

Abstract
Protein-protein interactions (PPIs) mediated by the polo-box domain (PBD) of polo-like kinase 1 (Plk1) serve important roles in cell proliferation. Critical elements in the high affinity recognition of peptides and proteins by PBD are derived from pThr/pSer-residues in the binding ligands. However, there has been little examination of pThr/pSer mimetics within a PBD context. Our current paper compares the abilities of a variety of amino acid residues and derivatives to serve as pThr/pSer replacements by exploring the role of methyl functionality at the pThr β–position and by replacing the phosphoryl group by phosphonic acid, sulfonic acid and carboxylic acids. This work sheds new light on structure activity relationships for PBD recognition of phosphoamino acid mimetics.
Keywords: Plk1, polo-like kinase, polo-box domain, phosphoamino acid mimetic
1. Introduction
The central roles that protein-protein interactions (PPIs) play in biochemical processes1,2 and their frequent reliance on binding “hot spots”3–6 have made them attractive therapeutic targets.7–14 Many components of cellular signal transduction employ sub-classes of PPIs that involve the recognition of either phosphotyrosyl (pTyr) or phosphothreonyl (pThr)/phosphoserine (pSer)–containing sequences.15–19 Because phosphoryl groups serve as key elements of high affinity interactions, these phospho-dependent PPIs are potentially amenable to disruption by small-to-moderate size molecules.
The polo-like kinases (Plks) are a subfamily of serine-threonine kinases that play critical roles in cellular proliferation.20–24 Of the known Plks (Plk1 – Plk5), Plk1 has been identified as an anticancer target due to its ability to promote tumorigensesis. 25–27 In addition to its catalytic domain, Plk1 contains a C-terminal polo-box domain (PBD), which recognizes pThr/pSer-containing sequences and directs the enzyme to specific sub-cellular locations.28 While significant effort has been devoted to developing inhibitors targeting the catalytic activity of Plk1,29–36 targeting the PBD may afford an alternative PPI-directed approach to down-regulating Plk1 function.25,37–43
The five-mer peptide “Pro-Leu-His-Ser-pThr” (1, Figure 1) represents a high affinity PBD-binding sequence derived from the pT78 region of the polo-box interacting protein 1 (PBIP1), which offers a starting point for the design of PBD-directed inhibitors.38 Although the phosphoryl moiety provides an essential PBD recognition motif, it presents particular challenges to bioavailability due to its dianionic charge and the lability of the phosphoryl ester bond to phosphatases. Similar problems are also encountered in the development of inhibitors directed against pTyr-binding domains. However, unlike the latter family of compounds, for which a large number of pTyr mimetics have been examined,44,45 significantly fewer pThr/pSer mimetics have been reported.46–50 Our current paper reports the synthesis and PBD-binding affinities of peptides containing a variety of pThr/pSer mimetics, many of which have not yet been examined within the context of PBD-binding peptides.
Figure 1.

Structures of peptides discussed in the text.
2. Results and discussion
2.1 Design of phosphomimetic-containing peptides
Recently, we discovered that introduction of long-chain arylalkyl groups onto the δ1 nitrogen (N3) of the histidine imidazole ring of parent peptide 1 could significantly increase Plk1 PBD-binding affinity.42 From the X-ray co-crystal structure of a high affinity peptide containing a C6H5(CH2)8–His adduct (2a, Figure 1), we found that the potency enhancement occurred through new binding interactions in a previously occluded hydrophobic channel on the PBD surface. Although our original His-adduct-containing peptides were obtained in small quantities as synthetic by-products, we devised a route to synthesize protected δ1- alkyl-containing amino acid analogues of type 3 (Figure 2) that allowed direct Fmoc-based solid-phase synthesis His-modified peptides such as 2a.51 In our current study, we replaced the pThr phosphate group in 2a with phosphonic acid (2b), sulfonic acid (2c) and carboxylic acid (2d) functionality. We also replaced the pThr residue with pSer (2e), the β,β-bis-methyl variant of pSer (2f) and p(allo-Thr) (2g) as well as with Glu (2h), Gln (2i) and Asp (2j) residues (Figure 1) and determined their Plk1 PBD binding affinities.
Figure 2.

Protected amino acids used to prepare peptides 2a – 2g.
2.2 Preparation of protected amino acid analogues and their use in solid-phase peptide synthesis
Peptides 2a – 2k were prepared by solid-phase techniques using Fmoc-based protocols. The C-terminal residues of peptides 2a – 2g were incorporated using the protected amino acid analogues 4a – 4g, respectively (Figure 2). Commercially-available monobenzyl phosphoryl esters (4a, 4e and 4g) were used to introduce pThr, pSer and p(allo-Thr) respectively. Reagent 4b, which was used to introduce the (2S, 3R)-2-amino-3-methyl-4-phosphonobutyric acid (Pmab) residue, was prepared according to literature procedures.50 The remaining amino acid analogues (4c, 4d and 4f) were synthesized as outlined below.
2.2.1 Preparation of N-Fmoc (2S,3R)-2-amino-3-methyl-4-((2,2,2-trichloroethoxy)sulfonyl)butanoic acid (4c)
Preparation of peptide 2c, containing the sulfonic acid-based pThr mimetic, (2S, 3R)-2-amino-3-methyl-4-sulfonylbutanoic acid (Smab), was achieved using reagent 4c, which has a sulfonic acid moiety. Mitsunobu treatment of known 550 with thioacetic acid, triphenylphosphane, and diisopropyl diazodicarboxylate (DIAD) in dry THF gave 6 in good yield (Scheme 1). The N-benzyl carbamate group of 6 was converted to acetyl amide (7) using sodium iodide and acetyl chloride. Oxidative chlorination of 7 to the corresponding sulfonyl chloride was achieved in good yield using N-chlorosuccinimide (NCS) in aqueous HCl – MeCN 52 and this was followed by treatment with aqueous sodium hydroxide to provide the free sulfonic acid (8). Finally, removal of N-acetyl and O-methyl groups by refluxing in 6N HCl yielded the free amino acid, which was converted to the N-Fmoc-protected 4c using Fmoc-OSu (Scheme 1).
Scheme 1.

Reagents and conditions: a) AcSH (2.0 eq.), PPh3 (2.0 eq.), DIAD (2.0 eq.), THF, 0 °C – rt, 3 h, 98%; b) NaI (10.0 eq.), AcCl (10.0 eq.), MeCN, reflux, 7 h, 85%; c) 1) NCS (4.0 eq.), 2N HCl/MeCN (v/v, 1/4), 10 °C, 1 h; 2) 0.1N NaOH/dioxane (1: 1, v/v), rt, 0.5 h; d) 1) 6N HCl, reflux, 12 h; 2) FmocOSu, 0.1N NaOH/dioxane, overnight, 14% for four steps.
2.2.2 Preparation of N-Fmoc (2S,3S)-2-amino-5-(tert-butoxy)3-methyl-5-oxoentanoic acid (4d)
Introduction of the C-terminal pThr mimicking residue in peptide 2d employed the protected amino acid 4d. The synthesis of 4d is shown in Scheme 2. Conversion of the protected D-serine analogue 9 to 14 was according to literature procedures (Scheme 2).53 Compound 14 was first converted from the ethyl ester to the benzyl ester (15) and then subjected to Jones’ oxidation to afford the protected amino acid 16. The desired reagent 4d was obtained by removing the N-Boc group (4M HCl in dioxane) and installing the N- Fmoc group (Fmoc-OSu) (Scheme 2).
Scheme 2.

Reagents and conditions: a) 2,2-dimethoxypropane (10 eq.), BF3 Et2O (0.1eq.), acetone, rt, 2 h, 91%; b) 1) LiOH H2O (2.0 eq.), MeOH/H2O, rt, 4 h; 2) BOP (1.2 eq.), NH(OMe)Me HCl (1.25 eq.), Et3N (2.0 eq.), rt, 3 h, 70% for two steps; c) MeLi (1.7 eq.), THF, −78 °C, 1 h, 59%; d) triethyl phosphonoacetate (1.5 eq.), NaH (1.5 eq.), 0 °C, 24 h, 74%; e) 10% Pd•C, H2, MeOH/AcOEt, rt, 12 h, 97%; f) 1) LiOH H2O (3.0 eq.), THF/H2O, rt, 12 h.; 2) BnBr (1.5 eq.), NaHCO3 (2.0 eq.), DMF, rt, 24 h, 67% for two steps; g) 2.7 M Jones’ reagent (1.5 eq.), acetone, rt, 30 min, 89%; h) 1) 4M HCl in dioxane (10 eq.), 3 h; 2) Fmoc-OSu(1.5 eq.), NaHCO3 (2.0 eq.), THF – H2O (v/v 1:1), 12 h, 81% for two steps.
2.2.3 Preparation of N-Fmoc (2S)-2-amino-3-(benzyloxyphosphoryloxy)-3-methylbutanoic acid (4f)
The preparation of phosphopeptide 2f employed the protected amino acid 4f, which was prepared as shown in Scheme 3. Grignard reaction of 10 with MeMgI in diethyl ether at 0 °C proceeded to give 17, which was converted to 18 by oxidation with Jones’ reagent in acetone at 0 °C.54 Compound 18 was converted to its allyl ester and then the N-Boc group was exchanged for N-Fmoc protection (19) by first deprotecting using 1M HCl and then reacting with Fmoc-Osu. Following a similar strategy as previously reported for the synthesis of 4a,55 19 was converted first to the dibenzyl ester (20) and then to the desired reagent 4f (Scheme 3).
Scheme 3.

Reagents and conditions: a) Mg (6.0 eq.), MeI (7.5 eq.), Et2O, 0 °C, 30 min, 92%; b) Jones’ reagent (2.7 M, 1.5 eq.), acetone, 0 °C to rt, 1 h, 78%; c) 1) Allyl bromide (1.5 eq.), NaHCO3 (2.0 eq.), DMF, rt, 24 h, 60%; 2) 1M HCl in Et2O (10.0 eq.), rt, 3 h; 3) Fmoc-OSu (1.5 eq.), NaHCO3 (2.0 eq.), THF/H2O (1: 1 v/v), rt, 12 h, 85% for two steps; d) dibenzyl diisopropyl phosphoramidite (2.0 eq.), N-methylaniline trifluoroacetate (4.0 eq.), t-butyl hydroperoxide in n-octane (1.9 eq.), rt, 1 h, 90%; e) Pd(PPh3)4 (0.1 eq.), N-methylaniline (3.0 eq.), THF, rt, 1 h, 91%; 2) 10% Pd•C, 2, 2′-bipyridyl (0.5 eq.), H2, rt, 0.5 h, 60%.
2.3 Plk1 PBD-binding affinities of peptides 2a – 2k
2.3.1 Dianionic species
We determined the Plk1 PBD binding affinities using an ELISA-based 96-well assay that measured the ability of the synthetic peptides to compete with support-bound pT78-derived phosphopeptide for binding to Plk1 expressed in mitotic 293A cell lysates.38,42 Results are shown in Figures 3 and 4. As previously reported, addition of the C6H5(CH2)8–group to the histidine δ1-nitrogen (peptide 2a) significantly enhanced binding affinity relative to the parent parent peptide 1 (Figure 3). We first examined dianionic functionality. Consistent with previous results,38,42,43,50 high affinity was maintained when pThr was replaced by Pmab- (compare peptide 2a with 2b, Figure 3). This indicates that the phosphoryl ester oxygen in the pThr residue is not a critical component of recognition.
Figure 3.
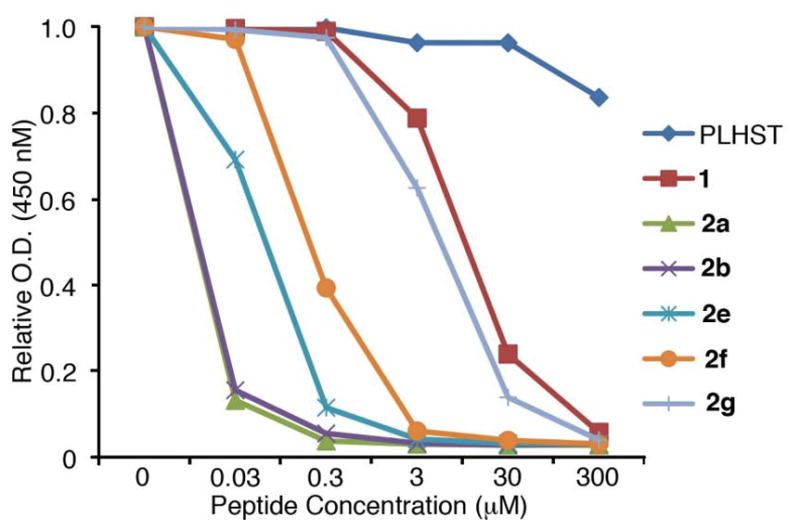
ELISA-based Plk1 PBD-binding results. Representative graphs are shown from three independent experiments.
Figure 4.
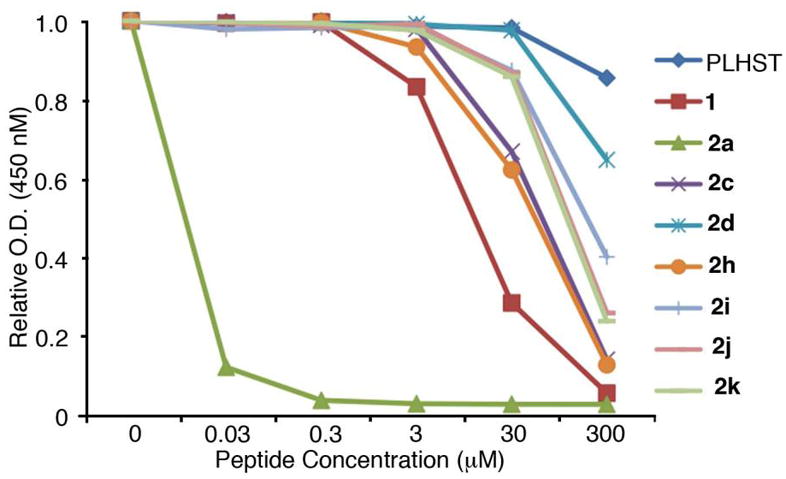
ELISA-based Plk1 PBD-binding results. Representative graphs are shown from three independent experiments.
Next, we directed our attention toward modifying the 3-position of pThr. We observed that deletion of the (3R)-methyl group to give pSer (peptide 2e) resulted in an approximate order-of-magnitude loss of affinity (Figure 3). This was in agreement with previous work that had shown an approximate 7-fold preference for pThr over pSer.56 However, it should be noted that high affinity pSer-containing peptides are known that include a Pro residue C-terminal to the pSer.57 Placement of a second methyl group at the 3-position (2f) led to a further loss of potency, but the 3,3-dimethyl analogue did retain moderately good affinity. In contrast, reversal of the pThr stereochemistry at the 3-position (p-alloThr, 2g), resulted in a nearly three orders-of-magnitude loss of affinity. The relatively good affinities of the des-methyl (pSer, 2e) and dimethyl (2f) analogues indicate that both the absence of the (3R)-methyl and the presence of a pro-(3S)-methyl are tolerated within the binding pocket. Therefore, the dramatic loss of affinity observed with 2g may be related to unfavorable conformational geometries of the p-alloThr residue.
2.3.2 Monoanionic and uncharged species
Unlike SH2 domains, in which recognition of tyrosyl phosphates involves salt bridges between both anionic phosphoryl oxygens and two conserved Arg residues,58 interaction of the PBD with the pThr phosphoryl group of 2a involves only a single salt bridge with Lys540 (PBD accession code 3RQ742). Additional PBD-phosphoryl interactions are hydrogen bonding in nature (with His538 and several water molecules). Therefore, while SH2 domain-directed pTyr mimetics often require maintenance of dianionic character for high affinity binding,45 it may be reasonable to expect that a single anionic charge could be sufficient for ligands to bind with high affinity to PBD domains. The Smab-containing peptide (2c) can be viewed as a monoanionic isostere of Pmab-containing 2b, in which a sulfonic acid moiety is employed rather than a phosphonic acid group. Sulfonic acids are known as monoanionic phosphoryl mimetics.59,60 However, peptide 2c exhibited almost four orders-of-magnitude loss of affinity relative to the reference peptide 2a (Figure 4). This unexpectedly large shift to the right in Figure 4 resulted in 2c binding less efficaciously than peptide 1, in which the His-adduct group is lacking.
Carboxylic acids can serve as monoanionic phosphoryl mimetics in a number of physiological contexts. Aryl-containing carboxylic acids have shown widespread value in the design of constructs targeting pTyr-dependent interactions.44 Glutamic acid has a side chain length that places its carboxylic acid group at a distance from the α-carbon that approximates the phosphoryl groups of both pSer (for example, see61–67) and pThr (for example, see61,68–71). Although the side chain length of Asp situates its carboxylic acid closer to the α-carbon than Glu, it has also frequently been used as a pSer mimetic (for example, see62–67). In our current work, we observed that the Glu-containing peptide (2h) bound with affinity equivalent to the sulfonic acid-containing 2c. This is somewhat unexpected, since the Smab residue in 2c exhibits greater structural similarity to pThr (or Pmab) than does 2h. The Asp-containing peptide (2j) bound with slightly less affinity than 2h and the uncharged Gln-containing peptide (2i) bound nearly as well as 2j. Based on the results presented in Figure 3 showing that a (3R)-methyl group enhances the binding of phosphate-containing species, we anticipated that a (3R)-methyl-containing Glu residue should serve as a better pThr mimetic than unmodified Glu. Therefore, it was surprising to find that introduction of a (3R)-methyl group onto the Glu residue in 2h resulted in a significant loss of affinity (peptide 2d, Figure 4). Peptide 2d bound with even less affinity than the non-phosphorylated peptide (2k), which contained no phosphoryl-mimicking functionality.
The differences in binding affinities of mono-anionic and uncharged species discussed above are minor in comparison to their shared greater than three orders-of-magnitude loss of affinity when compared to the parent pThr-containing peptide 2a. None of the sulfur or carbon-based phosphoryl mimetics was as effective as even the least potent phosphorus-based analogue (2g). It was noteworthy that 3-methyl functionality can play an important role on binding recognition of phosphoryl-containing species.
Our reasoning in selecting platform 2 as a display vehicle relied on the high affinity of the parent peptide 2a, which is approximately three orders-of-magnitude greater than 1. This added affinity reflects extended ligand-protein interactions outside the pThr binding pocket that are afforded by the C6H5(CH2)8-His adduct moiety. These interactions could potentially lessen the relative importance of binding within the pThr pocket. For example, while the non-phosphorylated version of 1 (PLHST) exhibits near negligible affinity, the corresponding non-phosphorylated form of 2 (peptide 2k) displays measurable affinity arising from a shift to the left in its binding curve that is consistent with the shift seen in going from 1 to 2a (Figure 4). It should also be noted that X-ray co-crystal data show that the binding orientation of 2a is nearly superimposable with 1.42 Therefore, the unexpected SAR data for certain series-2 pThr mimetics potentially indicate that something unexpected may be going on in the pThr binding pocket: Just what this “something” is, is not clear. In spite of these uncertainties, the current work represents the first report of PBD binding affinities of peptides containing a series of pThr/pSer mimetics. The SAR parameters derived from our work may be of value in the further design of Plk1 PBD-binding peptides, including those that do not contain the unusual His-adduct moiety.
3. Experimental
3.1. General experimental
All experiments involving moisture-sensitive compounds were conducted under dry conditions (positive argon pressure) using standard syringe, cannula, and septa apparatus. Solvents: All solvents were purchased anhydrous (Aldrich) and used directly. HPLC-grade hexanes, EtOAc, CH2Cl2, and MeOH were used in chromatography. TLC: analytical TLC was performed on Analtech pre-coated plates (Uniplate, silica gel GHLF, 250 microns) containing a fluorescence indicator; NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. Coupling constants are reported in Hertz, and peak shifts are reported in δ (ppm) relative to TMS. Low-resolution mass spectra (ESI) were measured with an Agilent 1200 LC/MSD-SL system. High resolution, accurate mass measurements for confirmation of elemental compositions were obtained by positive ion, ESI analysis on a Thermo Scientific LTQ-XL Orbitrap mass spectrometer with HPLC sample introduction using a short narrow-bore C18 reversed-phase column and standard CH3CN/H2O gradient. Reported m/z values are the average of 8 or more scans over the chromatographic peak of interest.
3.2. Synthesis of N-Fmoc-(2S, 3R)-2-aminuteso-3-methyl-4-sulfobutanoic acid 4c from 5
3.2.1. (2S, 3R)-methyl N-Cbz-2-aminuteso-3-methyl-4-acetylthio-butanoate (6)
Starting from readily prepared aminuteso acid 550 DIAD (0.95 mL, 4.83 mmol) was added at 0 °C under dry nitrogen to a solution of triphenylphosphine (1.27 g, 4.83 mmol) in dry THF (15 mL) in a flame-dried round-bottomed flask, and the mixture was stirred until a white solid appeared. Stirring was continued for 10 minutes at 0 °C, after which compound 5 (0.68 g, 2.42 mmol) in dry THF (5 mL) was added. After the mixture had been stirred for 45 minutes, thioacetic acid (0.35 mL, 4.83 mmol) was added and stirring was continued for 3 h. Diethyl ether was added, and the mixture was washed with H2O, followed by drying of the organic layer over MgSO4. The mixture was filtered and concentrated. The final residue was purified by silica gel column chromatography (EtOAc: hexanes; from 1: 10 to 1: 2) to afford product 6 (0.8 g) as s colorless gum (98%). [α]D18 24.6 (c 0.77, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.39 – 7.27 (m, 5H), 5.62 (d, J = 8.0 Hz, 1H), 5.10 (s, 2H), 4.40 (dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 1H), 3.75 (s, 3H), 3.04 (dd, J1 = 12.0 Hz, J2 = 8.0 Hz, 1H), 2.68 (dd, J1 = 12.0 Hz, J2 = 8.0 Hz, 1H), 2.32 (s, 3H), 2.76 – 2.12 (m, 1H), 1.00 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 195.6, 171.9, 156.2, 136.3, 128.6, 128.2, 67.2, 57.7, 52.4, 37.2, 31.7, 30.6, 16.3; ESI-MS calcd for C16H22NO5S (M+H)+: 340.1, found: 340.1.
3.2.2. (2S, 3R)-Methyl N-acetyl-2-amino-3-methyl-4-acetylthio-butanoate (7)
To a mixture of the 6 (0.322g, 0.95 mmol) and sodium iodide (1.42g, 9.49 mmol) in dry acetonitrile (19 mL) was slowly added at 0 °C acetyl chloride (0.67 mL, 9.49 mmol). The resulting mixture was heated at 60 °C for 7 h with stirring under a nitrogen atmosphere. After addition of saturated aqueous sodium hydrogen sulfite and saturated aqueous sodium hydrogen carbonate with ice cooling, the mixture was thoroughly extracted with chloroform. The extract was washed with brine, dried over potassium carbonate, and evaporated under reduced pressure. Purification of the product using silica gel column chromatography (EtOAc: hexanes; from 1: 2 to 2: 1) gave the product 7 (0.2 g) as a white foam (85%). [α]D18 30.14 (c 0.75, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.34 (d, J = 8.0 Hz, 1H), 4.60 (dd, J1 = 12.0 Hz, J2 = 8.0 Hz, 1H), 3.76 (s, 3H), 3.08 (dd, J1 = 16.0 Hz, J2 = 8.0 Hz, 1H), 2.62 (dd, J1 = 16.0 Hz, J2 = 8.0 Hz, 1H), 2.34 (s, 3H), 2.24 – 2.10 (m, 1H), 2.07 (s, 3H), 1.00 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 172.2, 170.3, 55.9, 52.5, 37.4, 32.0, 30.7, 23.4, 16.7; ESI-MS calcd for C10H18NO4S (M+H)+: 248.1, found: 248.1.
3.2.3. N-Fmoc-(2S, 3R)-2-aminuteso-3-methyl-4-sulfobutanoic acid (4c)
At 0 °C, to a mixture of 7 (0.093 g, 0.38 mmol) in MeCN and 2N HCl (1 mL: 0.2 mL, 5/1 v/v) was added N-chlorosuccinimide (0.201 g, 1.50 mmol) in portions. The mixture was stirred at < 20 °C for 10 to 30 minutes. The solution was diluted with Et2O and washed with 12% aqeuous NaCl (3x). The organic layer was dried (Na2SO4) and concentrated to give product as an off-white solid. The crude product was added 0.1N NaOH and dioxane (v/v 1/1). After 30 minutes, the mixture was concentrated and H2O was added. The aqueous solution was washed with dichloromethane. After all, acidified it by concentrated HCl and lyophilized to get comp. 8, then used it directly in next step. Compound 8 was refluxed in 6N HCl (3.8 mL) for 12 h. The H2O was added (4mL), and then the aqueous solution was washed with dichloromethane (3x). The final solution was lyophilized to dry and used in next step.
The residue obtained above and NaOH (35 mg, 0.88 mmol) were dissolved in H2O (3.5 mL) cooled in an ice bath. A solution of Fmoc-OSu (130 mg, 0.38 mmol) in dioxane (3.5 mL) was added in one portion. The mixture was stirred at room temperature (4 h), then the mixture was evaporated under reduced pressure and H2O (7.0 mL) was added. The aqueous solution was washed with Et2O and acidified with concentrated aqueous HCl. The aqueous solution was lyophilized to yield crude product, which was purified by preparation HPLC (flow rate10 mL/minutes, acetonitrile/H2O with 0.1% TFA, acetonitrile from 30% to 100% in 30 minutes; Retention time = 17.2 minutes) to provide pure 4c as white foam 20 mg (14% for 4 steps). [α]D18 3.0 (c 0.95, MeOH); 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J = 8.0 Hz, 2H), 7.66 (t, J = 8.0 Hz, 2H), 7.40 – 7.20 (m, 4H), 4.30 (d, J = 4.0 Hz, 2H), 4.20 (t, J = 8.0 Hz, 2H), 3.02 (dd, J1 = 16.0 Hz, J2 = 4.0 Hz, 1H), 2.80 – 2.70 (m, 1H), 2.62 – 2.50 (m, 1H), 1.19 (d, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CD3OD) δ 173.6, 158.9, 145.4, 145.3, 142.7, 128.9, 128.3, 126.4, 121.0, 68.3, 60.5, 55.1, 34.3, 17.5; ESI-MS calcd for C20H22NO7S (M+H)+: 420.1, found: 420.0.
3.3. Synthesis of N-Fmoc (2S,3S)-2-aminuteso-5-(tert-butoxy)3-methyl-5-oxoentanoic acid (4d) from 9
Conversion of the protected D-serine analogue 9 to 14 was according to literature procedures.53
3.3.1. (3S, 4S)-Benzyl N, O-isopropylidenyl-4-(N-tert-butyloxycarbonylamino)-3-methyl-5-oxo-pentanoate (15)
To a solution of 14 (0.612 g, 1.94 mmol) in THF-H2O (8 mL/2 mL) at 0 °C was added LiOH•H2O (244 mg, 5.82 mmol) and the mixture was stirred at room temperature until the starting material was consumed (TLC). To the mixture was added 1N HCl until pH = 2 – 3, and then the THF was evaporated and the residue was extracted with EtOAc (3x). The combined organic phases were washed with brine, dried (MgSO4) and filtered and concentrated and the crude product was used directly in next step.
The crude material from above was dissolved in DMF (10 mL) at room temperature under argon and the NaHCO3 (326 mg, 3.88 mmol) and BnBr (0.346 mL, 2.91 mmol) were added sequentially. The mixture was stirred until the starting material was consumed (TLC). The mixture was diluted with EtOAc and then washed with H2O, brine, dried (MgSO4) and filtered and concentrated. The residue was purified by silica gel column chromatography (EtOAc: hexanes; from 1: 15 to 1: 4) to afford product 15 (0.5 g) as colorless oil (67% for two steps). [Note: Because it was found that starting 14 was obtained as ratio a 3:1 of diastereomers, which were difficult to separate, the diastereomeric mixture was used in the synthesis of 15, resulting in a 3:1 diastereomeric product ration for 15]: 1H NMR (400 MHz, CDCl3) δ 7.38 – 7.29 (m, 5H), 5.12 (s, 2H), 3.90 (d, J = 8.0 Hz, 2H), 3.79 (t, J = 8.0 Hz, 1H), 2.62 – 2.47 (m, 2H), 2.27 – 2.04 (m, 1H), 1.67 – 1.41 (m, 15H), 0.93 (d, J = 8.0 Hz, 3H); ESI-MS calcd for C21H32NO5 (M+H)+: 378.2, found: 378.2.
3.3.2. N-Fmoc (2S,3S)-2-Amino-5-(tert-butoxy)3-methyl-5-oxoentanoic acid (4d)
To a stirred solution of 15 (0.242 g, 0.64 mmol) in acetone (5.0 mL) at 0 °C was added freshly prepared Jones’ reagent (2.7 M, 0.356 mL, 0.96 mmol). The mixture was allowed to warm to room temperature over 30 minutes, and then stirred at room temperature (1 h). Celite and isopropyl alcohol (1.48 mL) were added to the mixture and the resulting precipitate was removed by filtration and the filtrate was concentrated and extracted with EtOAc (3x). The combined extracts were washed with brine (2x), dried (Na2SO4) and concentrated in vacuo to give 16 (0.2 g) as colorless gum (89%).
Crude product 16 (0.16 g, 0.46 mmol) was dissolved in HCl in dioxone (4M, 1.1 mL, 4.55 mmol) and the reaction mixture was stirred at room temperature (3 h), then the solvent was removed by evaporation and the residue was dissolved in THF and H2O (4.5 mL: 4.5 mL, 1:1 v/v) and cooled in an ice bath. Sodium bicarbonate (69 mg, 0.82 mmol) and FmocOSu (0.23 g, 0.68 mmol) were added and the mixture was stirred at room temperature for (10 h). At this time, 1N HCl was added to adjust pH to 2–3, and then the THF was removed by evaporation. The resulting aqueous solution was extracted with EtOAc, and the combined organic extracts was washed with brine, dried (Na2SO4), filtered and the filtrate concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (dichloromethane: MeOH, from 20: 1 to 10: 1) to yield 4d (0.17 g) as a semisolid (81% for two steps). [As noted above, starting 15 was present as a diastereomeric mixture, 4d was also obtained as a diastereomeric mixture]: 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 4.0 Hz, 2H), 7.59 (t, J = 4.0 Hz, 2H), 7.45 – 7.28 (m, 9H), 5.44 (d, J = 8.0 Hz, 1H), 5.15 (s, 2H), 4.62 (dd, J1 = 8.0 Hz, J2 = 4.0 Hz, 1H), 4.42 (d, J = 8.0 Hz, 2H), 4.22 (t, J = 8.0 Hz, 1H), 2.77 – 2.22 (m, 3H), 0.96 (d, J = 8.0 Hz, 3H); ESI-MS calcd for C28H28NO6 (M+H)+: 474.2, found: 474.2.
3.4 Synthesis of N-Fmoc (2S)-2-amino-3-(benzyloxyphosphoryloxy)-3-methylbutanoic acid (4f) from 10
Conversion of the protected D-serine analogue 10 to the Boc-protected amino acid 18 was according to literature procedures.54
3.4.1. Allyl N-Fmoc (2S)-2-amino-3-hydroxyl-3-methylbutanoate (19)
To a solution of 18 (0.503 g, 2.16 mmol) in DMF (10 mL) at room temperature under argon were sequentially added NaHCO3 (0.362 g, 4.31 mmol) and allyl bromide (0.28 mL, 3.23 mmol) and the mixture was stirred until the starting material had been consumed (TLC). The mixture was diluted with EtOAc and the solution was washed with H2O, brine and dried by (MgSO4) and then filtered and the filtrate concentrated. The resulting residue was purified by silica gel column chromatography (EtOAc: hexanes; from 1: 10 to 1: 2) to afford a white foam (0.35 g, 60%). This was dissolved in a 1M solution of HCl in Et2O (12.44 mL, 12.44 mmol) and the reaction mixture stirred at room temperature (3 h). The solvent was then removed by evaporation and the residue was dissolved in a solution of THF: H2O (12 mL: 12 mL, 1:1 v/v) and cooled in an ice bath. Sodium bicarbonate (0.21 g, 2.49 mmol) and FmocOSu (0.63 g, 1.87 mmol) were added and the mixture was stirred at room temperature (10 h). The THF was then removed by evaporation and the aqueous phase was extracted with EtOAc and the combined organic layer was washed with brine and dried (Na2SO4), then filtered and concentrated under reduced pressure. The resulting residue was purified by silica gel column chromatography (EtOAc: hexanes; from 1: 10 to 1: 2) to yield product 19 (0.42 g) as a foam (85% for two steps). [α]D18 −17.0 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 4.0 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.41 (t, J = 8.0 Hz, 2H), 7.32 (t, J = 8.0 Hz, 2H), 6.0 – 5.86 (m, 1H), 5.72 (d, J = 8.0 Hz, 1H), 5.38 (d, J = 16.0 Hz, 1H), 5.29 (d, J = 8.0 Hz, 1H), 4.76 – 4.62 (m, 2H), 4.50 – 4.37 (m, 2H), 4.32 (d, J = 8.0 Hz, 1H), 4.24 (t, J = 8.0 Hz, 1H), 2.55 (s, 1H), 1.31 (s, 3H), 1.29 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 171.6, 156.5, 144.0, 143.9, 141.5, 131.4, 127.9, 127.3, 125.3, 125.2, 120.21, 120.19, 119.7, 72.1, 67.4, 66.4, 61.8, 47.4, 27.2, 26.6; ESI-MS calcd for C23H26NO5 (M+H)+: 396.2, found: 396.1.
3.4.2. N-Fmoc (2S)-2-Amino-3-(dibenzyloxyphosphoryloxy)-3-methylbutanoic acid (20)
A solution of 19 (0.2 g, 0.51 mmol), dibenzyl-diisopropyl-phosphoramidite (0.35 g, 1.01 mmol) and N-methylaniline-trifluoroacetate (0.45 g, 2.02 mmol) in DMF (5 mL) were stirred at room temperature under argon (1 h). Oxidation was performed by addition of t-butyl-hydroperoxide in n-octane (5.0 M, 0.192 mL, 0.96 mmol). The solution was diluted with H2O and extracted with EtOAc and the combined organic layer was washed with H2O and brine, dried (MgSO4), then filtered and the filtrate concentrated. The resulting residue was purified by silica gel column chromatography (EtOAc: hexanes; from 1:4 to 2:1) to provide 20 (0.3 g) as semisolid (90%). [α]D18 −8.4 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 4.0 Hz, 2H), 7.65 – 7.60 (m, 2H), 7.43 – 7.28 (m, 14H), 6.40 (d, J = 8.0 Hz, 1H), 5.92 – 5.80 (m, 1H), 5.31 (d, J = 16.0 Hz, 1H), 5.20 (d, J = 12.0 Hz, 1H), 5.10 – 4.96 (m, 4H), 4.62 (d, J = 4.0 Hz, 2H), 4.43 (d, J = 2.0 Hz, 1H), 4.40 (d, J = 8.0 Hz, 2H), 4.25 (t, J = 8.0 Hz, 1H), 1.65 (s, 3H), 1.60 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.2, 156.5, 144.1, 143.9, 141.5, 136.0, 131.6, 128.8, 128.7, 128.0, 127.9, 127.3, 125.4, 120.2, 119.3, 84.4, 69.5, 67.5, 66.4, 62.5, 47.4, 29.9, 26.5, 25.4; ESI-MS calcd for C37H38NO8P (M+H)+: 656.2, found: 656.2.
3.4.3. N-Fmoc (2S)-2-Amino-3-(benzyloxyphosphoryloxy)-3-methylbutanoic acid (4f)
To a solution of 20 (0.141 g, 0.22 mmol) in THF (1.1 mL) was added N-methylaniline (70 μL, 0.64 mmol) followed by the addition of Pd(PPh3)4 (0.012 g, 10.75 μmol). The mixture was protected from light and stirred under at room temperature under nitrogen (45 minutes). The mixture was then diluted with EtOAc and washed with saturated aqueous NH4Cl. The NH4Cl layer was back-extracted with EtOAc and the combined EtOAc extracts were dried (Na2SO4) filtered and concentrated. The resulting residue was purified by silica gel chromatography eluting first with dichloromethane:MeOH (50:1) to remove aniline impurities. Subsequent elution with dichloromethane:MeOH:AcOH(25:1:0.1) provided the free acid as foam (0.12 g, 91%). A portion of this material (0.06 g, 0.097 mmol) and 2,2′-dipyridyl (7.6 mg, 0.049 mmol) were dissolved in MeOH (1 mL) and 10%Pd/C (10 mg) was added and the flask was flushed with H2 and subsequently stirred under a balloon of H2. After the reaction had achieved completion (TLC), the mixture was filtered and evaporated to dryness. The resulting oily solid was purified by preparative HPLC (flow rate 10 mL/minutes, acetonitrile/H2O with 0.1% TFA, acetonitrile from 30% to 100% in 20 minutes: retention time = 21.9 minutes) and lyophilized to provide 4f as white foam 30 mg (60%). [α]D18 −1.06 (c 0.45, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.85 (brs, 2H), 7.74 (d, J = 8.0 Hz, 2H), 7.59 (t, J = 8.0 Hz, 2H), 7.41 – 7.27 (m, 9H), 6.00 (brs, 1H), 5.05 (d, J = 8.0 Hz, 2H), 4.50 – 4.30 (m, 3H), 4.20 (t, J = 8.0 Hz, 1H), 1.71 (s, 3H), 1.49 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 173.6, 156.6, 144.0, 143.8, 141.5, 135.8, 128.8, 128.6, 128.0, 127.4, 125.4, 124.9, 120.2, 85.2, 69.6, 67.6, 62.8, 62.1, 47.3, 26.1, 25.5; ESI-MS calcd for C27H29NO8P (M+H)+: 526.2, found: 526.2.
3.5. Solid-Phase Peptide Synthesis
Fmoc protected amino acids 4a, 4e, 4g, and other amino acids whose syntheses were not described above, were purchased from Novabiochem. Reagent 4b was synthesized according to literature.50 Reagent 3 was readily prepared as recently reported.51 Peptides were synthesized on NovaSyn®TGR resin (Novabiochem, cat. no. 01-64-0060) using standard Fmoc solid-phase protocols in N-Methyl-2-pyrrolidone (NMP). 1-O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (5.0 eq.), hydroxybenzotriazole (HOBT) (5.0 eq.) and N,N-diisopropylethylaminutese (DIPEA) (10.0 eq.) were used as coupling reagents. Aminuteso terminal acetylation was achieved using 1-acetylimidazole. Finished resins were washed with DMF, MeOH, dichloromethane and Et2O and then dried under vacuum (overnight), washed with DMF, MeOH, dichloromethane and Et2O and then dried under vacuum (overnight). Peptides were cleaved from the resin by treatment with TFA/H2O/triisopropylsilane (95/2.5/2.5) (4 h). The resins were then filtered and the filtrate was concentrated under vacuum, then precipitated with cold ether and the precipitate washed with cold ether. The resulting solid was dissolved in 50% aqueous acetonitrile 5 mL) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (21 mm dia × 250 mm, cat. no: 00G-4436-P0) with a linear gradient from 0% aqueous acetonitrile (0.1% trifluoroacetic acid) to 100% acetonitrile (0.1% trifluoroacetic acid) over 30 minutes at a flow rate of 10.0 mL/minute. Lyophilization gave product peptides as white powders. Peptides 2a – 2c and 2e – 2k were synthesized directly by standard Fmoc-based solid-phase protocols. Because peptide 2d is obtained with the benzyl ester intact arising from amino acid 4d, following HPLC purification the product was dissolved in MeOH and subjected to hydrogenolysis (10% Pd•C, H2) to afford the completely deprotected 2d.
3.6 ELISA-based PBD-binding inhibition assays
Peptide pull-down assays were carried out essentially as described previously.38,42 A biotinylated p-T78 peptide was first diluted with 1X coating solution (KPL Inc., Gaithersburg, MD) to a final concentration of 0.3 μM, and then 100 μL of the resulting solution was immobilized onto a 96-well streptavidin-coated plate (Nalgene Nunc, Rochester, NY). The wells were washed once with PBS plus 0.05% Tween20 (PBST), and incubated with 200 μL of PBS plus 1% BSA (blocking buffer) for 1 h to prevent non-specific binding. Mitotic 293A lysates expressing HA-EGFP-Plk1 were prepared in TBSN {20 mM Trish-Cl (pH 8.0), 150 mM NaCl, 0.5% NP-40, 5 mM EGTA, 1.5 mM EDTA, 20 mM p-nitrophenylphosphate and protease inhibitor cocktail (Roche)} buffer (~60 μg total lysates in 100 μL buffer), mixed with the indicated amount of peptide ligands and applied immediately onto the biotinylated p-T78 peptide-coated ELISA wells, and then incubated with constant rocking for 1 h at 25 °C. Following incubation, the ELISA plates were washed 4 times with PBST. To detect bound HA-EGFP-Plk1, the plates were probed for 2 h with 100 μL/well of anti-HA antibody at a concentration of 0.5 μg/mL in blocking buffer and then washed 5 times. The plates were further probed for 1 h with 100 μL/well of HRP-conjugated secondary antibody (GE Healthcare, Piscataway, NJ) at a 1:1,000 dilution in blocking buffer. The plates were washed 5 times with PBST and incubated with 100 μL/well of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (Sigma, St. Louis, MO) until a desired absorbance was achieved. The reactions were stopped by the addition of 100 μL/well of stop solution (Cell Signaling Technology, Danvers, MA) and the optical densities (O.D.) were measured at 450 nm using an ELISA plate reader (Molecular Devices, Sunnyvale, CA). Results are shown in Figures 3 and 4.
Table 1.
ESI-Mass Spectral Data and HPLC Purity of Synthetic Peptides.
| No | Expected (M + H)+ | Observed (M + H)+ | HPLC Purity |
|---|---|---|---|
| 2a | 863.4 | 863.3 | 100% |
| 2b | 861.4 | 861.3 | 100% |
| 2c | 861.4 | 861.4 | 100% |
| 2d | 825.5 | 825.4 | 90% |
| 2e | 849.4 | 849.3 | 100% |
| 2f | 877.5 | 877.4 | 98% |
| 2g | 863.5 | 863.4 | 96% |
| 2h | 811.5 | 811.4 | 100% |
| 2i | 810.5 | 810.4 | 98% |
| 2j | 797.4 | 797.4 | 99% |
| 2k | 767.5 | 767.4 | 100% |
Acknowledgments
This work was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health. We thank Drs. Christpher C. Lai and James A. Kelley of the CBL for obtaining the high-resolution mass spectra and for assistance in the interpretation of the data. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.del Sol A, Carbonell P. PLoS Comp Biol. 2007;3:2446–2455. doi: 10.1371/journal.pcbi.0030239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keskin O, Gursoy A, Ma B, Nussinov R. Chem Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 3.Clackson T, Wells J. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 4.Ofran Y, Rost B. PLoS Comput Biol. 2007;3:1169–1176. doi: 10.1371/journal.pcbi.0030119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreira IS, Fernandes PA, Ramos MJ. Proteins: Struct, Funct, Bioinf. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 6.Geppert T, Hoy B, Wessler S, Schneider G. Chem Biol. 2011;18:344–353. doi: 10.1016/j.chembiol.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Yin H, Hamilton AD. Angew Chem, Int Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 8.Wells JA, McClendon CL. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 9.Zinzalla G, Thurston DE. Future Med Chem. 2009;1:65–93. doi: 10.4155/fmc.09.12. [DOI] [PubMed] [Google Scholar]
- 10.Meireles LMC, Mustata G. Curr Top Med Chem. 2011;11:248–257. doi: 10.2174/156802611794072632. [DOI] [PubMed] [Google Scholar]
- 11.Garner AL, Janda KD. Curr Top Med Chem. 2011;11:258–280. doi: 10.2174/156802611794072614. [DOI] [PubMed] [Google Scholar]
- 12.Morelli X, Bourgeas R, Roche P. Curr Opin Chem Biol. 2011;15:475–481. doi: 10.1016/j.cbpa.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 13.Rechfeld F, Gruber P, Hofmann J, Kirchmair J. Curr Top Med Chem. 2011;11:1305–1319. doi: 10.2174/156802611795589610. [DOI] [PubMed] [Google Scholar]
- 14.Mullard A. Nat Rev Drug Discov. 2012;11:173–175. doi: 10.1038/nrd3680. [DOI] [PubMed] [Google Scholar]
- 15.Yaffe MB. Nat Rev Mol Cell Biol. 2002;3:177–186. doi: 10.1038/nrm759. [DOI] [PubMed] [Google Scholar]
- 16.Pawson T. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 17.Yaffe MB, Elia AEH. Curr Opin Cell Biol. 2001;13:131–138. doi: 10.1016/s0955-0674(00)00189-7. [DOI] [PubMed] [Google Scholar]
- 18.Yaffe MB, Smerdon SJ. Annu Rev Biophys Biomol Struct. 2004;33:225–244. doi: 10.1146/annurev.biophys.33.110502.133346. [DOI] [PubMed] [Google Scholar]
- 19.Mayer BJ. Methods Mol Biol. 2006;332:79–99. doi: 10.1385/1-59745-048-0:79. [DOI] [PubMed] [Google Scholar]
- 20.Barr FA, Sillje HHW, Nigg EA. Nat Rev Mol Cell Biol. 2004;5:429–441. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 21.Dai W. Oncogene. 2005;24:214–216. doi: 10.1038/sj.onc.1208270. [DOI] [PubMed] [Google Scholar]
- 22.Lowery DM, Lim D, Yaffe MB. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 23.van de Weerdt BCM, Medema RH. Cell Cycle. 2006;5:853– 864. doi: 10.4161/cc.5.8.2692. [DOI] [PubMed] [Google Scholar]
- 24.Archambault V, Glover DM. Nat Rev Mol Cell Biol. 2009;10:265–275. doi: 10.1038/nrm2653. [DOI] [PubMed] [Google Scholar]
- 25.Strebhardt K, Ullrich A. Nat Rev Cancer. 2006;6:321–330. doi: 10.1038/nrc1841. [DOI] [PubMed] [Google Scholar]
- 26.Eckerdt F, Yuan J, Strebhardt K. Oncogene. 2005;24:267–276. doi: 10.1038/sj.onc.1208273. [DOI] [PubMed] [Google Scholar]
- 27.Takai N, Hamanaka R, Yoshimatsu J, Miyakawa I. Oncogene. 2005;24:287–291. doi: 10.1038/sj.onc.1208272. [DOI] [PubMed] [Google Scholar]
- 28.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 29.Goh KC, Wang H, Yu N, Zhou Y, Zheng Y, Lim Z, Sangthongpitag K, Fang L, Du M, Wang X, Jefferson AB, Rose J, Shamoon B, Reinhard C, Carte B, Entzeroth M, Ni B, Taylor ML, Stuenkel W. Drug Dev Res. 2004;62:349–361. [Google Scholar]
- 30.McInnes C, Mezna M, Fischer PM. Curr Top Med Chem. 2005;5:181–197. doi: 10.2174/1568026053507660. [DOI] [PubMed] [Google Scholar]
- 31.Gumireddy K, Reddy MVR, Cosenza SC, Nathan RB, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. Cancer Cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 32.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA, Cheung M. Mol Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 33.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. Curr Biol. 2007;17:304–315. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 34.Lu LY, Yu X. Cell Div. 2009;4:12. doi: 10.1186/1747-1028-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reindl W, Yuan J, Kraemer A, Strebhardt K, Berg T. ChemBioChem. 2009;10:1145–1148. doi: 10.1002/cbic.200900059. [DOI] [PubMed] [Google Scholar]
- 36.Strebhardt K. Nat Rev Drug Discov. 2010;9:643–659. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 37.Reindl W, Yuan J, Kraemer A, Strebhardt K, Berg T. Chem Biol. 2008;15:459–466. doi: 10.1016/j.chembiol.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 38.Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wipf P, Arnold D, Carter K, Dong S, Johnston PA, Sharlow E, Lazo JS, Huryn DH. Curr Top Med Chem. 2009;9:1194–1205. doi: 10.2174/156802609789753590. [DOI] [PubMed] [Google Scholar]
- 40.Li L, Wang X, Chen J, Ding H, Zhang Y, Hu T-c, Hu L-h, Jiang H-l, Shen X. Acta Pharmacol Sin. 2009;30:1443–1453. doi: 10.1038/aps.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe N, Sekine T, Takagi M, Iwasaki J-i, Imamoto N, Kawasaki H, Osada H. J Biol Chem. 2009;284:2344–2353. doi: 10.1074/jbc.M805308200. [DOI] [PubMed] [Google Scholar]
- 42.Liu F, Park JE, Qian WJ, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR., Jr Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu F, Park JE, Qian WJ, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. ACS Chem Biol. 2012 doi: 10.1021/cb200469a. Ahead of Print (doi.org/10.1021/cb200469a) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burke TR, Jr, Lee K. Acc Chem Res. 2003;36:426–433. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- 45.Costantino L, Barlocco D. Cur Med Chem. 2004;11:2725–2747. doi: 10.2174/0929867043364315. [DOI] [PubMed] [Google Scholar]
- 46.Berkowitz DB, Bose M. J Fluorine Chem. 2001;112:13–33. [Google Scholar]
- 47.Otaka A, Mitsuyama E, Watanabe J, Watanabe H, Fujii N. Biopolymers. 2004;76:140–149. doi: 10.1002/bip.10570. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez MC, Diaz A, Guillin JJ, Blanco O, Ruiz M, Ojea V. J Org Chem. 2006;71:6958–6974. doi: 10.1021/jo061072x. [DOI] [PubMed] [Google Scholar]
- 49.Panigrahi K, Eggen M, Maeng JH, Shen Q, Berkowitz DB. Chemistry Biology. 2009;16:928–936. doi: 10.1016/j.chembiol.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu F, Park JE, Lee KS, Burke TR. Tetrahedron. 2009;65:9673–9679. doi: 10.1016/j.tet.2009.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian W, Liu F, Burke TR. J Org Chem. 2011;76:8885–8890. doi: 10.1021/jo201599c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nishiguchi A, Maeda K, Miki S. Synthesis. 2006:4131–4134. [Google Scholar]
- 53.Konno H, Takebayashi Y, Nosaka K, Akaji K. Heterocycles. 2010;81:79–89. [Google Scholar]
- 54.Yonezawa Y, Shimizu K, Yoon K-s, Shin C-g. Synthesis. 2000:634–636. [Google Scholar]
- 55.Vorherr T, Bannwarth W. Bioorg Med Chem Lett. 1995;5:2661–2664. [Google Scholar]
- 56.Elia AEH, Cantley LC, Yaffe MB. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 57.Johmura Y, Soung NK, Park JE, Yu LR, Zhou M, Bang JK, Kim BY, Veenstra TD, Erikson RL, Lee KS. Proc Natl Acad Sci USA. 2011;108:11446–11451. doi: 10.1073/pnas.1106223108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bradshaw JM, Waksman G. Adv Protein Chem. 2003;61:161–210. doi: 10.1016/s0065-3233(02)61005-8. [DOI] [PubMed] [Google Scholar]
- 59.Kotoris CC, Chen MJ, Taylor SD. Bioorg Med Chem Lett. 1998;8:3275–3280. doi: 10.1016/s0960-894x(98)00598-8. [DOI] [PubMed] [Google Scholar]
- 60.Beaulieu PL, Cameron DR, Ferland J-M, Gauthier J, Ghiro E, Gillard J, Gorys V, Poirier M, Rancourt J, Wernic D, Llinas-Brunet M, Betageri R, Cardozo M, Hickey ER, Ingraham R, Jakes S, Kabcenell A, Kirrane T, Lukas S, Patel U, Proudfoot J, Sharma R, Tong L, Moss N. J Med Chem. 1999;42:1757–1766. doi: 10.1021/jm980676t. [DOI] [PubMed] [Google Scholar]
- 61.Koitabashi N, Aiba T, Hesketh GG, Rowell J, Zhang M, Takimoto E, Tomaselli GF, Kass DA. J Mol Cell Cardiol. 2010;48:713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee S, Shuman JD, Guszczynski T, Sakchaisri K, Sebastian T, Copeland TD, Miller M, Cohen MS, Taunton J, Smart RC, Xiao Z, Yu LR, Veenstra TD, Johnson PF. Mol Cell Biol. 2010;30:2621–2635. doi: 10.1128/MCB.00782-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang GL, Shi X, Haefliger S, Jin J, Major A, Iakova P, Finegold M, Timchenko NA. J Clin Invest. 2010;120:2549–2562. doi: 10.1172/JCI41933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farghaian H, Chen Y, Fu AWY, Fu AKY, Ip JPK, Ip NY, Turnley AM, Cole AR. J Biol Chem. 2011;286:19724–19734. doi: 10.1074/jbc.M110.205781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, Bayer KU. J Biol Chem. 2011;286:31272–31281. doi: 10.1074/jbc.M111.233668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maheswaranathan M, Gole HKA, Fernandez I, Lassegue B, Griendling KK, San MA. J Biol Chem. 2011;286:35430–35437. doi: 10.1074/jbc.M111.268284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mielgo A, Seguin L, Huang M, Camargo MF, Anand S, Franovic A, Weis SM, Advani SJ, Murphy EA, Cheresh DA. Nat Med. 2011;17:1641–1645. doi: 10.1038/nm.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moss NM, Wu YI, Liu Y, Munshi HG, Stack MS. J Biol Chem. 2009;284:19791–19799. doi: 10.1074/jbc.M109.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Romanova LY, Holmes G, Bahte SK, Kovalchuk AL, Nelson PJ, Ward Y, Gueler F, Mushinski JF. J Cell Sci. 2010;123:1567–1577. doi: 10.1242/jcs.060996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ng YW, Raghunathan D, Chan PM, Baskaran Y, Smith DJ, Lee CH, Verma C, Manser E. Structure. 2010;18:879–890. doi: 10.1016/j.str.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 71.Weidenfeld-Baranboim K, Koren L, Aronheim A. Biochem J. 2011;436:661–669. doi: 10.1042/BJ20101031. [DOI] [PubMed] [Google Scholar]