sequence similarity to E. coli FabR. To assess whether these homologs repress fabAB expression in P. aeruginosa, β-galactosidase activities were measured in wild-type, ΔPA4890 and ΔPA1539 mutant strains containing a chromosomally-integrated fabA′-lacZ fusion. The addition of OA repressed fabA transcription in all three strains tested (Fig. 4). In the ΔPA4890 strain, however, OA repressed fabA expression to a lesser extent than in wild-type and the ΔPA1539 strain. However, deletion of PA4890 did not affect fabA expression in the absence of OA, indicating that only PA4890 is involved in negative regulation of UFA synthesis in the presence of OA. In addition, gel shift assays utilizing purified protein showed that PA4890 binds to the putative FabR-binding site located upstream of fabA [22]. Later, PA4890 was renamed DesT, a repressor desBC operon expression. Our data suggest that DesT may also negatively regulate UFA synthesis via regulation of fabAB operon expression.
Figure 4. Effects of FabR homologs on fabA′-lacZ expression.
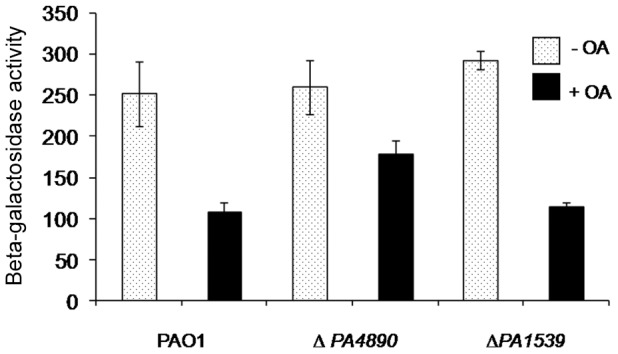
β-galactosidase activities were measured in wild-type PAO1 and ΔPA4890 and ΔPA1539 mutant strains containing a chromosomal fabA′-lacZ fusion. Cells were grown in LB medium. Where indicated, 0.05% oleic acid (OA) was added to cells with 0.05% Brij-58.
Positive regulation of fabAB expression
Our previous studies indicated that expression of the fabAB operon was up-regulated in biofilm-grown cells, probably because in biofilms bacteria grow under anaerobic conditions (data not shown). Furthermore, lacZ expression in a wild-type strain containing a chromosomally integrated fabA′-lacZ fusion was repressed by 40%–50% in the presence of 0.05% of OA (Fig. 2). In other words, the addition of exogenous UFA resulted in repression of fabAB transcription, indicating that its expression may be regulated by unknown activating factors. This situation is reminiscent of the one found in E. coli where in the presence of exogenous UFAs FadR binds acyl-CoAs and the acyl-CoA-FadR complex then binds to its cognate binding site, resulting in repression of fabA transcription. These results suggested the existence of a FadR-like fabAB regulatory gene in P. aeruginosa.
Deletion of candidates to identify putative regulatory genes
PA1611 was considered as a candidate for a fabAB regulatory gene, because of its location upstream of fabA and because it encodes a two-component regulatory system (a probable sensor kinase/response regulator hybrid) and thus, may sense exogenous UFAs similar to what has been demonstrated in B. subtilis and other Gram-positive bacteria. However, deletion of PA1611 had no effect on expression of a fabA′-lacZ fusion (data not shown). The PA1627 gene product is 26% homologous to E. coli FadR, which is known to positively regulate UFA synthesis, but deletion of PA1627 had no effect on transcription of a fabA′-lacZ fusion (data not shown). Although UFA synthesis is increased when cells are grown under low temperature growth conditions in several bacteria, including E. coli, PA1611 and PA1627 deletion mutants grown at 16°C had similar levels of fabA transcription when compared to wild-type PAO1 (data not shown). Taken together, it can be concluded that neither PA1611 nor PA1627 are involved in regulation of fabAB transcription. Previously, all unidentified transcriptional regulators of the GntR family were deleted in PAO1 containing a chromosomally inserted fabA′-lacZ fusion by employing a high-throughput Gateway cloning technology [10]. Significant changes in lacZ expression levels in those knockout mutants would be indicative of positive regulation of fabAB transcription. However, none of the mutations did affect fabA expression and, therefore, transcription factors of the GntR family do not seem to be involved in the regulation of fabAB expression in cells grown either at 37°C or 16°C.
mariner-based random insertional mutagenesis of a fabA(Ts) strain to identify putative regulatory genes
mariner-based random mutagenesis of potential fabAB activators in strain PAO1 fabA(Ts), which is a conditional mutant requiring OA supplementation only at 42°C, will create unconditional mutants requiring OA supplementation at all temperatures. OA auxotrophs were identified by observing cell growth on LB without OA. Transposon insertions were transferred back to the fabA(Ts) strain to verify linkage between transposon insertion and the unconditional OA auxotrophy phenotype. Transposon insertion sites in mutants classified as OA auxotrophs were determined by nested PCR and DNA sequencing. mariner insertions causing OA auxotrophy due to inactivation of fabAB were excluded from further consideration. Several transposon insertions causing the desired phenotype were found and insertion sites were localized to the genes encoding the σ54 factor rpoN; PA0286 (desA), encoding aerobic fatty acid desaturase; PA1629, encoding a probable enoyl-CoA hydratase/isomerase; PA4760, encoding a putative heat shock protein; PA4233, encoding a probable major facilitator superfamily (MFS) transporter; and several other genes – e.g. PA0358, PA3649, and PA4476 - encoding proteins of unknown function (Table 3).
Table 3. mariner insertions in genes causing unconditional UFA auxotrophy in fabA(Ts) PAO1.
| Mutant | Gene with Mariner (TnM) insertion | Function |
| # 6 | rpoN::TnM | σ54 transcription factor |
| # 16 | PA4760::TnM | Heat shock protein DnaJ |
| # 25 | PA1629::TnM | Probable enoyl-CoA hydratase/isomerase |
| # 30 | PA0358::TnM | Hypothetical protein |
| # 39 | PA3649::TnM | Hypothetical protein (63% similar to E. coli yaeL) |
| # 44 | PA4476::TnM | Hypothetical protein |
| # 75 | PA4233::TnM | Probable major facilitator superfamily (MFS) transporter |
| # 77 | PA0286 (desA)::TnM | Fatty acid desaturase DesA |
To assess whether any given mutation generated by mariner transposon insertion directly affected fabAB expression, chromosomal DNA fragments of Gmr auxotrophic integrants were transferred into the parental strain containing a chromosomally integrated fabA′-lacZ fusion and βGal activities were measured in cells grown in the presence of OA. These experiments showed that all transformants had similar levels of fabA expression when compared to the wild-type, indicating that the products of the mutated genes affected fabAB expression indirectly (data not shown). These data also indicated that a considerable number of genes and metabolic pathways seem to be affect fabAB expression.
To directly screen for mutants affecting fabA transcription, the PAO1 fabA(Ts) strain containing a chromosomally integrated fabA′-lacZ fusion was used for mariner-based random mutagenesis. mariner insertion in an activator encoding gene would not only create an unconditional mutant requiring OA supplementation at all temperatures, but also result in lighter blue colonies caused by decreased fabA′-lacZ expression. A significant decrease in βGal activities was observed in 14 mutants among the 62 OA auxotrophs that were identified in this screen. Among them, only three mutants were identified as ones contained mariner insertions in the PA2222-PA2223 locus encoding hypothetical protein and hypothetical outer membrane protein by nested PCR amplification and sequencing (data not shown). However, since the PA2222-PA2223 operon could not be deleted, a further characterization of this operon was not pursued. However, for future studies transposon-generated PA2222-PA2223 mutants could be employed to further study the role of these genes in fabAB regulation.
Use of a surrogate host strain to identify potential fabAB regulators
The complex regulation of fabAB operon expression in P. aeruginosa may hamper the identification of directly acting fabAB activator(s) by genetic means. To avoid this complexity and interference with other factors, an attempt was made to identify activating factors in a Δlac E. coli strain containing a chromosomally-integrated fabA′-′lacZY translational fusion. This experimental approach is illustrated in Fig. 1. A genomic library of P. aeruginosa PAO1 was transformed into the E. coli strain with the fabA′-′lacZY gene fusion. Potential activators would stimulate fabA′-′lacZ expression and transformants encoding such factors were therefore selected as dark blue colonies on X-Gal indicator medium. Linkage of this phenotype in transformants to plasmids encoding activating factors was confirmed by re-transforming the plasmids into the same E. coli host cells. Twenty-one isolates exhibited a blue colony phenotype. Restriction analyses of plasmids isolated from these transformants indicated the presence of the same PstI-EcoRI fragment, but different flanking fragments. Besides the 2.9 kb fragment of pUC18 vector, the restriction enzymes released 0.4 kb, 1.2 kb, and 2.5 kb fragments. Among these, the 0.4 kb and 1.2 kb fragment were present in all plasmids, indicating that this region is responsible for activating fabA expression. These plasmids were classified into two groups and one plasmid from each group was selected for sequencing by using M13 forward and reverse primers. Two types of inserts were identified, those shown in the upper part of Fig. 5A (50%) and those shown in the lower part of the figure (50%). The only intact gene contained on both types of plasmids was anr (anaerobic NO3 − regulator), which presumably activated fabA′-′lacZY transcription. To assess a possible role of Anr in regulation of fabAB expression in P. aeruginosa, the anr gene was deleted from PAO1 containing a chromosomal pfabA′-lacZ fusion and βGal activities were measured in wild-type PAO1 and its Δanr mutant. The presence of the anr deletion reduced lacZ expression only by ∼25% compared to the wild-type (Fig. 5B). In addition, this effect could only be complemented when anr + was present on the high-copy number plasmid pUCP20, but not on the low-copy number plasmid pVLT35, even with IPTG induction (Fig. 5B).
Figure 5. Effect of Anr in regulation of fabAB expression.
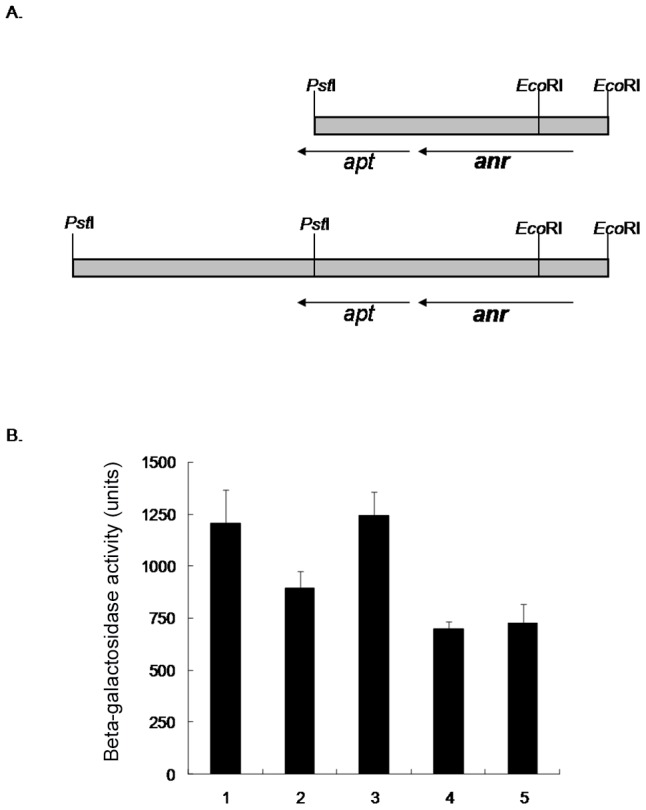
A) anr is responsible for activation of fabA′-′lacZY transcription in E. coli. The restriction maps shown here are from cloned chromosomal DNA fragments isolated from blue, putative activator-expressing colonies. Plasmids isolated from ten blue colonies were sequenced by using M13 forward and reverse primers to verify the presence of the indicated genes. B) Effects of anr on fabA expression in P. aeruginosa. β-galactosidase activities were measured in wild-type and Δanr PAO mutants containing a chromosomally integrated pfabA′-lacZ fusion. Columns: 1, PAO1 (wild-type); 2, PAO1010 (PAO1 Δanr); 3, PAO1010 with pPS1684 (pUCP20 with anr +); 4, PAO1010 with pPS1682 (pVLT35 with anr+) in the absence of IPTG; 5, PAO1010 with pPS1682 in the presence of IPTG.
Biochemical attempts aimed at identification of fabAB regulatory proteins
Since previous experimentation suggested that the 30 bp sequence within the fabA upstream region has a regulatory function in fabAB expression, fragments containing this sequence were used for identification of DNA binding proteins. Affinity chromatography strategies using streptavidin beads and DIG failed to obtain 30 bp-bound protein on SDS-PAGE gel. When CNBr-activated Sepharose beads were used as a ligand to immobilize DNA binding protein, distinct band on SDS-PAGE gel was successfully collected, subjected to in-gel trypsin digestion, and peptide mass fingerprinting. However, there was no good matched protein. Unfortunately, none of the strategies were successful in identifying regulatory proteins, indicating that the experimental conditions employed in these studies were possibly not correct. fabA expression is highly upregulated in biofilm-type grown cells, suggesting that anaerobic growth or biofilm-type growth might be proper conditions for identification of fabAB regulatory proteins.
Analysis of a number of stable secondary structures of fabA 5′ untranslated region (UTR)
DINAMelt analysis (http://www.bioinfo.rpi.edu/applications/hybrid/hybrid2.php) of the fabA 5′ untranslated region (UTR) revealed that this region can assume a number of stable secondary structures. Two of these structures are shown in Fig. 6. These structures exhibit some of the criteria of riboregulatory elements [23]: i) they are found in the 5′ UTR; ii) they contain a number of stem-loop structures, one of which is conserved for ligand binding and the others are variable. It is noteworthy that the structure shown in Fig. 6A contains a stem-loop followed by a stretch of U residues, a situation found in typical rho-independent transcriptional terminators. This stem-loop structure is not present in the second structure shown in Fig. 6B.
Figure 6. Secondary structures of the fabA 5′ untranslated region (UTR) by DINAMelt analysis.

A) Terminator-containing structure. B) Antiterminator-containing structure. The red and blue lines indicate G-C and A-T complementary hydrogen bonds, respectively. The green and black boxes indicate ribosome-binding site (RBS) and terminator, respectively.
Discussion
Previous work demonstrated that UFA biosynthesis in P. aeruginosa is governed by two pathways, depending on the oxygen availability: 1) two aerobic fatty acid desaturase pathways consisting of DesA and DesBC, and 2) the anaerobic FabAB pathway. The fabAB-encoded proteins play an essential and dominant role in the UFA biosynthesis under both aerobic and anaerobic conditions.
While the existence of and the mechanisms of UFA synthesis via the anaerobic pathway were known at the onset of the studies presented here, little was known about the regulation of expression of the fabA and fabB genes. It was known that, unlike E. coli where fabA and fabB map to two distinct locations on the chromosome, these two genes form an operon in P. aeruginosa. It was also known that the expression of this operon was up-regulated in biofilm-grown cells and repressed by addition of fatty acids, especially oleic acid, to the growth medium. While this study did not reveal any specific regulatory protein(s) involved in regulation of fabAB operon expression or the exact molecular mechanisms governing regulation of expression of this operon, they revealed some previously unknown findings.
First, RT-PCR analyses indicated that the fabAB operon is transcribed from at least two promoters. It is co-transcribed with the upstream, seemingly unrelated PA1612-PA1611 operon, but an additional promoter located in the PA1611-fabA intergenic region which also contributes to fabAB expression.
Second, the DesT (FabR) repressor binds to a palindromic sequence in the PA1611-fabA intergenic region, but seems to play only a modest role in the fabAB operon expression.
Third, a 30 bp sequence present in the PA1611-fabA intergenic region is a regulatory element involving the positive regulation of fabA. The position of this sequence at an appropriate position upstream of a putative -10 promoter consensus sequence indicates that it may be an activator-binding site.
Fourth, while E. coli FadR belongs to the GntR family of transcriptional regulators, deletion of none of the 25 genes encoding the GntR family of regulators in P. aeruginosa affected fabA transcription. Therefore, the P. aeruginosa FadR-like activator, if it exists, must belong to a different family of regulators. Additionally, deletion of the immediate upstream gene PA1611 encoding a hybrid sensor kinase/response regulator protein did not adversely affect fabA transcription, at least not under the conditions employed in the present studies.
Fifth, numerous other cellular factors including RpoN, DesA and Anr seem to play at least minor roles in regulation of fabAB transcription, possibly through modulation of intracellular fatty acid levels or other metabolites. RpoN is a global regulator which regulates expression of nitrogen assimilation gene and fermentation gene expression (reviewed in [24]). However, there is no evidence that rpoN regulates UFA biosynthesis in P. aeruginosa. If it does, it may be due to global effects. According to previous study, PA0286 was shown to encode DesA, an aerobic fatty acid desaturase [3]. This gene might be closely linked to the regulation of the fabAB operon by affecting cellular UFA levels. Enoyl-CoA hydratase/isomerases are involved in fatty acid metabolism. These hydratase activities catalyze the hydratation of 2-trans-enoyl-CoA into 3-hydroxyacyl-CoA and the isomerase activities shift the 3- double bond of the intermediates of UFA oxidation to the 2-trans position. The anr gene product, which senses low oxygen, supports anaerobic growth by activating numerous genes. P. aeruginosa is able to survive in an anaerobic environment. Since P. aeruginosa grows as a biofilm-type in the anaerobic CF lung mucus, anaerobic conditions may be an important factor to regulate metabolic pathways for robust growth and establishment of persistent infections. The anaerobic survival mode is supported by denitrification of nitrate or nitrite [25]-[27]. In addition, the arginine deaminase (ADI) pathway plays a key role in catabolizing L-arginine to L-ornithine, with the formation of ATP from ADP, resulting in the growth of P. aeruginosa under anaerobic condition in the absence of terminal electron acceptors such as molecular oxygen or nitrate (reviewed in [28]). Since Anr globally regulates expression of many genes, P. aeruginosa fabAB regulation via Anr may be indirect via other Anr-dependent factors. An alternate explanation might be that Anr activity may be inhibited by another factor(s) which may be highly expressed under aerobic growth, but Anr expression from a high-copy number plasmid may be able to saturate this “anti-Anr” factor and thus complement the deletion mutant. Since Δanr mutants are unable to grow anaerobically, additional experiments should be performed with cells grown under microaerophilic conditions to further assess the potential role of Anr in fabAB gene expression. In conclusion, use of gene fusion technology revealed Anr as an activator of fabA′-lacZ expression in E. coli, but it played only a minor and probably indirect role in P. aeruginosa. While none of these findings does yet provide a clear picture of the molecular mechanisms governing transcription of the fabAB operon, they indicate that regulation of P. aeruginosa fabAB operon expression is very complex and most likely quite different from what has been described in E. coli.
Sixth, DINAMelt analysis suggests that fabAB expression may be regulated not via protein-binding, but via a yet-to-be discovered mechanism. One possibility lies in small regulatory RNA (srRNA)-mediated regulation of fabAB expression. srRNA can regulate gene expression at the transcriptional and translational levels. Intracellular metabolites such as amino acids, sugars and nucleotides can bind to cis-acting metabolite-sensing regulatory RNA elements and control gene expression, which are called riboswitches (reviewed in [29]). Various types of riboswitches are present in bacteria. Generally, non-coding regulatory RNA elements are found in the 5′-untranslated region (5′-UTR) and can give rise to three-dimensional conformational alternate changes in response to changes intracellular metabolite signals. Recently, it was found that besides metabolites, metals such as intracellular Mg2+ can regulate the Mg2+ transporter MgtA of Salmonella enterica serovar Typhimurium by the metal-sensing 5′-UTR of the mgtA gene [29]. Furthermore, changes in environmental conditions such as temperature can result in a conformational change of regulatory RNA, thus functioning as a “thermometer”. For example, virulence genes are highly expressed at 37°C by a PrfA transcriptional activator, which is thermally regulated by the 5′UTR of mRNAs of prfA in Listeria monocytogenes [30]. In addition, the ROSE (repressor of heat-shock gene expression) element placed in the 5′UTR region of heat-shock genes in many Gram-negative bacteria senses temperature changes in order to control their gene expression [31]. E. coli rpoS mRNA translation is regulated by the DsrA small RNA which disrupts a sequestering helix of a ribosome binding site during temperature downshift [32].
P. aeruginosa may employ a regulatory RNA for controlling anaerobic UFA synthesis. One probable pathway is that a metabolite-sensing non-coding RNA may recognize intracellular or exogenous UFA and change its structure, resulting in fabAB gene repression. This is consistent with the observation that OA supplementation repressed fabA expression up to 50% (Fig. 2). Another possible explanation is that low growth temperature might be an important controlling factor. Since cells should maintain the membrane fluidity for normal function required for transport and movement, they adapt to changes in environmental conditions, especially temperature. Upon exposure to low temperatures, cell membranes become solid-state, but they preserve their fluidity by increasing UFA levels. According to previous study, fabA expression is highly up-regulated in cells grown at 16°C compared to cells grown at at 37°C, suggesting positive regulation by low temperature [14]. Therefore, the 5′-UTR of fabA may sense low temperature and coordinate an adaptation process caused by a temperature downshift. However, having said all of this, searches of the fabAB upstream sequences have yet to reveal possible srRNAs.
As mentioned above, the fabAB operon is transcribed from at least two promoters, P1 and P2, resulting in transcription of two mRNAs, mRNA I and mRNA II (Fig. 7). In the absence of exogenous UFAs and under anaerobic, as well as perhaps low temperature conditions, the fabAB operon is transcribed from both of these promoters and the respective transcripts terminate at a single transcriptional terminator which is located immediately downstream of the fabAB operon (Fig. 7A). Transcription from P2 requires an activator protein which binds to the 30 bp region. Maximal transcription and translation ensures an adequate supply of FabA and FabB proteins for UFA synthesis. At the mRNA level, this situation is characterized by a riboregulatory element configuration on mRNA I containing an antitermination element.
Figure 7. A working model for P. aeruginosa fabAB regulation.

In the presence of exogenous UFAs and/or aerobic or high (37°C) temperature conditions, the proposed activator probably binds an UFA-CoA ligand and the activator-UFA-CoA complex dissociates from the 30 bp activation element resulting in loss or of fabAB transcription from P2 (Fig. 7B). At the mRNA level, this situation is characterized by a riboregulatory element configuration that contains a terminator. The result is a low level transcription and translation of fabA and fabB, a desirable situation when oxygen levels and temperatures are high, exogenous UFAs are available and the overall cellular demand for UFAs is either low or can be satisfied by the activities of the aerobic desaturase pathways.
While many of our experimental results support this model, several questions do remain unanswered. For example, why were repeated genetic and biochemical attempts aimed at identification of the proposed activator protein unsuccessful? Why does purified DesT bind to the PA1611-fabA intergenic region, but there is little to no effect on fabA expression in lacZ fusion or microarray experiments? A possible explanation for this may be that most of the experiments described in this study were performed with cells grown aerobically and at 37°C, possibly conditions during which synthesis and/or activity of an activator protein is repressed or DesT activity is masked. This notion is supported by the fact that fabA′-lacZ expression was significantly higher in cells grown at 16°C when compared to cells grown at 37°C. Future experiments should therefore be performed with cells grown under anaerobic or microaerophilic conditions, as well as with cells grown at lower temperatures. Although DINAMelt analysis suggests the possibility that a riboregulatory element may be involved in regulation of fabAB operon expression, there is currently no experimental evidence supporting this possibility. Future efforts should therefore also include experiments that would address the existence of a 5′ UTR RNA with a high degree of secondary structure. The experiments would consist of in-line probing [33], [34] and RNase H cleavage assays of RNA-DNA hybrids [35] in the presence and absence of substrate to determine if the structure not only exists, but also undergoes a conformational change upon addition of the substrate or changes in an environmental cue. The nature of the substrate(s) or environmental cue(s), of course, remains speculative. Substrates could be metabolites such as a UFA-CoAs and environmental cue signals such as low temperature or low oxygen levels.
Studies on anaerobic UFA synthesis by the P. aeruginosa FabAB pathway and identification of its regulatory signals are significant because many infections caused by this bacterium, especially cystic fibrosis lung infections, are biofilm-type infections during which this bacterium's physiology is adapted to an anaerobic lifestyle. Such studies have therefore the potential for discovering new targets and drugs for treatment of biofilm-type infections.
Supporting Information
The sequence of fabA - PA1611 intergenic region. The last four PA1611 codons and first eight fabA codons are shown. The conserved 30 bp sequence is boxed. A putative −10 region with good homology to the TATAAT consensus is indicated in underlined bold-faced letters. The DesT binding site is marked inverted arrows.
(TIF)
The fabA upstream sequences are conserved in the three Pseudomonas spp., P. aeruginosa , P. putida , and P. syringae . The PA1611, PP4173 and PSPT02212 genes encode conserved hybrid sensor kinase/response regulatory proteins. Also highly conserved is the shaded 30 bp sequence. A lollipop structure indicates the transcriptional terminators of the respective fabAB operons.
(TIF)
FabR binding sites in the fabA or fabB upstream regions of several bacteria. The sequences in the gray box indicate conserved FabR binding sites. P.p., P. putida; P.s., P. syringae; P.a., P. aeruginosa; E.c., E. coli; Y.p., Y. pestis; V.c., Vibrio cholerae. Only the P.a. and E.c. sequences were experimentally shown to bind FabR (E.c.) and its P.a. homolog DesT.
(TIF)
Locations of binding sites for E. coli FadR or P. aeruginosa FadR-like activator and FabR in the E. coli fabA and P. aeruginosa PAO1 fabAB upstream regions.
(TIF)
Funding Statement
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2010-0004068). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Hoang TT, Schweizer HP (1999) Characterization of Pseudomonas aeruginosa enoyl-acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J Bacteriol 181: 5489–5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoang TT, Schweizer HP (1997) Fatty acid biosynthesis in Pseudomonas aeruginosa: cloning and characterization of the fabAB operon encoding b-hydroxyacyl-acyl carrier protein dehydratase (FabA) and b-ketoacyl-acyl carrier protein synthase I (FabB). J Bacteriol 179: 5326–5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu K, Choi KH, Schweizer HP, Rock CO, Zhang YM (2006) Two aerobic pathways for the formation of unsaturated fatty acids in Pseudomonas aeruginosa . Mol Microbiol 60: 260–273. [DOI] [PubMed] [Google Scholar]
- 4. Cronan Jr JE, Subrahmanyam S (1998) FadR, transcriptional co-ordination of metabolic expediency. Mol Microbiol 29: 937–943. [DOI] [PubMed] [Google Scholar]
- 5. Campbell JW, Cronan Jr JE (2001) Escherichia coli FadR positively regulates transcription of the fabB fatty acid biosynthetic gene. J Bacteriol 183: 5982–5990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henry MF, Cronan Jr JE (1992) A new mechanism of transcriptional regulation: release of an activator triggered by small molecule binding. Cell 70: 671–679. [DOI] [PubMed] [Google Scholar]
- 7. Nunn WD, Giffin K, Clark D, Cronan Jr JE (1983) Role for fadR in unsaturated fatty acid biosynthesis in Escherichia coli . J Bacteriol 154: 554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Aalten DM, DiRusso CC, Knudsen J, Wierenga RK (2000) Crystal structure of FadR, a fatty acid-responsive transcription factor with a novel acyl coenzyme A-binding fold. Embo J 19: 5167–5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang YM, Marrakchi H, Rock CO (2002) The FabR (YijC) transcription factor regulates unsaturated fatty acid biosynthesis in Escherichia coli . J Biol Chem 277: 15558–15565. [DOI] [PubMed] [Google Scholar]
- 10. Choi KH, Schweizer HP (2005) An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol 5: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sambrook J, Russell DW (2001) Molecular cloning, Third edn. Cold Spring harbor, NY: Cold Spring Harbor Laboratory Press.
- 12. Kulasekara HD, Ventre I, Kulasekara BR, Lazdunski A, Filloux A, et al. (2005) A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol 55: 368–380. [DOI] [PubMed] [Google Scholar]
- 13. Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212: 77–86. [DOI] [PubMed] [Google Scholar]
- 14. Choi KH, Kumar A, Schweizer HP (2006) A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. J Microbiol Methods 64: 391–397. [DOI] [PubMed] [Google Scholar]
- 15. Miller VL, Mekalanos JJ (1988) A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR . J Bacteriol 170: 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. West SE, Schweizer HP, Dall C, Sample AK, Runyen-Janecky LJ (1994) Construction of improved Escherichia-Pseudomonas shuttle vectors derived from pUC18/19 and sequence of the region required for their replication in Pseudomonas aeruginosa . Gene 148: 81–86. [DOI] [PubMed] [Google Scholar]
- 17. Casadaban MJ, Chou J, Cohen SN (1980) In vitro gene fusions that join an enzymatically active b-galactosidase segment to amino-terminal fragments of exogenous proteins: Escherichia coli plasmid vectors for the detection and cloning of translational initiation signals. J Bacteriol 143: 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Larson TJ, Schumacher G, Boos W (1982) Identification of the glpT-encoded sn-glycerol-3-phosphate permease of Escherichia coli, an oligomeric integral membrane protein. J Bacteriol 152: 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller JH (1992) A Short Course in Bacterial Genetics. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
- 20. Barton HA, Johnson Z, Cox CD, Vasil AI, Vasil ML (1996) Ferric uptake regulator mutants of Pseudomonas aeruginosa with distinct alterations in the iron-dependent repression of exotoxin A and siderophores in aerobic and microaerobic environments. Mol Microbiol 21: 1001–1017. [DOI] [PubMed] [Google Scholar]
- 21. McCue L, Thompson W, Carmack C, Ryan MP, Liu JS, et al. (2001) Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res 29: 774–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramanian C, Rock CO, Zhang Y-M (2010) DesT coordinates the expression of anaerobic and aerobic pathways for unsaturated fatty acid biosynthesis in Pseudomonas aeruginosa. J. Bacteriol 192: 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winkler WC, Breaker RR (2005) Regulation of bacterial gene expression by riboswitches. Annu Rev Microbiol 59: 487–517. [DOI] [PubMed] [Google Scholar]
- 24. Reitzer L (2003) Nitrogen assimilation and global regulation in Escherichia coli . Annu Rev Microbiol 57: 155–176. [DOI] [PubMed] [Google Scholar]
- 25. Carlson CA, Ingraham JL (1983) Comparison of denitrification by Pseudomonas stutzeri, Pseudomonas aeruginosa, and Paracoccus denitrificans . Appl Environ Microbiol 45: 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies KJ, Lloyd D, Boddy L (1989) The effect of oxygen on denitrification in Paracoccus denitrificans and Pseudomonas aeruginosa . J Gen Microbiol 135: 2445–2451. [DOI] [PubMed] [Google Scholar]
- 27. Zumft WG (1997) Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev 61: 533–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu CD, Winteler H, Abdelal A, Haas D (1999) The ArgR regulatory protein, a helper to the anaerobic regulator ANR during transcriptional activation of the arcD promoter in Pseudomonas aeruginosa . J Bacteriol 181: 2459–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cromie MJ, Shi Y, Latifi T, Groisman EA (2006) An RNA sensor for intracellular Mg2+ . Cell 125: 71–84. [DOI] [PubMed] [Google Scholar]
- 30. Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, et al. (2002) An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes . Cell 110: 551–561. [DOI] [PubMed] [Google Scholar]
- 31. Chowdhury S, Maris C, Allain FH, Narberhaus F (2006) Molecular basis for temperature sensing by an RNA thermometer. Embo J 25: 2487–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Repoila F, Majdalani N, Gottesman S (2003) Small non-coding RNAs, co-ordinators of adaptation processes in Escherichia coli: the RpoS paradigm. Mol Microbiol 48: 855–861. [DOI] [PubMed] [Google Scholar]
- 33. Soukup GA, Breaker RR (1999) Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5: 1308–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winkler WC, Cohen-Chalamish S, Breaker RR (2002) An mRNA structure that controls gene expression by binding FMN. Proc Natl Acad Sci . U S A 99: 15908–15913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fuchs RT, Grundy FJ, Henkin TM (2006) The SMK box is a new SAM-binding RNA for translational regulation of SAM synthetase. Nat Struct Mol Biol 13: 226–233. [DOI] [PubMed] [Google Scholar]
- 36. Holloway BW (1955) Genetic recombination in Pseudomonas aeruginosa . J Gen Microbiol 13: 572–581. [DOI] [PubMed] [Google Scholar]
- 37. Choi KH, Gaynor JB, White KG, Lopez C, Karkhoff-Schweizer RR, et al. (2005) A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2: 443–448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The sequence of fabA - PA1611 intergenic region. The last four PA1611 codons and first eight fabA codons are shown. The conserved 30 bp sequence is boxed. A putative −10 region with good homology to the TATAAT consensus is indicated in underlined bold-faced letters. The DesT binding site is marked inverted arrows.
(TIF)
The fabA upstream sequences are conserved in the three Pseudomonas spp., P. aeruginosa , P. putida , and P. syringae . The PA1611, PP4173 and PSPT02212 genes encode conserved hybrid sensor kinase/response regulatory proteins. Also highly conserved is the shaded 30 bp sequence. A lollipop structure indicates the transcriptional terminators of the respective fabAB operons.
(TIF)
FabR binding sites in the fabA or fabB upstream regions of several bacteria. The sequences in the gray box indicate conserved FabR binding sites. P.p., P. putida; P.s., P. syringae; P.a., P. aeruginosa; E.c., E. coli; Y.p., Y. pestis; V.c., Vibrio cholerae. Only the P.a. and E.c. sequences were experimentally shown to bind FabR (E.c.) and its P.a. homolog DesT.
(TIF)
Locations of binding sites for E. coli FadR or P. aeruginosa FadR-like activator and FabR in the E. coli fabA and P. aeruginosa PAO1 fabAB upstream regions.
(TIF)