

Abstract
Objective:
To evaluate the prevalence of aquaporin-4 (AQP4) antibody in Thai patients with idiopathic inflammatory demyelinating CNS diseases (IIDCDs) and to analyze the significance of the autoantibody to distinguish neuromyelitis optica (NMO) and other NMO spectrum disorders (ONMOSDs) from other IIDCDs, especially multiple sclerosis (MS).
Methods:
We retrospectively evaluated 135 consecutive patients with IIDCDs seen at the MS clinic at Siriraj Hospital, Bangkok, Thailand, and classified them into NMO, ONMOSDs, optic-spinal MS (OSMS), classic MS (CMS), and clinically isolated syndrome (CIS) groups in this order with accepted diagnostic criteria. The patients' coded sera were tested separately for AQP4 antibody. Then the relations between the clinical diagnosis and the AQP4 antibody serologic status were analyzed.
Results:
Among the 135 patients, 53 (39.3%) were AQP4 antibody–positive. Although the AQP4 antibody–positive group had features of NMO, such as female predominance, long cord lesions (>3 vertebral bodies), and CSF pleocytosis, only 18 patients (33% of 54) fully met Wingerchuk 2006 criteria except for AQP4 antibody–seropositive status. We also detected some AQP4 antibody–positive patients in the OSMS (4 of 7), CMS (11 of 46), and CIS (1 of 16) groups. These patients had been misdiagnosed with MS because they often had brain lesions and never underwent spinal cord MRI examination or lacked long cord lesions.
Conclusions:
AQP4 antibody was highly prevalent (almost 40%) in Thai patients with IIDCDs. Moreover, only one-third of AQP4 antibody–positive patients fully met Wingerchuk 2006 criteria, and many were misdiagnosed with MS. A sensitive AQP4 antibody assay is required in this region because the therapy for NMO is different from that for MS.
Neuromyelitis optica (NMO) is an idiopathic inflammatory demyelinating CNS disease (IIDCD) that typically develops into severe optic neuritis and longitudinally extensive transverse myelitis.1–3 The relation between NMO and multiple sclerosis (MS) has been controversial, but since the discovery of aquaporin-4 (AQP4), an NMO-specific autoantibody,4,5 clinical and laboratory features that distinguish NMO from MS have become clear.4–6 In addition, treatment strategies for the 2 diseases are different.7,8 AQP4 antibody studies showed that besides definite NMO defined by Wingerchuk 2006 criteria,9 there are other AQP4 antibody–positive patients with NMO spectrum disorders (NMOSDs).10–13
The ratios of NMO to MS are much higher in Asian countries than in Western countries.14,15 Japanese reports showed that approximately 20% of patients with IIDCDs have NMO.15 The ratio of NMO to MS in southeastern Asia appears even higher,16 and one might misdiagnose some AQP4 antibody–positive patients with MS and treat them as such. Thus, we expect that detailed analyses with AQP4 antibody tests in a large number of patients with IIDCDs would have therapeutic implications, but there have been no such studies in any southeast Asian country.
The objectives of this study were to analyze the relation between clinical diagnosis and AQP4 antibody serologic status in Thai patients with IIDCDs by applying accepted diagnostic criteria and a sensitive AQP4 antibody assay.
METHODS
Patients and study design.
A total of 141 consecutive Thai patients with suspected IIDCD visiting the MS clinic at Siriraj Hospital, Mahidol University, Bangkok, Thailand, during the period from May 1, 2009, to February 28, 2010, participated in the study. We made a clinical diagnosis in each patient with the use of the diagnostic criteria and process described below.
Separately from the clinical diagnoses, 2 of us (S.S. and N.P.) collected serum samples of the patients, coded them, and sent them to the laboratory at the Department of Neurology, Tohoku University Graduate School of Medicine, Sendai, Japan, for AQP4 antibody testing. Hence one of us (T.T.) did the AQP4 antibody assay without knowledge of the clinical diagnoses. Then, after we determined the AQP4 antibody serologic status of each patient, we analyzed the relation between the serologic status and the clinical diagnosis.
Standard protocol approvals, registration, and patient consents.
The study received approval from an institutional review board/ethics committee. All participants gave written informed consent.
Diagnostic criteria and process of clinical diagnosis.
Two neurologists (S.S. and N.P.) reviewed the medical records of the 141 patients with suspected IIDCDs and made a clinical diagnosis in each patient with diagnostic criteria. The neurologists were blinded to each other's decision and reached consensus if a discrepancy occurred. The diagnostic criteria and the process of diagnosis were as follows.
NMO: first, we studied whether the patients met Wingerchuk 2006 criteria,9 with the exception of AQP4 antibody status. We diagnosed those who fulfilled all of the following 4 criteria with NMO: optic neuritis, acute myelitis, contiguous spinal cord MRI lesion extending over >3 vertebral bodies (VBs), and onset brain MRI not meeting Paty criteria for MS.17 We further evaluated patients who were “not NMO” in this step in step 2.
Other NMO spectrum disorders (ONMOSDs): we diagnosed patients who fulfilled any one of the following 3 criteria with ONMOSD: recurrent optic neuritis without brain lesions, acute myelitis with long spinal cord lesion (>3 VBs) (with or without brain lesions), or optic neuritis or myelitis without long spinal cord lesion (>3 VBs) with brain MRI findings compatible with those seen in NMO, such as symmetric diffuse white matter lesions, symmetric diencephalic lesions, and periaqueductal lesions.1,12,18–20 We further evaluated patients who were “not ONMOSD” in this step in step 3.
Optic-spinal MS (OSMS): we diagnosed patients who had optic neuritis and acute myelitis with spinal cord MRI lesion not extending over >3 VBs and with normal brain MRI with OSMS. We further evaluated patients who were “not OSMS” in this step in step 4.
Classic MS (CMS): we diagnosed patients who fulfilled McDonald 2005 criteria21 with CMS. We further evaluated patients who were “not CMS” in this step in step 5.
Clinically isolated syndrome (CIS): we diagnosed patients with the first neurologic event caused by inflammatory demyelination who did not meet the brain MRI findings of McDonald 2005 criteria for dissemination in time with CIS.22 We diagnosed patients who were “not CIS” in this step as not having an IIDCD.
AQP4 antibody testing.
We incubated human AQP4-transfected human embryonic kidney 293 cells with diluted serum and washed and incubated them with fluorescein-conjugated goat antihuman immunoglobulin G (IgG). We photographed fixed cells using confocal microscopy and scored antibody seropositivity through comparison with mock-transfected cells that did not express AQP4. We performed assays as described previously.23 We coded all samples at the time of the assays.
Statistical analysis.
We used SPSS version 14.0 (SPSS, Chicago, IL) to perform the statistical analysis. We applied Student t test for quantitative data and χ2 and Fisher exact tests for qualitative data. We considered p < 0.05 as statistically significant.
RESULTS
AQP4 antibody seropositivity in total patients enrolled and in each patient group.
Of the 141 Thai patients, 5 were later proved in the clinic to have other diseases (one each with neuro-Behçet disease, stroke, pachymeningitis, myasthenia gravis, and antiphospholipid syndrome), and one patient presenting with unilateral optic neuritis was lost to follow-up. These patients were excluded from the analysis (figure 1). Among the remaining 135 patients with IIDCDs, 53 (39.3%) were seropositive for AQP4 antibody and the remaining 82 were seronegative. AQP4 antibody positivity was seen in OSMS, CMS, and CIS as well as in NMO and ONMOSD (figure 1).
Figure 1. Clinical diagnostic process and aquaporin-4 (AQP4) antibody seropositivity in the present study.

We examined whether individual patients fulfilled the diagnostic criteria for neuromyelitis optica (NMO), other neuromyelitis optica spectrum disorders (ONMOSD), opticospinal multiple sclerosis (OSMS), classic multiple sclerosis (CMS), and clinically isolated syndrome (CIS) described in the Diagnostic criteria and process of clinical diagnosis section in this order. When a patient did not fulfill the diagnostic criteria for one disease, that disease was excluded, and the next diagnostic criteria were applied until certain criteria were met.
Of the 19 AQP4 antibody–positive patients with ONMOSDs, we diagnosed 2 with Sjögren syndrome.24 Both patients showed spinal cord MRI lesions extending over 7 VBs before developing sialoadenitis with parotid gland enlargement. Tests for SSA and SSB antibodies were positive, and the parotid gland biopsy revealed histopathology consistent with Sjögren syndrome.
Comparison of clinical, MRI, and laboratory findings in AQP4 antibody–positive patients and AQP4 antibody– negative patients (excluding those with inadequate data and other diseases).
AQP4 antibody–positive patients included a higher percentage of women, had higher Expanded Disability Status Scale scores,25 and had more acute attacks than AQP4 antibody–negative patients. The first clinical manifestations were similar in the 2 groups (table 1).
Table 1.
Comparison of AQP4 antibody–positive and AQP4 antibody–negative Thai patients with idiopathic inflammatory demyelinating CNS diseasesa
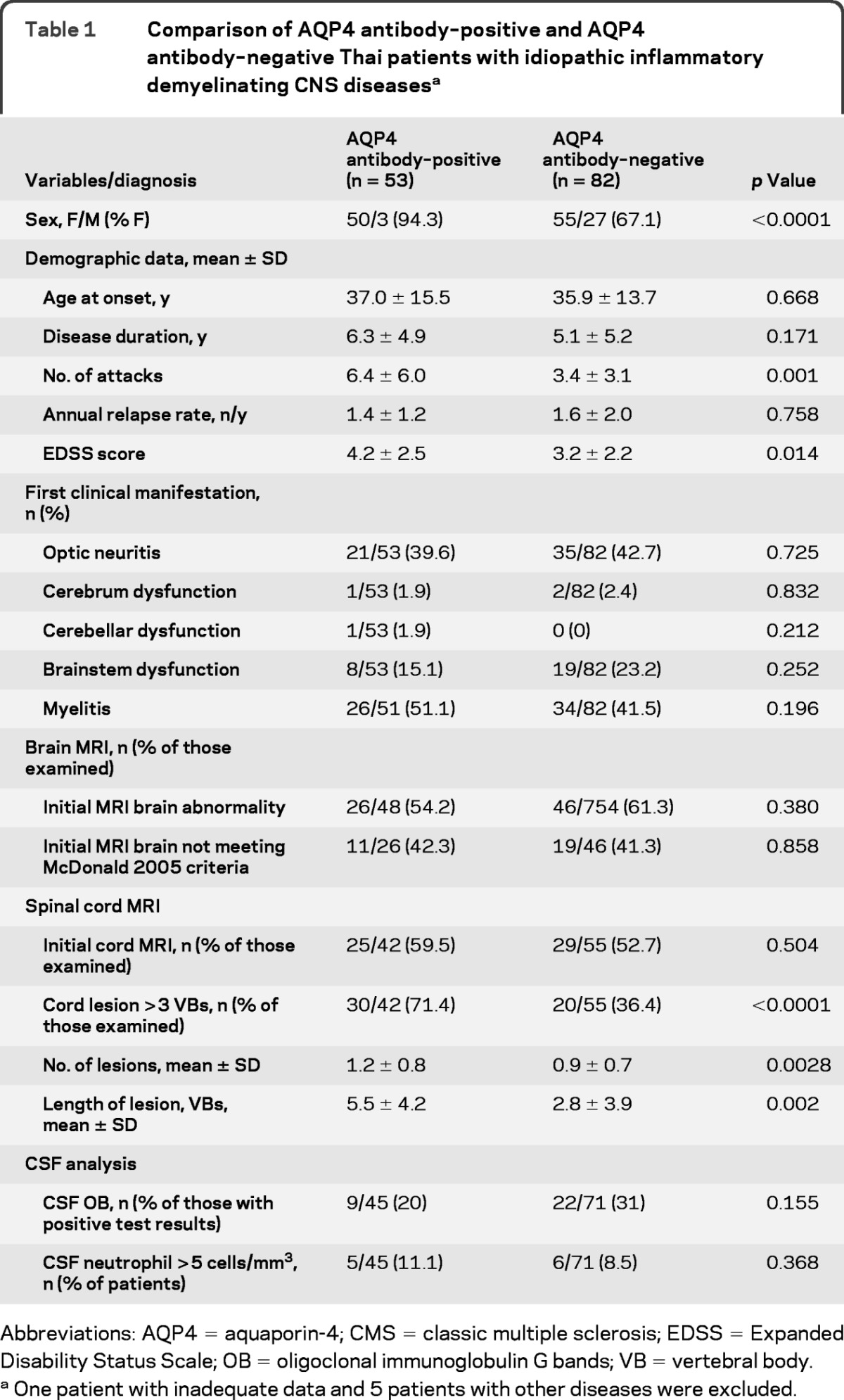
Abbreviations: AQP4 = aquaporin-4; CMS = classic multiple sclerosis; EDSS = Expanded Disability Status Scale; OB = oligoclonal immunoglobulin G bands; VB = vertebral body.
One patient with inadequate data and 5 patients with other diseases were excluded.
There was no difference in the percentages of patients who fulfilled the brain MRI findings for McDonald 2005 criteria in the 2 groups. Spinal cord MRI demonstrated a higher rate of long spinal cord lesions (>3 VBs) in the AQP4 antibody–positive group. Cord lesions in the AQP4 antibody–positive group were longer than those in the AQP4 antibody–negative group. AQP4 antibody–positive patients had slightly more cord lesions than seronegative patients.
CSF analysis showed higher numbers of white blood cells in the CSF in the seropositive group than in the seronegative group (53.7 ± 205.8 vs 19.8 ± 57.1 cells/μL). The percentage of patients with positive test results for CSF oligoclonal IgG bands (OBs) was similar in the 2 groups. Serologic autoimmune screening tests including antinuclear antibody (ANA), anti–double-stranded DNA, anti-thyroglobulin, and perinuclear antineutrophil cytoplasmic antibody did not show any significant differences in the 2 groups.
Clinical features of AQP4 antibody–negative CMS.
We compared 35 patients with AQP4 antibody–negative CMS with 18 patients with AQP4 antibody–positive NMO. The group with AQP4 antibody–negative CMS had less female preponderance (71.4% vs 100%) and fewer (0.7 ± 0.7 vs 1.5 ± 0.8) and shorter (1.1 ± 1.0 vs 6.4 ± 3.3 VBs) cord lesions than the group with AQP4 antibody–positive NMO. Of the 35 patients with AQP4 antibody–negative CMS, CSF samples were available for 29 patients and 17 (58.6%) were OB-positive by the isoelectric focusing method; this frequency of OB was much higher than the 20% in patients with AQP4 antibody–positive NMO.
Clinical characteristics of patients who were diagnosed with CMS by the diagnostic criteria but were AQP4 antibody–positive.
We judged 11 (20.4%) of the 54 AQP4 antibody–positive patients as not NMO because they did not fully meet Wingerchuk 2006 criteria. We then diagnosed them with CMS in the present diagnostic process. All but one were women, and the onset age ranged from 16 to 43 years (table 2, figure 2).
Table 2.
Clinical and laboratory data of patients who were diagnosed with CMS but were AQP4 antibody–positive
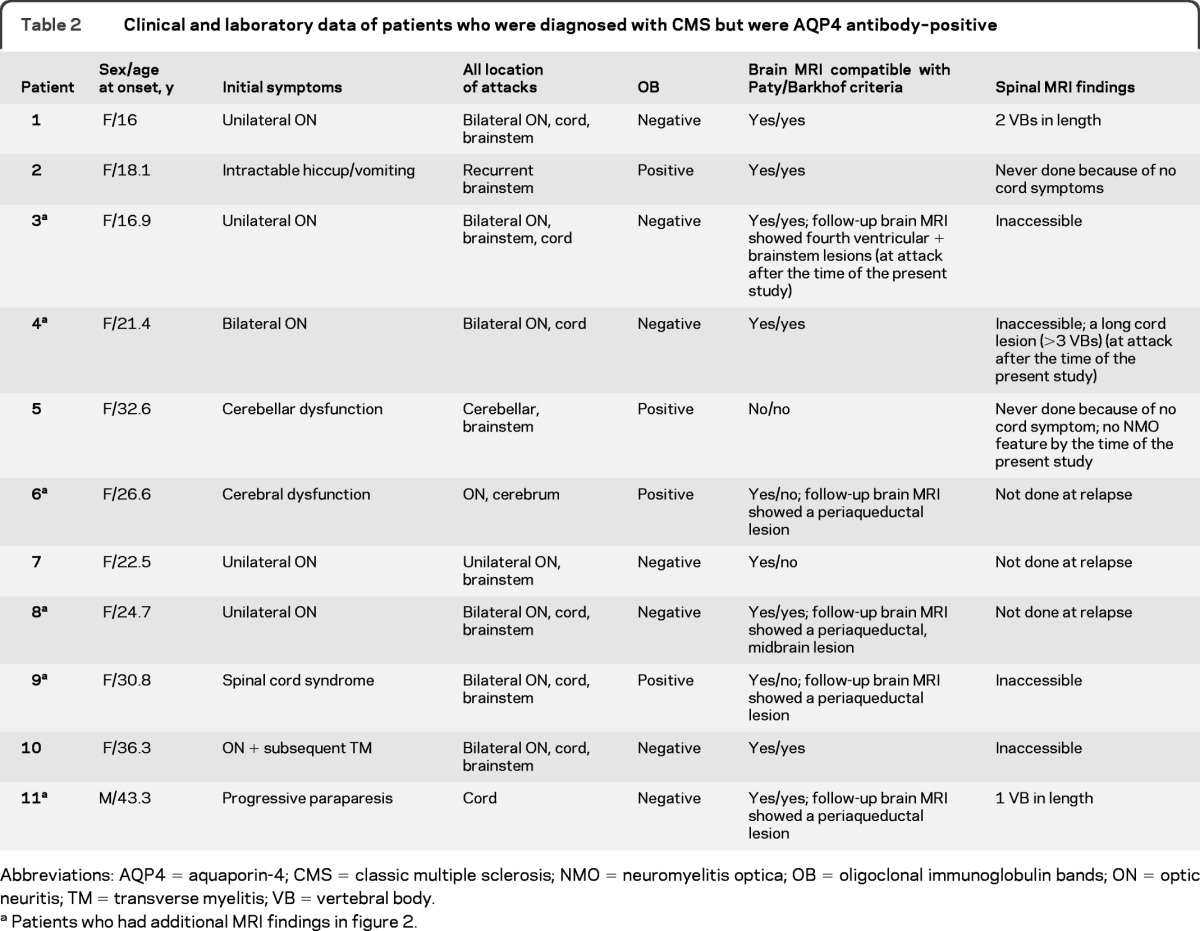
Abbreviations: AQP4 = aquaporin-4; CMS = classic multiple sclerosis; NMO = neuromyelitis optica; OB = oligoclonal immunoglobulin bands; ON = optic neuritis; TM = transverse myelitis; VB = vertebral body.
Patients who had additional MRI findings in figure 2.
Figure 2. MRI findings.

VB = vertebral body.
Of these patients, patient 1 had initial symptoms of unilateral optic neuritis with brain MRI abnormalities compatible with IIDCD and a medium-length spinal cord MRI lesion (2 VBs). She experienced a subsequent attack of bilateral optic neuritis with poor visual recovery (<20/200). Patient 2 experienced intractable hiccup/vomiting lasting for 3 weeks. She had brain MRI lesions compatible with McDonald 2005 criteria but had never undergone spinal cord MRI. Patient 3 had bilateral optic neuritis, and brainstem and spinal cord symptoms. Her spinal cord MRI was inaccessible. Follow-up brain MRI out of the time of the study showed a lesion near the fourth ventricle. Patient 4 had an attack of bilateral optic neuritis followed by a myelitis attack, and brain MRI findings compatible with IIDCD. However, her spinal cord MRI was inaccessible. Patient 5 had recurrent neurologic attacks and was OB-positive. She had never had spinal cord MRI. Patient 6 had a recurrent neurologic deficit and was OB-positive. She had spinal cord MRI more than 30 days after the onset. Follow-up brain MRI showed a periaqueductal lesion. Patient 7 had optic neuritis with poor visual recovery (<20/200) and brainstem dysfunction and was OB-negative. Her spinal cord MRI was performed later than 30 days after the onset. Patient 8 had recurrent attacks including one attack with paraplegia, sensory impairment, and bowel and bladder dysfunction, consistent with transverse myelitis, and was OB-negative. Her spinal cord MRI was performed later than 30 days after the onset. Subsequent brain MRI showed a midbrain lesion. Patient 9 had spinal cord syndrome, optic neuritis, and brainstem dysfunction. Brain MRI was suggestive of IIDCD. Her spinal cord MRI was inaccessible. Subsequent brain MRI showed periaqueductal involvement. Patient 10 had initial symptoms of optic neuritis with subsequent spinal cord dysfunction. Brain MRI was suggestive of IIDCD. She had never had spinal cord MRI. Patient 11 was diagnosed with probable primary progressive MS with a history of gradually progressive paraparesis over the 1-year period and brain MRI suggestive of IIDCD and was OB-negative. His spinal cord MRI showed only one short lesion (1 VB).
Taken together, these results show that all patients except patient 5 had certain clinical or MRI features that are relatively common in or unique to NMO, such as bilateral optic neuritis, recurrent optic neuritis with poor recovery, intractable hiccup/vomiting, periaqueductal lesions, or transverse myelitis. However, except for these features of NMO, there were no differences between patients with AQP4 antibody–negative and antibody– positive CMS (table 3).
Table 3.
Comparison of patients with AQP4 antibody–positive and AQP4 antibody–negative CMS
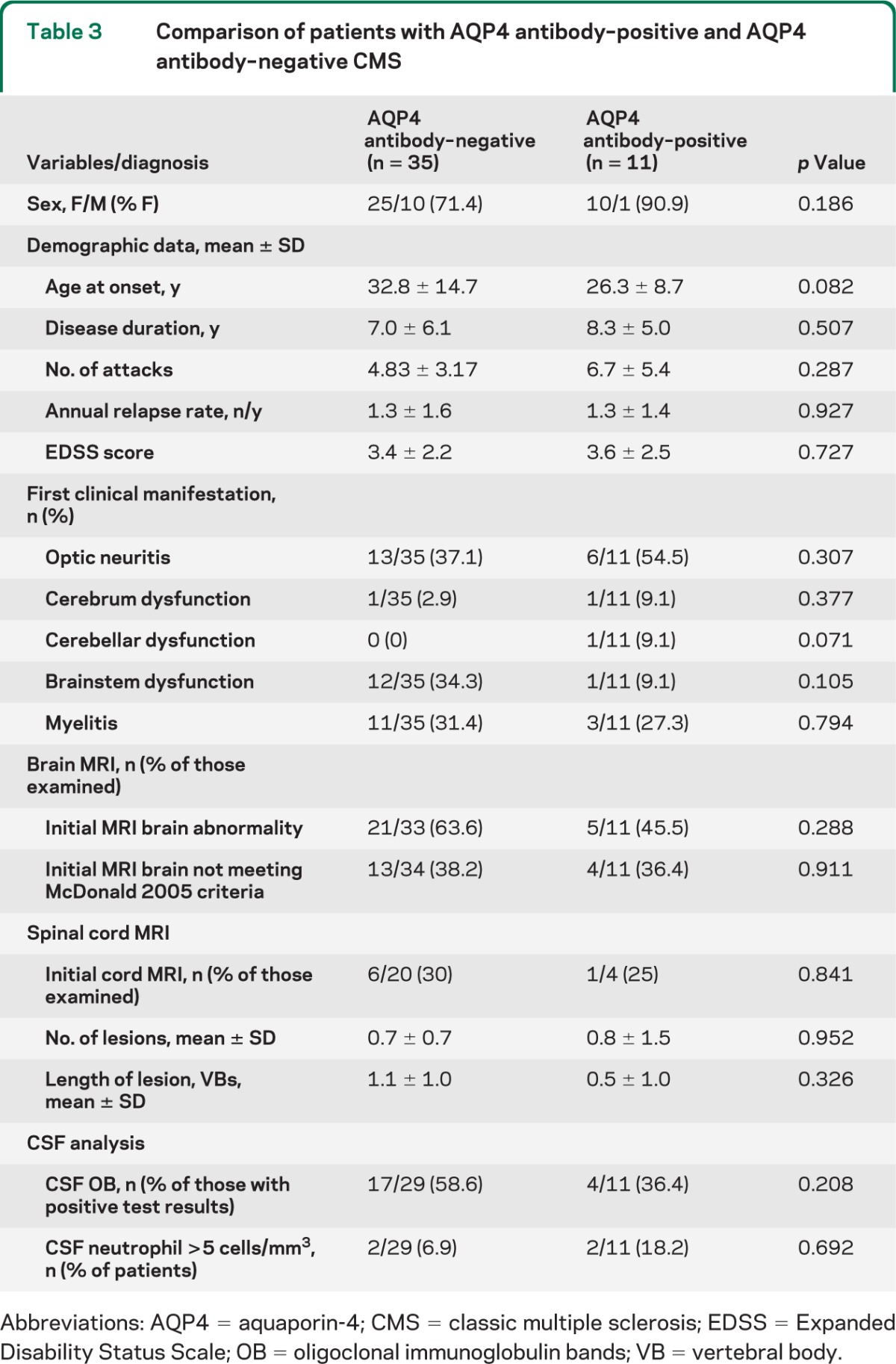
Abbreviations: AQP4 = aquaporin-4; CMS = classic multiple sclerosis; EDSS = Expanded Disability Status Scale; OB = oligoclonal immunoglobulin bands; VB = vertebral body.
Clinical characteristics of patients who were diagnosed with NMO but were AQP4 antibody–negative (AQP4 antibody–negative NMO).
Five patients met all 4 Wingerchuk 2006 criteria and were diagnosed with NMO, but they were AQP4 antibody–negative. One of them presented with unilateral optic neuritis followed by subsequent attacks of optic neuritis, myelitis, or brainstem lesions. Brain MRI at onset was normal, but spinal cord MRI showed entire cord involvement. The patient was OB-positive and positive for ANA (titer 1:640). Two other patients had recurrent attacks of optic neuritis and transverse myelitis (>3 VBs) with normal brain MRI results at onset. Both were OB-negative. The remaining 2 patients had optic neuritis, brainstem lesions, and myelitis. Brain MRI showed a few white matter spotty lesions and did not meet Paty criteria, but spinal cord MRI revealed long cord lesions (>3 VBs). Both were OB-negative.
DISCUSSION
The AQP4 antibody seroprevalence in our Thai patients with IIDCDs was 39.3%. This figure is remarkably higher than those in Western countries where cases of NMO constitute less than 10% of total IIDCDs and is still higher than available Asian data, such as those in Japanese cohorts15 and the 33.1% in 236 consecutive Korean patients with IIDCD seen at a major referral center.26 The very high prevalence of the NMO-specific autoantibody in the present study is probably due to the low prevalence of MS in this region.16 In addition, because MS is a much milder disease, access issues might disproportionately inflate the proportion of cases of NMO in this non-population-based study. However, the high proportion of AQP4 antibody–positive patients in this Thai study has important clinical implications.
AQP4 is a dominant water channel in the CNS and is abundantly expressed on astrocytic endfeet.27 Neuropathologic and CSF analyses in NMO demonstrated massive destruction of astrocytes in acute NMO lesions, but there were no such changes in MS.28,29 More recent experimental studies clearly showed that AQP4 antibody derived from patients with NMO, predominantly of the IgG1 subclass, activated the complement pathway efficiently and caused astrocytic necrosis in cell culture and NMO-like lesions in animals with experimental autoimmune encephalomyelitis.30 Purified IgG from AQP4 antibody–negative patients did not induce those morphologic changes. These findings clearly indicate that AQP4 antibody is pathogenic and NMO defined by AQP4 antibody is a clinical entity distinct from MS.
The clinical and laboratory features of our AQP4 antibody–positive patients were similar to those reported previously: high female/male ratio (16:1), longitudinally extensive transverse myelitis with high Expanded Disability Status Scale scores and frequent relapse (about 1.0/year on average), and CSF pleocytosis.31 Of the patients who fulfilled Wingerchuk 2006 criteria, nearly 80% were AQP4 antibody–positive. Meanwhile, in contrast with previous studies, age at onset and frequency of brain MRI abnormalities in those meeting McDonald Criteria and CSF OB were not different between AQP4 antibody–positive and AQP4 antibody–negative Thai patients with IIDCD. For 11 patients who we diagnosed with CMS but who were AQP4 antibody–positive, in retrospect, all but one had clues that they might have had NMO. In most of these, spinal cord MRI was not done or was inaccessible. Thus, we should incorporate brain and spinal cord MRI in the initial diagnostic workup to differentiate NMO from CMS. Moreover, regardless of clinical, MRI, or laboratory findings, we should maintain a high index of suspicion for NMO, especially in Asian countries, where NMO is a common cause of IIDCDs.
The AQP4 antibody is highly sensitive and specific for detecting NMO, but extensive analyses of AQP4 antibody–positive patients revealed that in addition to those with typical NMO, there are AQP4 antibody–positive patients with recurrent optic neuritis or longitudinally extensive myelitis alone or certain brain lesions.10–13,32,33 We classified those patients as ONMOSD in the present study. Our definition of ONMOSD in the present study, however, is different from that of NMOSD originally proposed by investigators at the Mayo Clinic in that both positive serology and clinical features are used to define this entity rather than clinical features only. It is likely that other investigators would have suspected that many of the patients diagnosed with CMS and CIS in the present study had ONMOSD and established this diagnosis by testing for AQP4 antibody. Some of our patients with ONMOSD may have pre-NMO, which first presented with optic neuritis or longitudinally extensive myelitis. Although none of them have fulfilled Wingerchuk 2006 criteria for definite NMO to date, we need longitudinal data for each patient to clarify the evolution of NMO.
In Asian countries, we have commonly termed cases of recurrent optic neuritis and myelitis OSMS.14,34 However, so-called OSMS comprises NMO and MS with opticospinal presentation.35 Because therapy for NMO is different from that for MS,36–38 the differential diagnosis is important. In the present study, we defined OSMS as MS with selective opticospinal involvement (optic neuritis and acute myelitis with a spinal cord MRI lesion not extending over >3 VBs and with normal brain MRI) to distinguish it from NMO. With the application of the criteria for OSMS and other diseases in the sequence described in Methods, in the present study, in addition to NMO and ONMOSD, we detected AQP4 antibody–positive patients in all other groups (57% in OSMS, 24% in CMS, and 6% in CIS). At the time of this study, those patients did not meet the criteria for NMO or ONMOSD in the absence of AQP4 antibody serologic status mainly because they had brain lesions, some of which met the MRI findings in the McDonald 2005 criteria,21,39 and they never underwent MRI, had a delayed spinal cord MRI examination, or lacked long cord lesions. Alternatively, spinal cord lesions may be shorter and AQP4 antibody may be undetectable in patients receiving immunosuppressive drugs.40 Moreover, some AQP4 antibody–positive patients do not have any known features of NMO or ONMOSD at the time of diagnosis such as patient 5 in table 2. We may misdiagnose such patients with MS and probably treat them with MS disease-modifying drugs. Unlike acute exacerbations in MS, those in NMO are often refractory to high-dose corticosteroids and need plasma exchange to hasten recovery.38 Interferon-β is not efficacious in preventing relapse in NMO.7,8 Conversely, we diagnosed 5 patients with AQP4 antibody–negative NMO. One of them had been treated with interferon-β over 10 months. Two other patients were treated with azathioprine, which might have lowered AQP4 antibody levels below the detection limit as mentioned previously.40
In the present study, the male/female ratio for patients with AQP4 antibody–negative CMS was 2.5, which was lower than that in our previous report (6.2), whereas positive OB tests (58.6%) in these patients were more frequent than in patients in the previous study (21%)31 and in patients with AQP4 antibody–positive CMS in the present study. We did our previous study without AQP4 antibody status, and thus we might have misdiagnosed many patients with AQP4 antibody–positive NMO or ONMOSD as CMS. Our AQP4 antibody–negative, OB-positive patients with CMS were very similar to patients with MS in Western countries.
Our study revealed that no single criterion can perfectly distinguish NMO from MS, and we must consider all clinical, CSF, and MRI findings as well as AQP4 antibody status in determining the optimal treatment for a patient, which is quite different for NMO than for MS. However, AQP4 antibodies are highly specific and positive test results should weigh strongly in favor of a diagnosis of NMO.
GLOSSARY
- ANA
antinuclear antibody
- AQP4
aquaporin-4
- CIS
clinically isolated syndrome
- CMS
classic multiple sclerosis
- IIDCD
idiopathic inflammatory demyelinating CNS disease
- IgG
immunoglobulin G
- MS
multiple sclerosis
- NMO
neuromyelitis optica
- NMOSD
neuromyelitis optica spectrum disorder
- OB
oligoclonal immunoglobulin G band
- ONMOSD
other neuromyelitis optica spectrum disorder
- OSMS
optic-spinal multiple sclerosis
- VB
vertebral body.
Footnotes
Editorial, page 812
AUTHOR CONTRIBUTIONS
Dr. Siritho: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data, statistical analysis. Dr. Nakashima: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, study supervision, and obtaining funding. Dr. Takahashi: analysis or interpretation of data, contribution of vital reagents/tools/patients, and acquisition of data. Dr. Fujihara: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, study supervision, and obtaining funding. Dr. Prayoonwiwat: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, contribution of vital reagents/tools/patients, acquisition of data, study supervision, and obtaining funding.
DISCLOSURE
Dr. Siritho has received funding for travel and speaker honoraria from Merck Serono, Bayer Schering Pharma, and Eisai Inc. and has received research support from Merck Serono and sanofi-aventis. Dr. Nakashima has received funding for travel and speaker honoraria from Bayer Schering Pharma and Biogen Idec; serves on the editorial board of Multiple Sclerosis International; and has received research support from Mitsubishi Tanabe Pharma Corporation and Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology. Dr. Takahashi reports no disclosures. Dr. Fujihara serves on scientific advisory boards for Bayer Schering Pharma, Biogen Idec, and Merck Serono; has received funding for travel and speaker honoraria from Bayer Schering Pharma, Biogen Idec, Eisai Inc., Mitsubishi Tanabe Pharma Corporation, Astellas Pharma Inc., Takeda Pharmaceutical Company Limited, and Asahi Kasei Kuraray Medical Co., Ltd.; serves on the editorial board of Clinical and Experimental Neuroimmunology; receives publishing royalties for Clinical Practice Guide of Orthopedic Surgery (Bunkodo, 2007); and has received research support from Bayer Schering Pharma, Biogen Idec, Asahi Kasei Kuraray Medical Co., Ltd, The Chemo-Sero-Therapeutic Research Institute (KAKETSUKEN), Teva Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Teijin Pharma, Eisai Inc., and Kowa Pharmaceuticals America, Inc. and Grants-in-Aid for Scientific Research from the Ministry of Education, Science and Technology and the Ministry of Health, Labor and Welfare of Japan. Dr. Prayoonwiwat has received funding for travel and speaker honoraria from Bayer Schering Pharma, Eisai Inc., Pfizer Inc, Novartis, and sanofi-aventis and has received research support from Bayer Schering, Merck Serono, and sanofi-aventis.
REFERENCES
- 1. O'Riordan JI, Gallagher HL, Thompson AJ, et al. Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry 1996;60:382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114 [DOI] [PubMed] [Google Scholar]
- 3. Cree BA, Goodin DS, Hauser SL. Neuromyelitis optica. Semin Neurol 2002;22:105–122 [DOI] [PubMed] [Google Scholar]
- 4. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112 [DOI] [PubMed] [Google Scholar]
- 5. Weinshenker BG, Wingerchuk DM. Neuromyelitis optica: clinical syndrome and the NMO-IgG autoantibody marker. Curr Top Microbiol Immunol 2008;318:343–356 [DOI] [PubMed] [Google Scholar]
- 6. Weinshenker BG, Wingerchuk DM, Nakashima I, Fujihara K, Lennon VA. OSMS is NMO, but not MS: proven clinically and pathologically. Lancet Neurol 2006;5:110–111 [DOI] [PubMed] [Google Scholar]
- 7. Palace J, Leite MI, Nairne A, Vincent A. Interferon beta treatment in neuromyelitis optica: increase in relapses and aquaporin 4 antibody titers. Arch Neurol 2010;67:1016–1017 [DOI] [PubMed] [Google Scholar]
- 8. Shimizu J, Hatanaka Y, Hasegawa M, et al. IFNβ-1b may severely exacerbate Japanese optic-spinal MS in neuromyelitis optica spectrum. Neurology 2010;75:1423–1427 [DOI] [PubMed] [Google Scholar]
- 9. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489 [DOI] [PubMed] [Google Scholar]
- 10. Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815 [DOI] [PubMed] [Google Scholar]
- 11. Weinshenker BG. Neuromyelitis optica in Western countries: establishing diagnostic criteria and characterization of the spectrum. Neurol Asia 2008;13:161–166 [Google Scholar]
- 12. Misu T, Fujihara K, Nakashima I, Sato S, Itoyama Y. Intractable hiccup and nausea with periaqueductal lesions in neuromyelitis optica. Neurology 2005;65:1479–1482 [DOI] [PubMed] [Google Scholar]
- 13. Magana SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 2009;72:712–717 [DOI] [PubMed] [Google Scholar]
- 14. Kira J. Multiple sclerosis in the Japanese population. Lancet Neurol 2003;2:117–127 [DOI] [PubMed] [Google Scholar]
- 15. Osoegawa M, Kira J, Fukazawa T, et al. Temporal changes and geographical differences in multiple sclerosis phenotypes in Japanese: nationwide survey results over 30 years. Mult Scler 2009;15:159–173 [DOI] [PubMed] [Google Scholar]
- 16. Chong HT. Multiple sclerosis in South East Asia and diagnostic criteria for Asians. Neurol Asia 2008;13:145–146 [Google Scholar]
- 17. Paty DW, Oger JJ, Kastrukoff LF, et al. MRI in the diagnosis of MS: a prospective study with comparison of clinical evaluation, evoked potentials, oligoclonal banding, and CT. Neurology 1988;38:180–185 [DOI] [PubMed] [Google Scholar]
- 18. Filippi M, Rocca MA. MR imaging of Devic's neuromyelitis optica. Neurol Sci 2004;25(suppl 4):S371–S373 [DOI] [PubMed] [Google Scholar]
- 19. Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964–968 [DOI] [PubMed] [Google Scholar]
- 20. Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390–396 [DOI] [PubMed] [Google Scholar]
- 21. Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald criteria.” Ann Neurol 2005;58:840–846 [DOI] [PubMed] [Google Scholar]
- 22. Tintore M, Rovira A, Martinez MJ, et al. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. AJNR Am J Neuroradiol 2000;21:702–706 [PMC free article] [PubMed] [Google Scholar]
- 23. Takahashi T, Fujihara K, Nakashima I, et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235–1243 [DOI] [PubMed] [Google Scholar]
- 24. Vitali C, Bombardieri S, Moutsopoulos HM, et al. Assessment of the European classification criteria for Sjögren's syndrome in a series of clinically defined cases: results of a prospective multicentre study: the European Study Group on Diagnostic Criteria for Sjögren's Syndrome. Ann Rheum Dis 1996;55:116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 1983;33:1444–1452 [DOI] [PubMed] [Google Scholar]
- 26. Kim W, Park MS, Lee SH, et al. Characteristic brain magnetic resonance imaging abnormalities in central nervous system aquaporin-4 autoimmunity. Mult Scler 2010;16:1229–1236 [DOI] [PubMed] [Google Scholar]
- 27. Misu T, Fujihara K, Kakita A, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007;130:1224–1234 [DOI] [PubMed] [Google Scholar]
- 28. Misu T, Takano R, Fujihara K, Takahashi T, Sato S, Itoyama Y. Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: an astrocytic damage marker. J Neurol Neurosurg Psychiatry 2009;80:575–577 [DOI] [PubMed] [Google Scholar]
- 29. Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010;75:208–216 [DOI] [PubMed] [Google Scholar]
- 30. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 2009;66:617–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siritho S, Prayoonwiwat N. A retrospective study of multiple sclerosis in Siriraj Hospital, Bangkok, Thailand. Can J Neurol Sci 2007;34:99–104 [DOI] [PubMed] [Google Scholar]
- 32. de Seze J, Arndt C, Jeanjean L, et al. Relapsing inflammatory optic neuritis: is it neuromyelitis optica? Neurology 2008;70:2075–2076 [DOI] [PubMed] [Google Scholar]
- 33. Yanagawa K, Kawachi I, Toyoshima Y, et al. Pathologic and immunologic profiles of a limited form of neuromyelitis optica with myelitis. Neurology 2009;73:1628–1637 [DOI] [PubMed] [Google Scholar]
- 34. Misu T, Fujihara K, Nakashima I, et al. Pure optic-spinal form of multiple sclerosis in Japan. Brain 2002;125:2460–2468 [DOI] [PubMed] [Google Scholar]
- 35. Nakashima I, Fukazawa T, Ota K, et al. Two subtypes of optic-spinal form of multiple sclerosis in Japan: clinical and laboratory features. J Neurol 2007;254:488–492 [DOI] [PubMed] [Google Scholar]
- 36. Watanabe S, Misu T, Miyozawa I, et al. Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler 2007;13:968–974 [DOI] [PubMed] [Google Scholar]
- 37. Jacob A, Matiello M, Weinshenker BG, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 2009;66:1128–1133 [DOI] [PubMed] [Google Scholar]
- 38. Wingerchuk DM. Diagnosis and treatment of neuromyelitis optica. Neurologist 2007;13:2–11 [DOI] [PubMed] [Google Scholar]
- 39. Barkhof F, Filippi M, Miller DH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997;120:2059–2069 [DOI] [PubMed] [Google Scholar]
- 40. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008;131:3072–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]