

Abstract
Hepcidin regulation is linked to both iron and inflammatory signals and may influence iron loading in nonalcoholic steatohepatitis (NASH). The aim of this study was to examine the relationships among HFE genotype, serum hepcidin level, hepatic iron deposition and histology in nonalcoholic fatty liver disease (NAFLD). SNP genotyping for C282Y (rs1800562) and H63D (rs1799945) HFE mutations was performed in 786 adult subjects in the NASH Clinical Research Network (CRN). Clinical, histologic, and laboratory data were compared using nonparametric statistics and multivariate logistic regression. NAFLD patients with C282Y, but not H63D mutations, had lower median serum hepcidin levels (57 vs 65 ng/ml, p=0.01) and higher mean hepatocellular (HC) iron grades (0.59 vs 0.28, p<0.001), compared to wild type (WT) subjects. Subjects with hepatic iron deposition had higher serum hepcidin levels than subjects without iron for all HFE genotypes (p<0.0001). Hepcidin levels were highest among patients with mixed HC/reticuloendothelial system cell (RES) iron deposition. H63D mutations were associated with higher steatosis grades and NAFLD activity scores (OR≥1.4, CI >1.0≤2.5, p≤0.041), compared to WT, but not with either HC or RES iron. NAFLD patients with C282Y mutations had less ballooning or NASH (OR ≤0.62, 95% CI >0.39<0.94, p≤0.024) compared to WT subjects.
Conclusions
Presence of C282Y mutations in patients with NAFLD is associated with greater HC iron deposition and decreased serum hepcidin levels and there is a positive relationship between hepatic iron stores and serum hepcidin level across all HFE genotypes. These data suggest that body iron stores are the major determinant of hepcidin regulation in NAFLD regardless of HFE genotype. A potential role for H63D mutations in NAFLD pathogenesis is possible through iron-independent mechanisms.
Keywords: NAFLD, Steatohepatitis, HFE, hepcidin, iron
INTRODUCTION
Hemochromatosis gene (HFE) mutations may exacerbate chronic liver diseases through increased iron accumulation and subsequent oxidative stress (1). Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in North America and much of the developed world; children and possibly as many as a third of all US adults are affected (2). A number of previous studies, especially those conducted in predominantly Caucasian populations, reported that HFE mutations were enriched among NAFLD patients compared to controls, suggesting these genotypes may confer increased risk of NAFLD (3-8). However these studies have been limited by inadequate power, small sample size, lack of standardized pathology and lack of data on iron distribution. A recent meta-analysis of 1,727 NAFLD Caucasian cases and 4,275 controls concluded that HFE mutations were not more prevalent among NAFLD than non-NAFLD subjects (9). Several studies have found that HFE mutations are associated with higher hepatic and/or serum iron indices in NASH patients (4-8, 12, 14). Some studies have also examined the relationship between presence of HFE mutations and severity of fibrosis in NASH (3-14). However, carriage of HFE mutations has not been identified as an independent risk factor for advanced fibrosis among patients with NAFLD or NASH in the majority of reports (6, 9-14).
We have recently reported that more than one-third of US patients enrolled in the Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) had stainable hepatic iron on liver biopsy in one of the following three histologic patterns: hepatocellular only (HC, 7.4%), reticuloendothelial system cells only (RES, 10.7%), or a mixed HC/RES pattern (16.3%)(15). In addition, RES iron was associated with advanced histologic features, including a higher mean NAFLD Activity Score (NAS). By contrast, an HC only pattern of iron deposition was associated with milder histologic and clinical features compared to the other groups, and the mixed HC/RES iron group had intermediate histologic severity. We proposed that the pattern of hepatic iron deposits in NAFLD is due to differential expression of the body iron regulatory hormone, hepcidin by genetic factors and inflammatory signals (15).
Hepcidin regulates iron absorption and recycling by binding to and initiating internalization and degradation of the sole cellular iron exporter ferroportin, thus down-regulating iron efflux from the enterocyte, macrophage and hepatocyte (16). Elevated body iron stores or plasma transferrin levels result in hepcidin up regulation via the SMAD/BMP/HJV pathway (17) or through the HFE/TFR1/TFR2 complex, respectively (18-20). Hemochromatosis patients with mutations in the HFE, HJV and TFR2 genes have been shown to have inappropriately low urinary hepcidin levels allowing unregulated iron absorption resulting in hepatic iron loading in the classic hemochromatosis parenchymal HC pattern (21, 22). Thus, HC iron deposition in NAFLD, similar to that seen hemochromatosis, could be associated with decreased circulating hepcidin levels due to carriage of hemochromatosis mutations such as HFE, HJV, TFR2 or other iron-regulatory genes such as those involved in the SMAD/BMP pathway.
The goal of this study was to investigate the relationship between the two common HFE mutations, serum hepcidin levels, hepatic iron deposition and histologic features of NASH in the large, well characterized NASH CRN cohort.
MATERIALS AND METHODS
Patients
A total of 888 adult (age≥18 years) subjects enrolled into the NASH CRN Database and PIVENS therapeutic trial, based on inclusion criteria described elsewhere (23, 24), between October 2004 and February 2008, having biopsy-proven NAFLD (defined as >5% steatosis) and hepatic iron staining results were evaluated in the present study. From this group, 787 subjects with available DNA that had previously consented to provide DNA for genetic analysis were genotyped for the two common HFE gene mutations. Patients with known hemochromatosis (defined as presence of hepatic iron index [hepatic iron (μmol/g)/age (years)] ≥1.9 or removal of more than 4 g of iron by phlebotomy), C282Y homozygosity for the HFE gene or unexplained hepatic iron overload (≥3+ stainable iron on liver biopsy) were excluded from all NASH CRN studies. One subject without hepatic iron deposition and a normal serum ferritin level was C282Y homozygous and was excluded from the study. Demographic information such as age, sex, ethnicity, and race and medical history to identify co-morbidities and medications were obtained from patient interviews during screening. A physical exam including body weight and height measures was performed. Laboratory data including hepatic, hematologic, metabolic, lipid and serum iron and hepcidin levels were analyzed. Total dietary consumption of iron, vitamin C, tea and coffee were determined from the Block 98 food frequency questionnaire; (NutritionQuest, Berkeley, CA); alcohol consumption was determined from the AUDIT-C questionnaires completed during study visits closest to the time of biopsy (25).
HFE genotyping
Genotyping for the two common HFE mutations C282Y (rs1800562) and H63D (rs1799945) was performed using a real time genotyping assay. Briefly, 10 ng of genomic DNA was plated into 384 well plates using a Beckman Coulter Biomek FX robotic workstation. Each 5 μl reaction containing DNA, fluorescently labeled MGB-Eclipse probes (Epoch Bioscience, Bothell WA) and 0.3 u JumpStart Taq (Sigma-Aldrich, St. Louis, MO, USA) were analyzed on an ABI HT7900. Primer sequences are as follows:
C282Y (rs1800562):Forward 5’AATAAATCATAAGGGCTGGATAACCTTGGCT3’, Reverse 5’ CAGTCACATACCCCAGATCACAATGAG3’
H63D (rs1799945):Forward 5’AATAAATCATAAGTGGATGACCAGCTGTTCGT3’, Reverse 5’AATAAATCATAACTGGAAACCCATGGAGTTC3’
Serum hepcidin assay
Serum hepcidin levels were determined by ELISA (Intrinsic Life Sciences, San Diego, CA) (26). The lower limit of detection in this assay is 5 ng/ml. Ten subjects did not have serum available to be assayed. Subjects with undetectable serum hepcidin levels (15/777, 1.9%) were included in the analysis at the detectable limit of 5 ng/ml, in the following proportions: C282Y/H63D (2/13, 15.4%), C282Y/WT (2/89, 2.2%), H63D/WT (3/174, 1.7%), WT/WT (8/483, 1.7%) and H63D/H63D (0/17, 0%).
Histological assessment
Histologic features of NAFLD and iron accumulation were assessed by the Pathology Committee of the NASH CRN in a centralized consensus review format as previously described (15, 27). In addition to generating individual scores that comprise the composite for the NAFLD Activity Score (NAS), fibrosis stage, and other histologic variables, a pattern-based diagnosis is also rendered. Iron is scored for visibility at various microscopic magnifications, localization in cellular compartments (HC, RES, parenchymal, portal/septal), and zonality when HC.
Statistical analysis
Baseline demographic, clinical, and laboratory characteristics were recorded as number and percentage for categorical data and means and standard deviation or median and interquartile range for continuous data. Continuous variables including laboratory measures were not normally distributed and were analyzed using the Wilcoxon rank sum or Kruskal-Wallis with Dunn’s post-hoc tests. Differences in mean histological scores such as HC and RES iron grade, steatosis grade, fibrosis stage, lobular inflammation grade and NAS between HFE genotypes and WT/WT subjects were analyzed using multivariate ordinal logistic regression after adjustment for confounding variables such as sex, age, BMI and presence of diabetes, history of GI bleeding or iron overload, dietary or supplemental iron and vitamin C. Multivariate logistic regression analysis was used to investigate the independent association of HFE genotype and HC iron phenotype to the most severe categories of each histologic feature and NASH diagnosis after adjustment for sex, age, BMI, ALT, HOMA-IR and presence of diabetes. When presence of HC iron was used as the dependent variable, the presence of any HFE mutation as a dichotomous variable (Y/N) was included as an independent variable in the model. Conversely, each HFE genotype (C282Y/WT and C282Y/H63D or H63D/WT and H63D/H63D) was modeled individually including the presence of any HC iron as a dichotomous independent variable (Y/N). The relationship between hepcidin and HC or RES grade was investigated using linear regression analysis. All analyses were performed using STATA (version 9, College Station, TX, USA). Nominal, two-sided P-values were used and were considered to be statistically significant if p<0.05.
RESULTS
Patient characteristics
A total of 786 adult NASH CRN subjects with biopsy proven NAFLD and available DNA and serum for HFE genotyping and serum hepcidin measurements were included in the study. The characteristics of the study cohort are shown in Table 1, including the number and proportion of the total cohort with each HFE genotype. C282Y (11%) and H63D (23%) heterozygote frequencies were similar to that previously reported in the US by us and others (8, 9). C282Y/H63D and H63D/H63D genotypes were both present in about 2% of subjects. There were no differences between genotypes in clinical data such as age, sex, BMI, and the prevalence of obesity or diabetes. The majority of patients with HFE mutations were Caucasian (279/296, 94%) and 42% of all Caucasians had at least one HFE mutation. As expected, there were lower proportions of other racial groups with HFE mutations compared to Caucasians especially for the C282Y mutation.
Table 1.
Patient characteristics according to HFE genotype
| Characteristic | Total Patients | WT/WT | C282Y/WT | H63D/WT | H63D/H63D | C282Y/H63D |
|---|---|---|---|---|---|---|
| Number | 786 (100) | 490 (62) | 89 (11) | 177 (23) | 17 (2) | 13 (2) |
| Age (yrs) | 49.4 ± 1.4 | 48.5 ± 11.6 | 47.5 ± 11.2 | 49.4 ± 11 | 51.2 ± 12.4 | 49.5 ± 11.7 |
| Male (No.) | 281 (36) | 175 (62) | 26 (9) | 65 (23) | 8 (3) | 7 (3) |
| BMI (kg/m2) | 34.5 ± 1.1 | 34.2 ± 6.4 | 34.0 ± 5.6 | 34.5 ± 6.7 | 36.7 ± 6.9 | 35.3 ± 5.3 |
| Obese (≥30 BMI) | 574 (73) | 354 (62) | 65 (11) | 131 (23) | 14 (2) | 10 (2) |
| Diabetes mellitus | 228 (29) | 140 (61) | 26 (11) | 53 (23) | 4 (2) | 5 (2) |
| Race (No.) | ||||||
| White | 671 (85) | 392 (58) | 86 (13)† | 167 (25)† | 15 (2) | 11 (2) |
| Black | 24 (3) | 20 (83) | 2 (8) | 1 (4)† | 1 (4) | 0 (0) |
| Asian | 40 (5) | 37 (93) | 1 (3)† | 2 (5)† | 0 (0) | 0 (0) |
| American Indian or Alaska Native | 53 (7) | 34 (64) | 2 (4) | 9 (17) | 5 (9) | 3 (6) |
| Other | 38 (8) | 21 (55) | 2 (5) | 10 (26) | 4 (11) | 1 (3) |
| Ethnicity (No.) | ||||||
| Non- Hispanic | 703 (89) | 429 (61) | 87 (12) | 160 (23) | 16 (2) | 11 (2) |
| Hispanic | 83 (11) | 61 (73) | 2 (2) | 17 (21) | 1 (1) | 2 (2) |
Values are N (%) or mean ± SD
P < 0.05 vs WT/WT from Fisher’s exact test for categorical variables
Relationship between laboratory data and HFE genotype
Differences in serum hepcidin, aminotransferases and serum iron studies between patients with different HFE genotypes are shown in Table 2. C282Y heterozygotes had significantly lower median serum hepcidin (p=0.039) and AST levels (p=0.016) compared to WT patients. C282Y/H63D patients had higher serum iron markers including iron (p=0.039), percent transferrin-iron saturation (TS) (p=0.001) and lower total iron binding capacity (TIBC) (p=0.0007), and just missed statistical significance for hepcidin (p=0.0501). H63D homozygotes had higher TS compared to WT subjects (p=0.033). No other laboratory tests including fasting insulin, glucose, lipid or HOMA-IR levels were significantly different between groups (data not shown).
Table 2.
Comparison of laboratory values of patients with different HFE genotypes
| Characteristic | WT/WT (n=490) | C282Y/WT (n=89) | H63D/WT (n=177) | H63D/H63D (n=17) | C282Y/H63D (n=17) |
|---|---|---|---|---|---|
| Serum hepcidin (ng/ml) | 67 (38-99) | 58 (29-86)† | 65 (39-86) | 57 (25-83) | 38 (10-72) |
| Liver enzymes | |||||
| AST (U/L) | 43 (30-63) | 37 (28-49)† | 43 (30-62) | 36 (32-50) | 41 (23-69) |
| ALT (U/L) | 57 (37-87) | 55 (35-76) | 62 (42-91) | 53 (42-75) | 37 (33-66) |
| Serum Iron Studies | |||||
| Iron (ug/ml) | 85 (64-107) | 83 (67-106) | 88 (69-111) | 97 (76-112) | 94 (89-110)† |
| Ferritin (ng/ml) | 370 (329-416) | 129 (75-278) | 155 (77-308) | 110 (71-363) | 185 (45-277) |
| TIBC (μg/dL) | 146 (81-288) | 356 (326-415) | 380 (337-412) | 350 (302-374) | 315 (286-335)† |
| Transferrin-sat (%) | 23 (17-30) | 24 (16-33) | 24 (18-29) | 28 (22-33)† | 30 (26-36)† |
Values are median (IQR)
P< 0.05 vs WT/WT from Wilcoxon rank-sum test.
Relationship between HFE genotype, serum hepcidin level and pattern and degree of hepatic iron deposition
The proportion of subjects with each hepatic iron phenotype according to HFE genotype is shown in Figure 1A. Subjects with C282Y mutations (i.e., C282Y/WT or C282Y/H63D genotypes) were significantly more likely to have HC iron deposition either alone or in a mixed pattern compared to WT/WT subjects (p<0.05); 12% of C282Y/WT heterozygotes had stainable iron only in HC while 34% had any HC iron compared to 6% (p=0.03) and 21% (p=0.007) among WT/WT subjects, respectively. Subjects with H63D mutations were not more likely to have HC iron deposits than WT/WT patients. The proportion of subjects with C282Y mutations and either HC iron only or HC/RES mixed iron patterns, (both 20% of the total subjects within each pattern) was significantly greater than the proportion of subjects with C282Y mutations and either no stainable iron (12%) or RES only iron (6%) (p<0.05 for all except HC only vs RES only p=0.06, Fig 1B). There were no differences in the proportion of subjects with H63D mutations between any of the iron staining patterns (range 24%-29%).
Figure 1.
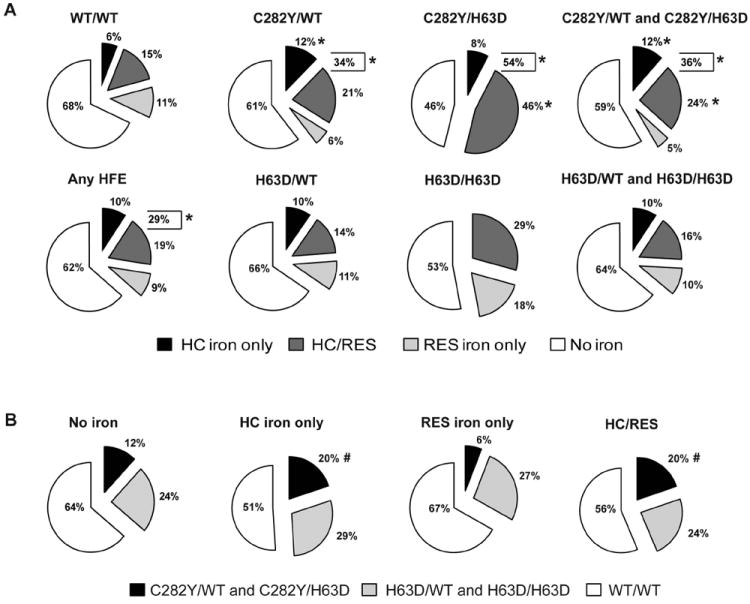
A) Proportion of subjects with different hepatic iron phenotypes according to their HFE mutation status. The percentage of the total subjects for each HFE genotype or combination of genotypes having stainable hepatic iron in each of the three patterns HC, RES or mixed HC/RES are shown in the pie charts. The percentage of the total is labeled for each pattern. The combined total percentage of subjects having any HC iron (ie., HC only plus HC/RES) are shown in brackets. B) Proportion of subjects with combined C282Y, H63D or WT/WT genotypes according to their hepatic iron phenotypes. The percentage of the total subjects with each iron staining pattern and C282Y/WT plus C282Y/H63D, H63D/WT plus H63D/H63D or WT/WT genotypes are shown in the pie charts. * p<0.05 compared to WT/WT; # p<0.05 compared to subjects without stainable iron.
C282Y/WT and C282Y/H63D subjects also had a higher grade of HC iron compared to those with WT/WT genotype (mean grade 0.51 and 1.15, respectively vs 0.28); the C282Y/WT or C282Y/H63D genotype was independently associated with increased HC iron deposition after adjustment for other potential contributing factors selected a priori including age at biopsy, sex, BMI, history of GI bleeding or iron overload, dietary or supplemental iron and vitamin C, (OR ≥2.4, 95% CI >1.4≤23.4, p≤0.001;see Figure 2). The H63D/WT or H63D/H63D genotypes were not associated with either the presence or grade of HC iron. There were no significant differences in the presence or grade of RES iron for any HFE genotype compared to WT/WT.
Figure 2.
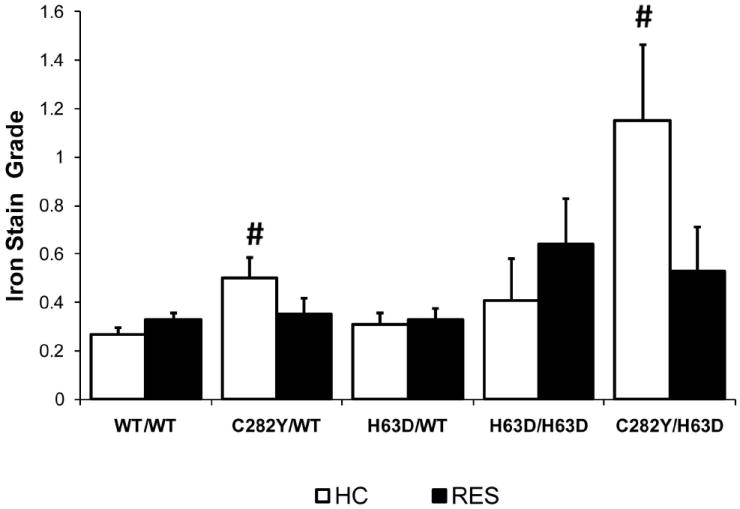
Mean histological HC and RES iron grade according to HFE genotype. Differences in mean HC iron grade for each HFE genotype compared to WT/WT were determined using ordinal regression modeling after adjustment for other potential contributing factors selected a priori including age at biopsy, sex, BMI, history of GI bleeding or iron overload, dietary or supplemental iron and vitamin C, (OR >2.4, 95% CI >1.4<23.4, # p≤0.001). Standard deviations are indicated by the error bars.
We sought to investigate the relationship between serum hepcidin levels and HFE mutations among NAFLD subjects with and without hepatic iron deposition. As shown in Figure 3, median hepcidin values were significantly lower among C282Y subjects, compared to WT subjects with, [82 ng/ml, IQR (64-108) vs 98 (72-131), p=0.04] or without stainable hepatic iron [35 ng/ml, IQR (13-68) vs 55 (30-81), p=0.0003]. In all HFE genotype groups, subjects with stainable hepatic iron had significantly higher serum hepcidin levels than subjects without stainable iron (p<0.0001). There was a significant positive association between hepcidin and both HC (beta 0.37, p<0.001) and RES iron grade (beta 0.42, p<0.001). We also found that serum hepcidin was significantly different across different hepatic iron phenotype groups (p<0.0001) (see Figure 4). In post-hoc pairwise comparisons, subjects without iron staining [51 ng/ml, IQR (28-78)] had significantly lower serum hepcidin compared to subjects with HC [70 ng/ml, IQR (55-107)], RES [86 ng/ml, IQR (65-117)] and mixed HC/RES iron distribution [100 ng/ml, IQR (77-140)] (p<0.0001 for all).
Figure 3.
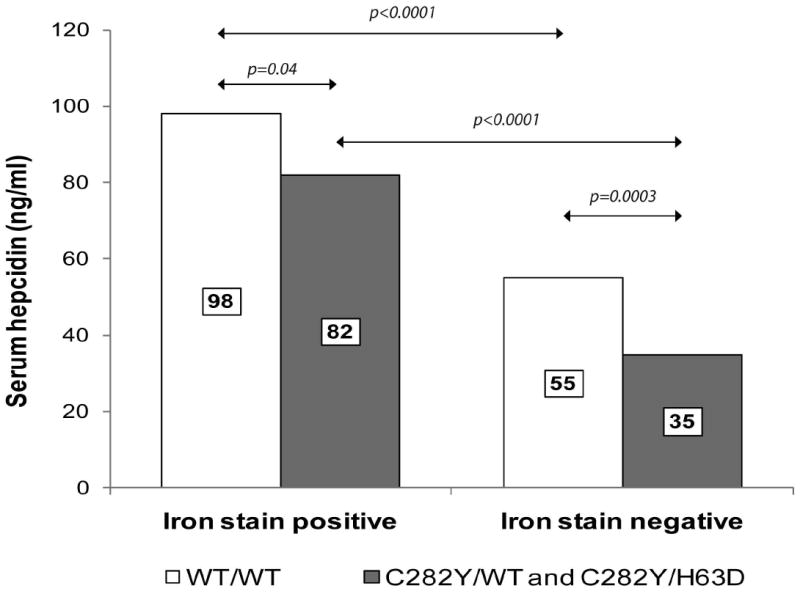
Comparison of the median serum hepcidin values of NAFLD subjects with C282Y and WT HFE genotypes with or without stainable hepatic iron. Significant differences between groups are indicated by arrows (Wilcoxon rank-sum test). Median levels of each group are labeled.
Figure 4.
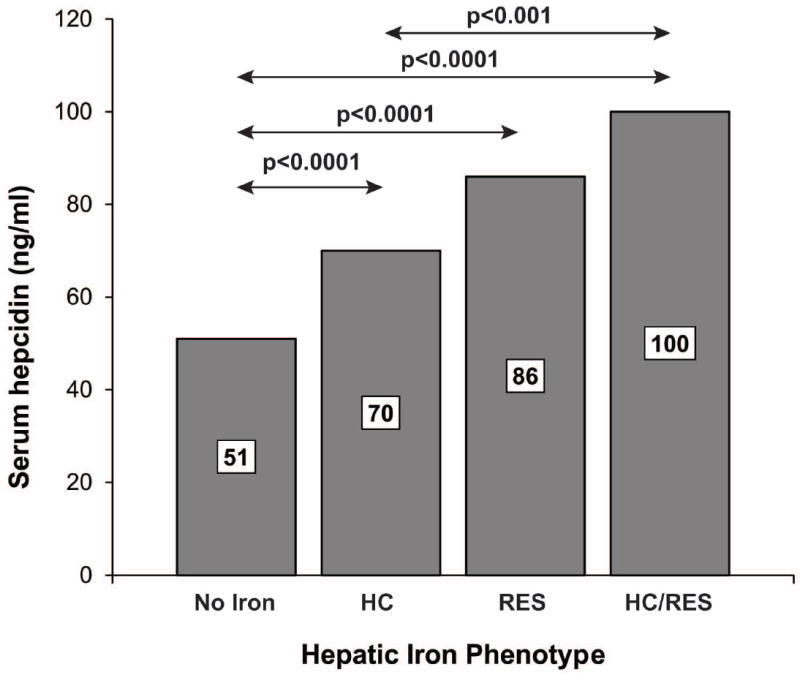
Comparison of the median serum hepcidin values of NAFLD subjects with different hepatic iron phenotypes. There was a significant difference between groups (Kruskal-Wallis test, p<0.0001). Significant post-hoc pairwise comparisons between groups are indicated by arrows (Dunn’s test). Median levels of each group are labeled.
Histologic differences between HFE genotypes
Differences in mean histological scores such as steatosis grade, fibrosis stage, lobular and portal inflammation grade and the NAFLD Activity Score (NAS) between patients with HFE mutations and WT subjects were analyzed using ordinal logistic regression after adjustment for sex, age, BMI and presence of diabetes (Table 3). Differences in the proportion of subjects with a definitive diagnosis of NASH between HFE genotypes were analyzed using logistic regression after adjustment for sex, age, BMI and presence of diabetes (Table 3). Subjects with any HFE mutation had a higher steatosis grade than WT subjects (1.9 ± 0.8 vs 1.8 ± 0.8, OR 1.5, 95% CI 1.2-2.1, p=0.001). Subjects with H63D mutations had higher steatosis grades than WT subjects (2.0 ± 0.8 vs 1.8 ± 0.8, OR 1.7, 95% CI 1.2-2.3, p=0.001) and higher NAS (4.7 ± 1.6 vs 4.5 ± 1.6 or 4.4 ± 1.6, OR ≥1.4, 95% CI >1.0≤2.5, p≤0.041) than C282Y or WT genotypes, respectively. Subjects with C282Y mutations had lower ballooning scores compared to WT subjects (0.9 ± 0.9 vs 1.1 ± 0.9, OR 0.62, 95% CI 0.42-0.93, p=0.022) and were less likely to have a definitive NASH diagnosis (49% vs 61%, OR >0.60, 95% CI ≥0.37≤0.99, p≤0.05) compared to H63D or WT genotypes. The proportion of subjects with a definitive diagnosis of NASH was similar between those with H63D mutations and WT subjects. There was also no significant difference in mean fibrosis among HFE genotype groups.
Table 3.
Histologic score differences between HFE genotypes
| Histologic feature | WT/WT | ANY HFE | C282Y/WT and C282Y/H63D | H63D/WT and H63D/H63D |
|---|---|---|---|---|
| Steatosis (0-3) | 1.8 ± 0.8 | 1.9 ± 0.8‡ | 1.9 ± 0.8 | 2.0 ± 0.8‡ |
| Lobular inflammation (0-3) | 1.6 ± 0.7 | 1.6 ± 0.7 | 1.5 ± 0.7 | 1.7 ± 0.7 |
| Portal inflammation (0-2) | 1.1 ± 0.6 | 1.1 ± 0.6 | 1.1 ± 0.6 | 1.1 ± 0.6 |
| Fibrosis (0-4) | 1.6 ± 1.3 | 1.6 ± 1.2 | 1.4 ± 1.2 | 1.6 ± 1.2 |
| Hepatocellular ballooning (0-2) | 1.1 ± 0.9 | 1.0 ± 0.9 | 0.9 ± 0.9† | 1.1 ± 0.9# |
| NAS (0-8) | 4.5 ± 1.6 | 4.6 ± 1.6 | 4.4 ± 1.6 | 4.7 ± 1.6†,# |
| Definitive NASH | 298 (61) | 168 (57) | 50 (49)* | 118 (61) |
Values are mean ± SD except definitive NASH is N (%)
P=0.001 vs WT/WT;
P< 0.05 vs WT/WT;
P< 0.05 vs C282Y/WT and C282Y/H63D from ordinal logistic regression after adjustment for sex, age, BMI and presence of diabetes.
P≤ 0.05 vs WT/WT or H63D/WT and H63D/H63D from logistic regression after adjustment for sex, age, BMI and presence of diabetes.
To investigate if the differences in histologic features of NAFLD between different HFE genotypes were independent of the effects of hepatic iron deposition we employed multivariate logistic regression analysis adjusting for potential confounders including sex, age, BMI, ALT, HOMA-IR and presence of diabetes. The presence of HC iron was modeled with the presence of any HFE mutation as a dichotomous independent variable (Y/N). Moreover, each HFE genotype (C282Y/WT and C282Y/H63D or H63D/WT and H63D/H63D) was modeled individually including the presence of any HC iron as a dichotomous independent variable (Y/N). These results are presented in Table 4. Presence of H63D mutations was an independent risk factor for the highest grade of steatosis, lobular inflammation and a NAS ≥5 (p≤0.045). HC iron was negatively associated with ballooning, NAS ≥5 and a definitive diagnosis of NASH (p≤0.048).
Table 4.
Independent risk of iron phenotype and HFE genotype for advanced histologic features of NAFLD and diagnosis of NASH
| Phenotype | OR | 95% Conf. Int. | P value |
|---|---|---|---|
| Steatosis Grade 3 | |||
| H63D mutation | 1.74 | 1.19 – 2.55 | 0.005 |
| Lob. Inflammation Grade ≥2 | |||
| H63D mutation | 1.45 | 1.02 – 2.07 | 0.040 |
| Hep. Ballooning Grade 2 | |||
| HC iron positive | 0.59 | 0.39 – 0.88 | 0.011 |
| NAS ≥5 | |||
| HC iron positive | 0.50 | 0.33 – 0.76 | 0.001 |
| H63D mutation | 1.44 | 1.01 – 2.06 | 0.045 |
| Definitive NASH | |||
| HC iron positive | 0.69 | 0.46 – 1.00 | 0.048 |
HC iron phenotype and HFE genotype was modeled individually including variables any HFE (Y/N) and any HC iron (Y/N), respectively. Each model included potential confounding variables age, sex, BMI, ALT, HOMA-IR and presence of diabetes
DISCUSSION
Here we report that the hemochromatosis C282Y mutation is associated with lower serum hepcidin levels and increased iron deposition specifically in hepatocytes compared to WT subjects. All subjects with hepatic iron deposition had higher hepcidin levels compared to subjects without iron staining for all HFE genotypes, suggesting an appropriate physiologic increase in hepcidin in response to the presence of iron occurs in these subjects, despite the presence of HFE mutations.
Hereditary hemochromatosis with phenotypic expression is characterized by marked and unabated multi-organ hepatic iron loading and is usually associated with HFE C282Y homozygosity; this mutation results in inappropriately low levels of the iron regulatory hormone, hepcidin. Therefore, HFE has been considered a plausible candidate as a disease modifying gene in many liver diseases due to the potential to contribute to increased hepatic iron loading and exacerbation of liver injury due to iron-related oxidative stress. Several studies in various liver diseases have found increased hepatic and/or serum iron parameters in subjects with heterozygous HFE mutations, particularly C282Y (1). A few recent studies have determined either serum hepcidin or prohepcidin levels using either ELISA (28, 29) or mass spectrometry (30, 31) in small cohorts of NAFLD subjects. Hepcidin, but not prohepcidin, was reportedly elevated in NAFLD vs healthy control subjects (28-30). To our knowledge, the present study represents the first detailed investigation to include HFE genotype status, serum hepcidin levels, hepatic iron deposition patterns and clinical and histological features in a large and uniformly evaluated cohort of NAFLD patients.
We believe the current observations provide novel mechanistic insights linking carriage of the HFE C282Y mutation to increased HC iron via decreased levels of hepcidin in NAFLD. Decreased serum hepcidin levels were specifically associated with iron deposition within hepatocytes, but not nonparenchymal RES cells. These data are in agreement with our previously proposed hypothesis that increased HC iron in NAFLD is due to increased iron absorption as a consequence of mutations in iron-regulatory genes such as HFE which result in inappropriately low hepcidin levels and lack of inhibition of iron absorption (15). While we found that C282Y/WT and C282Y/H63D contribute to decreased serum hepcidin levels and greater HC iron loading compared to WT mutations, we also found that HFE genotype did not affect the appropriate physiologic up-regulation of hepcidin when hepatic iron deposition was present. These data suggest that body iron stores drive hepcidin levels in patients with NAFLD, regardless of HFE genotype even among subjects with C282Y mutations and lower levels of serum hepcidin. Furthermore, we found that there was an association between pattern of iron staining and hepcidin level, suggesting that there may be a complex relationship between hepcidin signaling and hepatic iron phenotype; we aim to examine this mechanism further in future studies.
The overall prevalence of HFE genotypes in our study was similar to a recent large study of NAFLD patients from northern Italy (14), with the exception of a higher prevalence of C282Y/WT in our study (11.4% vs 5.8%), which was similar to previous US studies (8, 9). It is possible that the lower prevalence of C282Y/WT subjects in the study by Valenti et al could explain their lower frequency of subjects with a predominantly hepatocellular iron pattern and C282Y mutations (13%) compared to the 20% of subjects carrying C282Y mutations who had HC iron deposition in our study (14). The proportion of subjects with each HFE mutation and nonparenchymal RES iron deposition was very similar in both studies. Interestingly, in separate studies using similar cohorts of Italian NAFLD subjects, Valenti et al have shown that the C282Y/WT and H63D/H63D genotypes were independently associated with HC iron deposition using multivariate regression analysis (32), and that these combined genotypes had lower serum hepcidin levels than subjects with either H63D/WT or WT/WT genotypes with similar SF levels (31).
All known C282Y homozygotes were excluded from all NASH CRN studies thereby not allowing us to determine the effect of C282Y homozygosity on hepcidin level, degree of hepatic iron deposition or relationship to NAFLD severity. However, the degree of penetrance of C282Y heterozygosity to cause mild to moderate HC iron loading in NAFLD subjects (34%) appears to be similar to the penetrance of C282Y homozygosity to cause HC iron overload in HH patients; an estimated 38%-50% of C282Y homozygotes develop iron overload (33) with 1%-28% developing iron-related disease (34). While 61% of C282Y/WT subjects did not have stainable hepatic iron present, 20% of biopsies with HC iron staining were from patients with C282Y mutations. Thus, as has been proposed in hemochromatosis (35), additional modifying genes may alter the penetrance of C282Y in NAFLD. Genes with mutations that have been associated with altered iron metabolism in NAFLD patients include the β-globin (32) and alpha-1-antitrypsin genes (36). Additional studies are warranted to investigate the effects of the numerous potential hemochromatosis phenotype modifying genes upon iron metabolism in NAFLD patients.
Previous studies of HFE mutations in NAFLD patients have mainly focused on the relationship of HFE mutations and advanced fibrosis rather than other histologic features of NAFLD (3-11). Our results are in agreement with the majority of studies which failed to find an association between HFE mutations and advanced fibrosis (9). In fact, we found that C282Y genotypes were associated with lower ballooning scores and less frequent diagnosis of definitive NASH (which depends upon the presence of ballooning), but this association was not significant in multivariate regression analysis and probably represents multicollinearity with HC iron rather than an effect of the C282Y mutation per se (15). An interesting and unexpected finding in this study was the independent relationship between H63D mutations and several advanced histologic features of NAFLD unrelated to the presence of hepatic iron deposition; H63D genotypes were associated with the highest grade of steatosis and lobular inflammation independent of the presence of iron deposition, which was present in only 35.8% of subjects with H63D mutations.
Recent studies in Hfe deficient mice could explain our observation that H63D mutations may play a role in NAFLD pathogenesis independent of iron. Petrak et. al, compared the hepatic proteomic profiles of Hfe deficient mice to WT mice with equivalent hepatic iron stores obtained through dietary iron loading (37). A number of proteins were differentially expressed in the liver in the 2 groups of mice including proteins involved in TNFα signaling (glutathione-S-transferase P1) (38) and cholesterol and fatty acid metabolism (liver carboxylesterase 1) (39). Interestingly, three of the proteins upregulated in the Hfe deficient mice have been shown to be downregulated via PPARα activation (40), suggesting aberrant PPARα regulatory pathways in the Hfe deficient mice. PPARα is a nuclear receptor which regulates expression of genes important in fatty acid β-oxidation and has been shown to be important in decreasing hepatic steatosis (41). In another recent study in Hfe deficient mice, a high fat diet lead to development of NAFLD and upregulation of genes involved in lipid metabolism and down regulation of fatty acid β-oxidation genes, which was not observed in WT mice fed a high fat diet (42). High fat feeding also led to lower hepatic iron compared to normal chow in the Hfe (-/-) mice, suggesting that dysregulation of the lipogenic genes in these mice may be related to Hfe mutations independent of hepatic iron load (42). An alternate explanation is that HFE might be in linkage disequilibrium with one or more steatogenic genes. Effects of these genes could be masked by the effects of HC iron deposition in C282Y carriers, which we previously proposed may lead to a milder form of NAFLD without numerous metabolic abnormalities, thus explaining the lack of association of C282Y mutations with the higher steatosis and lobular inflammation grades that was observed in H63D genotypes.
This study did have limitations that should be noted. The small numbers of subjects with C282Y/H63D or H63D/H63D genotypes limit the conclusions when these genotypes were analyzed independently. For some analyses these genotypes were combined with either the C282Y/WT or H63D/WT genotypes, respectively. Some of the histologic differences we observed, especially for H63D mutations, although statistically significant and independent of confounding factors, were modest and may have limited clinical significance. Whether these observations represent a novel mechanism linking HFE protein to inflammation and lipid metabolism will need to be confirmed by detailed mechanistic studies.
In conclusion, our results suggest a mechanism whereby a genetic predisposition causing decreased hepcidin, results in a specific hepatic iron deposition pattern in NAFLD; C282Y mutations led to lower serum hepcidin levels and increased HC iron accumulation. However, hepcidin sensitivity to hepatic iron loading was preserved across all HFE genotypes, and even among C282Y heterozygous subjects with a blunted hepcidin response, suggesting that iron stores is the predominant regulator of hepcidin expression in NAFLD, regardless of HFE genotype. Our study also provides novel evidence that the HFE H63D mutation may contribute to NAFLD pathogenesis through as yet undefined mechanism(s) independent of iron loading.
Acknowledgments
Source of funding: The Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) is supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grants U01DK061718, U01DK061728, U01DK061731, U01DK061732, U01DK061734, U01DK061737, U01DK061738, U01DK061730, U01DK061713), and the National Institute of Child Health and Human Development (NICHD). This work was also supported by NIH grants R01DK087696 and K24DK002957 to KVK.
Several clinical centers use support from General Clinical Research Centers or Clinical and Translational Science Awards in conduct of NASH CRN Studies (grants UL1RR024989, M01RR000750, M01RR00188, UL1RR02413101, M01RR000827, UL1RR02501401, M01RR000065, M01RR020359).
This work was supported in part by the Intramural Research Program of the National Cancer Institute.
The authors would like to acknowledge the support of the BRI Genotyping Core facility. We would also like to thank Laura Wilson and Patricia Belt for assistance in preparation of the data.
Abbreviations
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NAS
NAFLD activity score
- HC
Hepatocellular
- RES
reticuloendothelial system
- SF
serum ferritin
- TS
transferrin saturation
Members of the Nonalcoholic Steatohepatitis Clinical Research Network
Baylor College of Medicine, Houston, TX: Stephanie H. Abrams, MD, MS; Leanel Angeli Fairly, RN
Case Western Reserve University Clinical Centers: MetroHealth Medical Center, Cleveland, OH: Arthur J. McCullough, MD; Patricia Brandt; Diane Bringman, RN (2004-2008); Srinivasan Dasarathy, MD; Jaividhya Dasarathy, MD; Carol Hawkins, RN; Yao-Chang Liu, MD (2004-2009); Nicholette Rogers, PhD, PA-C (2004-2008); Margaret Stager, MD (2004-2009); Judy Whitwell (2004-2009)
Cleveland Clinic Foundation, Cleveland, OH: Arthur J. McCullough, MD; Srinivasan Dasarathy, MD; Mangesh Pagadala,MD; Ruth Sargent, LPN; Lisa Yerian, MD; Claudia Zein, MD
California Pacific Medical Center, San Francisco, CA: Raphael Merriman, MD; Anthony Nguyen
Children’s National Medical Center, Washington DC (2007-2009): Parvathi Mohan, MD (2007-2009); Kavita Nair (2007-2009)
Cincinnati Children’s Hospital Medical Center, Cincinnati, OH: Stephanie DeVore, MSPH; Rohit Kohli, MD; Kathleen Lake, MSW; Stavra Xanthakos, MD
Columbia University, New York, NY: Yohaime Cosme, MD; Joel E. Lavine, MD, PhD; Ali Mencin, MD; Nadia Ovchinsky, MD
Duke University Medical Center, Durham, NC: Manal F. Abdelmalek, MD; Stephanie Buie; Anna Mae Diehl, MD; Marcia Gottfried, MD (2004-2008); Cynthia Guy, MD; Meryt Hanna (2010); Christopher Kigongo; Paul Killenberg, MD (2004-2008); Samantha Kwan, MS (2006-2009); Yi-Ping Pan; Dawn Piercy, FNP; Melissa Smith (2007-2010); Savita Srivastava, MD
Indiana University School of Medicine, Indianapolis, IN: Elizabeth Byam, RN; Naga Chalasani, MD; Oscar W. Cummings, MD; Marwan Ghabril, MD; Ann Klipsch, RN; Jean P. Molleston, MD; Linda Ragozzino, RN; Girish Subbarao, MD; Sweta Tandra, MD; Raj Vuppalanchi, MD
Johns Hopkins Hospital, Baltimore, MD: Caroline Devadason; Kimberly Pfeifer, RN; Ann Scheimann, MD; Michael Torbenson, MD
Mount Sinai Kravis Children’s Hospital, New York, NY: Nanda Kerkar, MD; Sreevidya Narayanappa; Frederick Suchy, MD (2010)
Northwestern University Feinberg School of Medicine/Children’s Memorial Hospital: Katherine Dunne; Mark H. Fishbein, MD; Katie Jacques; Ann Quinn, RD; Cindy Riazi, RN; Peter F. Whitington, MD
P:\Shared\Doc\NASH\credit roster Feb 24, 2011
Saint Louis University, St Louis, MO: Sarah Barlow, MD (2002-2007); Jose Derdoy, MD; Debra King, RN; Andrea Morris; Joan Siegner, RN; Susan Stewart, RN; Brent A. Neuschwander-Tetri, MD; Judy Thompson, RN
University of California San Diego, San Diego, CA: Cynthia Behling, MD, PhD; Jennifer Collins; Janis Durelle; Tarek Hassanein, MD (2004-2009); Joel E. Lavine, MD, PhD (2002-2010); Rohit Loomba, MD; Anya Morgan; Steven Rose, MD (2007-2009); Heather Patton, MD; Jeffrey B. Schwimmer, MD; Claude Sirlin, MD; Tanya Stein, MD (2005-2009)
University of California San Francisco, San Francisco, CA: Bradley Aouizerat, PhD; Kiran Bambha, MD (2006-2010); Marissa Bass; Nathan M. Bass, MD, PhD; Linda D. Ferrell, MD; Danuta Filipowski, MD (2005-2010); Shannon Fleck; Bo Gu (2009-2010); Bilal Hameed, MD; Camille Langlois; Mark Pabst; Monique Rosenthal (2005-2010); Philip Rosenthal, MD; Tessa Steel (2006-2008)
University of Washington Medical Center and Seattle Children’s Hospital, Seattle, WA: Melissa Coffey; Sarah Galdzicka; Karen Murray, MD; Matthew Yeh, MD, PhD
Virginia Commonwealth University, Richmond, VA: Sherry Boyett, RN, BSN; Melissa J. Contos, MD; Michael Fuchs, MD; Amy Jones; Velimir AC Luketic, MD; Puneet Puri, MD; Bimalijit Sandhu, MD (2007-2009); Arun J. Sanyal, MD; Carol Sargeant, RN, BSN, MPH; Kimberly Noble; Melanie White, RN, BSN (2006-2009)
Virginia Mason Medical Center, Seattle, WA: Sarah Ackermann; Kris V. Kowdley, MD; Jane Park; Tracey Pierce; Jody Mooney, MS; James Nelson, PhD; Cheryl Shaw, MPH; Alice Stead; Chia Wang, MD
Washington University, St. Louis, MO: Elizabeth M. Brunt, MD
Resource Centers
National Cancer Institute, Bethesda, MD: David E. Kleiner, MD, PhD
National Institute of Child Health and Human Development, Bethesda, MD: Gilman D. Grave, MD
National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD: Edward C. Doo, MD; Jay H. Hoofnagle, MD; Patricia R. Robuck, PhD, MPH; Averell Sherker, MD
Johns Hopkins University, Bloomberg School of Public Health (Data Coordinating Center), Baltimore, MD: Patricia Belt, BS; Frederick L. Brancati, MD, MHS (2003-2009); Jeanne M. Clark, MD, MPH; Ryan Colvin, MPH (2004-2010); Michele Donithan, MHS; Mika Green, MA; Rosemary Hollick (2003-2005); Milana Isaacson, BS; Wana K. Jin, BS; Alison Lydecker, MPH (2006-2008), Pamela Mann, MPH (2008-2009); Kevin P. May, MS; Laura Miriel, BS; Alice Sternberg, ScM; James Tonascia, PhD; Aynur Ünalp-Arida, MD, PhD; Mark Van Natta, MHS; Ivana Vaughn, MPH; Laura Wilson, ScM; Katherine Yates, ScM
Footnotes
Disclosures: No conflicts of interest exist
References
- 1.Bonkovsky HL, Lambrecht RW, Shan Y. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol. 2003;30:137–144. doi: 10.1016/s0741-8329(03)00127-7. [DOI] [PubMed] [Google Scholar]
- 2.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonkovsky HL, Jawaid Q, Tortorelli K, et al. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol. 1999;31:421–429. doi: 10.1016/s0168-8278(99)80032-4. [DOI] [PubMed] [Google Scholar]
- 4.George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology. 1998;114:311–318. doi: 10.1016/s0016-5085(98)70482-2. [DOI] [PubMed] [Google Scholar]
- 5.Fargion S, Mattioli M, Fracanzani AL, et al. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am J Gastroenterol. 2001;96:2448–2455. doi: 10.1111/j.1572-0241.2001.04052.x. [DOI] [PubMed] [Google Scholar]
- 6.Chitturi S, Weltman M, Farrell GC, et al. HFE mutations, hepatic iron, and fibrosis: ethnic-specific association of NASH with C282Y but not with fibrotic severity. Hepatology. 2002;36:142–149. doi: 10.1053/jhep.2002.33892. [DOI] [PubMed] [Google Scholar]
- 7.Valenti L, Dongiovanni P, Fracanzani AL, et al. Increased susceptibility to nonalcoholic fatty liver disease in heterozygotes for the mutation responsible for hereditary hemochromatosis. Dig Liver Dis. 2003;35:172–178. doi: 10.1016/s1590-8658(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 8.Nelson JE, Bhattacharya R, Lindor KD, et al. HFE C282Y mutations are associated with advanced hepatic fibrosis in Caucasians with nonalcoholic steatohepatitis. Hepatology. 2007;46:723–729. doi: 10.1002/hep.21742. [DOI] [PubMed] [Google Scholar]
- 9.Hernaez R, Yeung E, Clark JM, et al. Hemochromatosis gene and nonalcoholic fatty liver disease: A systematic review and meta-analysis. J Hepatol. 2011;55:1079–1085. doi: 10.1016/j.jhep.2011.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deguti MM, Sipahi AM, Gayotto LC, et al. Lack of evidence for the pathogenic role of iron and HFE gene mutations in Brazilian patients with nonalcoholic steatohepatitis. Braz J Med Biol Res. 2003;36:739–745. doi: 10.1590/s0100-879x2003000600009. [DOI] [PubMed] [Google Scholar]
- 11.Bugianesi E, Manzini P, D’Antico S, et al. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179–187. doi: 10.1002/hep.20023. [DOI] [PubMed] [Google Scholar]
- 12.Zamin I, Jr, Mattos AA, Mattos AZ, et al. Prevalence of the hemochromatosis gene mutation in patients with nonalcoholic steatohepatitis and correlation with degree of liver fibrosis. Arq Gastroenterol. 2006;43:224–228. doi: 10.1590/s0004-28032006000300013. [DOI] [PubMed] [Google Scholar]
- 13.Neri S, Pulvirenti D, Signorelli S, et al. The HFE gene heterozygosis H63D: a cofactor for liver damage in patients with steatohepatitis? Epidemiological and clinical considerations. Intern Med J. 2008;38:254–258. doi: 10.1111/j.1445-5994.2007.01474.x. [DOI] [PubMed] [Google Scholar]
- 14.Valenti L, Fracanzani AL, Bugianesi E, et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology. 2010;138:905–912. doi: 10.1053/j.gastro.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Nelson JE, Wilson L, Brunt EM, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology. 2011;53:448–457. doi: 10.1002/hep.24038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 17.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 18.Gao J, Chen J, Kramer M, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009;9:217–227. doi: 10.1016/j.cmet.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt PJ, Toran PT, Giannetti AM, et al. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008;7:205–214. doi: 10.1016/j.cmet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 22.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 23.Neuschwander-Tetri BA, Clark JM, Bass NM, et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology. 2010;52:913–924. doi: 10.1002/hep.23784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–1685. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson DA, Grant BF, Stinson FS, et al. Effectiveness of the derived Alcohol Use Disorders Identification Test (AUDIT-C) in screening for alcohol use disorders and risk drinking in the US general population. Alcohol Clin Exp Res. 2005;29:844–854. doi: 10.1097/01.alc.0000164374.32229.a2. [DOI] [PubMed] [Google Scholar]
- 26.Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood. 2008;112:4292–4297. doi: 10.1182/blood-2008-02-139915. [DOI] [PubMed] [Google Scholar]
- 27.Kleiner DE, Brunt EM, Van NM, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 28.Lee SH, Jeong SH, Park YS, et al. Serum prohepcidin levels in chronic hepatitis C, alcoholic liver disease, and nonalcoholic fatty liver disease. Korean J Hepatol. 2010;16:288–294. doi: 10.3350/kjhep.2010.16.3.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senates E, Yilmaz Y, Colak Y, et al. Serum levels of hepcidin in patients with biopsyproven nonalcoholic Fatty liver disease. Metab Syndr Relat Disord. 2011;9:287–290. doi: 10.1089/met.2010.0121. [DOI] [PubMed] [Google Scholar]
- 30.Zimmermann A, Zimmermann T, Schattenberg J, et al. Alterations in lipid, carbohydrate and iron metabolism in patients with non-alcoholic steatohepatitis (NASH) and metabolic syndrome. Eur J Intern Med. 2011;22:305–310. doi: 10.1016/j.ejim.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 31.Valenti L, Swinkels DW, Burdick L, et al. Serum ferritin levels are associated with vascular damage in patients with nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2011;21:568–575. doi: 10.1016/j.numecd.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Valenti L, Canavesi E, Galmozzi E, et al. Beta-globin mutations are associated with parenchymal siderosis and fibrosis in patients with non-alcoholic fatty liver disease. J Hepatol. 2010;53:927–933. doi: 10.1016/j.jhep.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 33.Whitlock EP, Garlitz BA, Harris EL, et al. Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2006;145:209–223. doi: 10.7326/0003-4819-145-3-200608010-00009. [DOI] [PubMed] [Google Scholar]
- 34.Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358:221–230. doi: 10.1056/NEJMoa073286. [DOI] [PubMed] [Google Scholar]
- 35.Levy JE, Montross LK, Andrews NC. Genes that modify the hemochromatosis phenotype in mice. J Clin Invest. 2000;105:1209–1216. doi: 10.1172/JCI9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valenti L, Dongiovanni P, Piperno A, et al. Alpha 1-antitrypsin mutations in NAFLD: high prevalence and association with altered iron metabolism but not with liver damage. Hepatology. 2006;44:857–864. doi: 10.1002/hep.21329. [DOI] [PubMed] [Google Scholar]
- 37.Petrak J, Myslivcova D, Halada P, et al. Iron-independent specific protein expression pattern in the liver of HFE-deficient mice. Int J Biochem Cell Biol. 2007;39:1006–1015. doi: 10.1016/j.biocel.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y, Fan Y, Xue B, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene. 2006;25:5787–5800. doi: 10.1038/sj.onc.1209576. [DOI] [PubMed] [Google Scholar]
- 39.Redinbo MR, Bencharit S, Potter PM. Human carboxylesterase 1: from drug metabolism to drug discovery. Biochem Soc Trans. 2003;31:620–624. doi: 10.1042/bst0310620. [DOI] [PubMed] [Google Scholar]
- 40.Chu R, Lim H, Brumfield L, et al. Protein profiling of mouse livers with peroxisome proliferator-activated receptor alpha activation. Mol Cell Biol. 2004;24:6288–6297. doi: 10.1128/MCB.24.14.6288-6297.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kallwitz ER, McLachlan A, Cotler SJ. Role of peroxisome proliferators-activated receptors in the pathogenesis and treatment of nonalcoholic fatty liver disease. World J Gastroenterol. 2008;14:22–28. doi: 10.3748/wjg.14.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan TC, Crawford DH, Jaskowski LA, et al. Altered lipid metabolism in Hfe-knockout mice promotes severe NAFLD and early fibrosis. Am J PhysiolGastrointest Liver Physiol. 2011;301:G865–G876. doi: 10.1152/ajpgi.00150.2011. [DOI] [PubMed] [Google Scholar]