

Abstract
The bovine brain A1 adenosine receptor (A1AR) is distinct from other A1ARs in that it displays the unique agonist potency series of N6-R-phenylisopropyladenosine (R-PIA) > N6-S-phenylisopropyladenosine (S-PIA) > 5′-N-ethylcarboxamidoadenosinea nd has a 5–10-fold higher affinity for both agonists and antagonists. The cDNA for this receptor has been cloned from a size-selected (2–4-kb) bovine brain library and sequenced. The 2.0-kb cDNA encodes a protein of 326 amino acid residues with a molecular mass of 36,570 daltons. The amino acid sequence fits well into the seven-transmembrane domain motif typical of G protein-coupled receptors. Northern analysis in bovine tissue using the full length cDNA demonstrates mRNAs of 3.4 and 5.7 kb with a tissue distribution consistent with A1AR binding. Subcloning of the cDNA in a pCMV5 expression vector with subsequent transfection into both COS7 and Chinese hamster ovary cells revealed a fully functional A1AR which could inhibit adenylylcyclase and retained the unique pharmacologic properties of the bovine brain A1AR
The A1AR was found to have a single histidine residue in each of transmembrane domains 6 and 7. Histidine residues have been postulated by biochemical studies to be important for ligand binding. Mutation of His-278 to Leu-278 (seventh transmembrane domain) dramatically decreased both agonist and antagonist binding by >90%. In contrast, mutation of His-251 to Leu-251 decreased antagonist affinity and the number of receptors recognized by an antagonist radioligand. In contrast, agonist affinity was not perturbed but the number of receptors detected by an agonist radioligand was also reduced. These data suggest that both histidines are important for both agonist and antagonist binding, but His-278 appears critical for ligand binding to occur.
A1 adenosine receptors are integral membrane proteins which bind extracellular adenosine and then initiate a transmembrane signal via G proteins1 to modulate the activity of a number of effector systems including adenylylcyclase (inhibition), K+ channels (opening), and Ca2+ channels (close) (1–3). These receptors are found in a diverse selection of cell types and tissues and are known to be dynamically regulated by a variety of pathophysiological conditions including agonist-induced desensitization (4, 5), antagonist-induced sensitization (6), and in altered thyroid states (7). Subpopulations of A1 adenosine receptors have been postulated to exist based on distinct pharmacological profiles and on absolute agonist and antagonist affinities (8–11).
One such subtype is the bovine A1AR which differs from the classical A1AR found in most tissues and species in that it binds antagonists such as XAC with subnanomolar affinities and has a unique agonist potency series of R-PIA > S-PIA > NECA (8–11). This contrasts with the typical A1AR which has 5–10-fold lower affinities for antagonists and agonists and a potency series of R-PIA > NECA > S-PIA (1, 2). We have previously documented that these unique properties of the bovine brain A1AR are displayed by the membranebound, solubilized, and purified receptor suggesting these characteristics are intrinsic to the receptor protein itself and not the environment in which it resides (10, 11). A1ARs of canine thyroid (12) and rat brain (13, 14) have recently been cloned. For both clones, the expressed receptor displays the pharmacological properties described above for the classical A1AR
Biochemical studies have documented that the A1AR is a glycoprotein which contains complex-type carbohydrate chains (10, 15), likely contains disulfide bonds (16, 17), and contains histidine residues in the ligand binding site (17). Klotz et al. (17) have suggested that there are at least 2 histidines in the receptor binding pocket and that agonists and antagonists interact with distinct transmembrane domain recognition sites.
In this paper, we report the cloning, expression, and characterization of the unique bovine brain A1AR, and through site-directed mutagenesis studies begin to probe the important amino acids in the receptor binding pocket
EXPERIMENTAL PROCEDURES
Materials
[γ·32P]ATP, [35S]dATP, [α-32P]ATP, and [3H]XAC were from Du Pont-New England Nuclear. Restriction enzymes and T4 DNA ligase were from either Boehringer-Mannheim or Promega. The pCMV5 expression vector was obtained from Dr. Marc Caron (Duke University) and was originally described by Dr. David Russell (University of Texas). [125I]APNEA was prepared as previously described (18). All cell culture supplies were from GIBCO
Library Screening
A 60-mer antisense oligonucleotide based on a region (amino acids 201–221) of the putative third intracellular loop of the protein encoded by the canine RDC7 clone (19) was synthesized and labeled with [γ·32P]ATP on the 5′-hydroxyl end by T4 polynucleotide kinase. This probe was used to screen approximately 1.1 × 106 recombinants from a size-selected (2–4-kb) bovine brain cDNA library (λZAP) generously provided by Richard Dixon (Merck, Sharp & Dohme Research Laboratories) and Robert Lefkowitz (Duke University). Duplicate nylon filters (Biotrans, ICN) were used for phage lifts and were prehybridized in 6 × TEN (1 × TEN = 15 mM Tris, 1 mM EDTA, 150 mM NaCl, pH 8.3)/6 × Denhardt’s solution, 0.1% SDS, 0.2 mg/ml salmon sperm DNA for 5 h at 45 °C. Hybridization with labeled probe (850,000 cpm/ml) was conducted in 6 × TEN, 6 × Denhardt’s, 0.1% SDS, 0.5 mg/ml sodium pyrophosphate for 16 h at 45 °C. Final wash conditions for primary and final round screening were 2 × TEN, 0.1% SDS at 45 °C for 10 min and 0.5 × TEN, 0.1% SDS at 55 °C for 5 min, respectively. Third round screening allowed for the isolation of a single clone, which was determined by analysis on a 1% agarose gel to contain an insert of ~2.0 kb. The insert was rescued from the phage vector using the R408 helper phage following the manufacturer’s protocol (Stratagene).
DNA Sequencing
The rescued insert contained in the pBluescript vector was used in sequence analysis. Both strands of the cDNA insert were sequenced using [35S]dATP and T7 DNA polymerase (Sequenase 2.0, U. S. Biochemical). The double-stranded DNA was denatured immediately prior to sequencing by the protocol supplied by U. S. Biochemical
Receptor Expression in Mammalian Cells
A Kpn/PstI fragment containing nearly the entire cDNA insert was excised from pBluescript and subcloned into the pCMV5 expression vector using T4 DNA ligase with the reaction performed at 15 °C for 15 h. For transient expression of the cDNA, ~1 × 106 COS7 cells were transfected with 15 μg of expression plasmid via the DEAE-dextran method (20). COS7 cells were grown in 75-cm2 flasks in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and gentamicin (0.01 mg/ml), and cells were harvested 48–72 h posttransfection for radioligand binding assays. CHO cells, grown in Ham’s F-12 media supplemented with 10% fetal bovine serum and penicillin (100 units/ml)/streptomycin (100 μg/ml) were employed for stable expression of the cDNA. Approximately 300,000 cells in 25-cm2 flasks were transfected with 30 μg of the pCMV5 expression vector containing the A1AR cDNA insert and 3 μg of pSVneo by a calcium phosphate precipitation and 15% glycerol shock procedure (21) with minor modifications. Cells were then maintained in the presence of 250 μg/m G418 (Geneticin, GIBCO), and, after 14 days of treatment, resistant colonies were selected and replated. Clonal cell lines expressing physiologic levels of A1AR, as determined by radioligand binding assays, were used in adenylylcyclase studies.
Site-directed Mutagenesis of the A1AR
The method for the creation of mutagenized cDNA was based on that described by Nelson and Long (22). Two oligonucleotides complementary to the sense strand of the cloned A1AR were synthesized in order to produce two mutant A1ARs. The first oligonucleotide (5′ AGG ATG AGC AAG GGC AGC CAG CTG AGG GC 3′) coded for the substitution of the histidine at position 251 (sixth transmembrane domain) with leucine. The second oligonucleotide (5′ GCC GAG TTG CCG AGT GAG AGG AAG ATG GCG ATG GTA 3′) was used to construct a mutated A1AR in which the histidine at position 278 (seventh transmembrane domain) was replaced with leucine. For both His-251 and His-278 mutants, an oligonucleotide complementary to the cDNA located 60 bp upstream from a unique BsmI site and the oligonucleotide coding for the His-Leu substitution were used as primers in a polymerase chain reaction using 100 ng of A1AR/pCMV5 plasmid as the template. The fragment created in the first-step polymerase chain reaction was gel-purified and then used as a primer in a second polymerase chain reaction (one cycle only) for the extension of the mutagenized cDNA with expression vector DNA used as template. This fragment was then amplified by continuing the polymerase chain reaction with the BsmI site oligonucleotide and an oligonucleotide complementary to a region of the cDNA located 60 bp downstream from a unique BstEII site used as primers. The final product of ~750 bp was extracted with phenol and chloroform and ethanol-precipitated. It was then digested with BsmI/BstEII and ligated into the A1AR/pCMV5 expression vector which had been similarly digested and gel-purified. Sequence of the mutant cDNA was verified by double strand sequencing. COS7 cells were transfected with His-251 and His-278 expression constructs as described above.
Radioligand Binding Assays
Membranes from COS7 cells were prepared as follows. Media was aspirated from the flask, and the cells were washed with 10 ml of lysis buffer (10 mM Tris, 5 mM EDTA, pH 7.4 at 22 °C). A fresh 5 ml of ice-cold lysis buffer was added to the flask, and cells were scraped and transferred to a dounce homogenizer on ice. The cells were homogenized with 20 strokes, spun at 38,500 × g for 5 min, and resuspended in 6 ml of 50/10/1 buffer (50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 8.26 at 5 °C). The cells were further diluted 3–4.5-fold with 50/10/1 buffer.
For saturation binding experiments, each assay tube contained 50 μl of H2O or 10 mM theophylline (to define nonspecific binding), 50 μl of radioligand, 50 μl of 50/10/1 buffer, and 100 μl (~5–10 μg of protein) of membrane suspension. In assays with mutagenized A1AR, 10 mM theophylline and 0.1 mM R-PIA gave similar results for nonspecific binding. For competition binding studies, each tube contained 50 μ1 of competing ligand, 50 μ1 of radioligand, 50 μl of 50/10/1 buffer, and 100 μl of the membrane suspension. All incubations were at 37 °C for 60 min and terminated by the addition of ice-cold 50/10/1 buffer containing 0.01% CHAPS and rapid filtration over 0.03% polyethylenimine-treated filters using a Brandel cell harvester. Data analysis was performed via a nonlinear least-squares fitting technique as previously described (23). Protein determinations were performed via the Bradford method (24)
Adenylyl cyclase Assay
Membranes from stably transfected CHO cells were prepared exactly as described above for COS7 cells. Adenylylcyclase assays were performed as previously described (25). cAMP accumulation experiments were performed using the cAMP radioimmunoassay kit as described by the manufacturer (Amersham). In both assays forskolin (50 μM) was used to activate the adenylylcyclase system.
Northern Analysis
RNA was prepared from bovine tissues that had been frozen in liquid nitrogen immediately upon dissection. Tissues were homogenized in ice-cold buffer consisting of 0.1 M Tris HCl, pH 7.5, 4 M guanidinium thiocyanate, and 1% β-mercaptoethanol with a Polytron set at full speed for a 30-s burst. Sodium lauryl sarcosinate was added to the homogenate to give a final concentration of 0.5%, and the suspension was layered onto CsCl (5.7 M CsCl, 0.01 M EDTA, pH 7.5) in an ultracentrifuge tube. Samples were centrifuged at 175,000 × g for 18 h. The resulting RNA pellet was resuspended in 10 mM Tris, 1 mM EDTA, pH 7.5, and 0.1% SDS; diluted with an equal volume of 40 mM Tris-HC1, pH 7.6, 1.0 M NaCl, 2 mM EDTA and 0.2% SDS; and then applied to an oligo(dT)-cellulose column (Bethesda Research Laboratories) according to standard procedures (21). Poly(A)+ RNA was eluted with 0.5 ml of water and ethanol-precipitated and electrophoresed on a 1% agarose and 6.6% formaldehyde gel (3-5 μg of RNA per lane). RNA was transferred from the gel to a Zeta-probe membrane (Bio-Rad) with 10 × SSC (1 × SSC = 150 mM NaC1, 15 mM sodium citrate, pH 7.0) over 12 h. The RNA was fixed to the membrane by UV cross-linking and incubated in a prehybridization solution of 50% formamide, 6 × SSPE (1 × SSPE = 150 mM NaCl, 10 mM NaH2PO4, 1 mM EDTA, pH 7.4), 5 × Denhardt’s solution, 0.5% SDS, and 0.25 mg/ml salmon sperm DNA at 42 °C overnight. The full-length bovine brain A1AR cDNA was labeled with [32P]ATP using the Amersham multiprime DNA labeling system and diluted in hybridization buffer (identical to prehybridization except for the omission of 5 × Denhardt’s solution and increase of salmon sperm DNA to 0.5 mg/ml) to a final activity of 9.5 × 105 cpm/ml. Hybridization was conducted at 42 °C for 24 h after which the membrane was washed twice in 2 × SSC, 0.5% SDS at 22 °C for 15 min. This was followed by three sequential washes in the 0.1 × SSC, 0.1% SDS for 15 min at 55, 60, and 66 °C.
RESULTS
Screening of a bovine brain cDNA library resulted in the cloning of an A1AR with a unique pharmacological profile compared to previously cloned A1ARs (12–14). The clone contained an insert of ~2.0 kb. The nucleotide sequence of this clone along with the deduced amino acid sequence of the longest open reading frame is shown in Fig. 1. As shown in Fig. 2, the encoded protein of 326 amino acids (36,570 daltons) possesses many of the structural characteristics of the G protein-coupled receptor family (26–28) and is highly homologous to the recently cloned A1ARs of canine thyroid gland (12) and rat brain (13, 14). Putative transmembrane segments 1, 2, and 6 of the bovine brain, rat brain, and canine thyroid A1ARs are 100% identical, while the bovine A1AR differs by a total of 7 (rat) and 8 (canine) amino acids in transmembrane domains 3, 4, 5, and 7. The majority of the differences in the translated proteins exist in extracellular loops 2 and 3 and the carboxyl terminus tail. Unlike the canine A1AR, the bovine A1AR contains two potential sites for glycosylation, both of which exist in the second extracellular loop. In comparison to many inhibitory G protein receptors, the third intracellular loop of the bovine A1AR is extremely short, composed of only 34 amino acids. This cytoplasmic loop contains potential phosphorylation sites for protein kinase C and protein kinase A. A single consensus sequence for protein kinase A phosphorylation is present in the second intracellular loop. These sites may represent important regions for A1AR regulation as a recent study has demonstrated the in vivo phosphorylation of the A1AR of DDT1 MF-2 smooth muscle cells in association with agonist-induced desensitization (29). The cytoplasmic carboxyl terminus of the bovine brain A1AR is also relatively short which is a feature shared by several other receptors that are linked to the inhibition of adenylylcyclase via the Gi protein (28). This region of the receptor also contains a cysteine at position 309 which represents a possible site for palmitoylation (30).
FIG. 1. Nucleotide and deduced amino acid sequence of the cloned bovine brain A1AR.

Translation of amino acids begins at the ATG providing the longest open reading frame. The stop codon is represented by @.
FIG. 2. Comparison of the deduced amino acid sequences of bovine, dog, and rat A1AR.
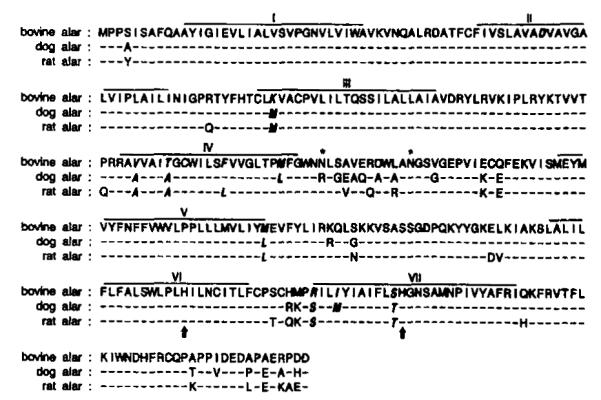
Dashes represent identical amino acids with differences noted in canine and rat sequences. Putative transmembrane domains (I–VII) are marked with a solid line. Italicized amino acids represent changes in the A1ARs in these transmembrane regions. Possible sites for glycosylation in extracellular loop 2 are marked by asterisk. His-251 and His-278 are marked by ↑.
To validate that the isolated cDNA did indeed code for the bovine brain A1AR, nearly the entire 2.0-kb insert was subcloned into the pCMV5 expression vector and the plasmid was transiently expressed in COS7 cells. As shown in Fig. 3A, membranes prepared from transfected COS7 cells displayed high affinity binding for the A1AR selective antagonist [3H] XAc (Kd = 0.37 ± 0.05 nM) with a Bmax of 2.56 ± 0.53 pmol/mg (n = 6). This high affinity for [3H]XAC is consistent with that described for the bovine A1AR (10, 11). Likewise, binding of the A1AR agonist, [125I]APNEA, was saturable and of high affinity with a Kd of 0.57 ± 0.16 nM and a Bmax of 1.00 ± 0.28 pmol/mg (n = 5). A representative saturation curve is shown in Fig. 3B. Neither ligand bound to membranes prepared from nontransfected COS7 cells.
FIG. 3. Representative curves depicting [3H]XAC (A) and [125I]APNEA (B) saturation binding assays with membranes prepared from COS7 cells transiently transfected with bovine A1AR cDNA.
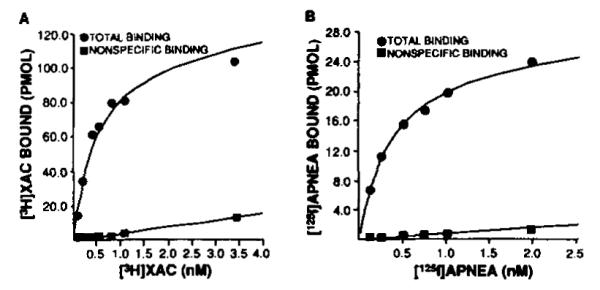
For both curves, nonspecific binding was defined with 10 mM theophylline. Assays were performed as described under “Experimental Procedures.” Each curve was replicated at least five times.
To document that the expressed receptor has the unique pharmacological profile expected, the ability of A1AR agonists to compete for [125I]APNEA binding sites was determined (Fig. 4). In five experiments, the IC50 values for R-PIA, S-PIA, and NECA were 0.37 ± 0.06, 5.05 ± 0.80, and 30.0 ± 7.3 nM, respectively. This potency order of R-PIA > S-PIA > NECA is identical to thaot bserved for the membrane, soluble, and purified forms of bovine cortex A1AR (10, 11). The A1AR is known to inhibit the enzyme adenylylcyclase. To document that the expressed A1AR was capable of functioning in this capacity, both cAMP accumulation and adenylylcyclase experiments in stably transfected CHO cells were performed. In these cells, the stably expressed receptor demonstrated high affinity [3H]XAC and [125I]APNEA binding as described above. Fig. 5 demonstrates a dose-dependent inhibition of adenylylcyclase by R-PIA. The ~30% attenuation is similar to that observed in previous studies of A1AR-mediated adenylylcyclase inhibition (4, 25). Similar results were obtained using cAMP accumulation as a determinant of A1AR functional activity (data not shown).
FIG. 4. Agonist competition uersus [126I]APNEA (~0.3 nM) in membranes prepared from COS7 cells transiently transfected with bovine brain A1AR cDNA.
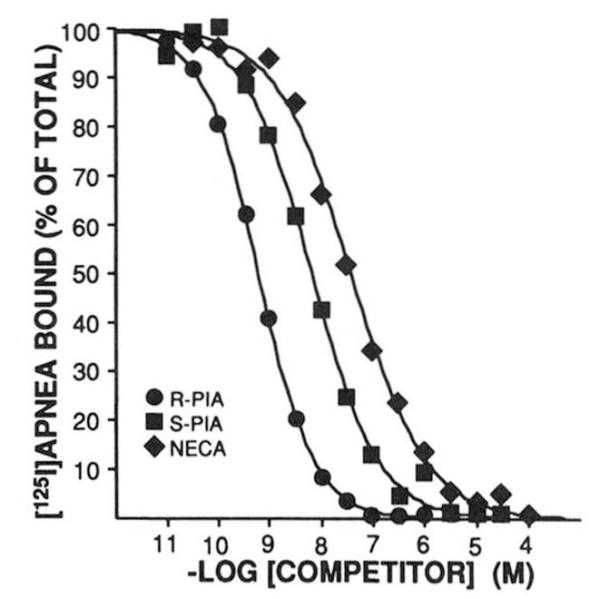
Competing ligand was added at the concentration shown on the abscissa. The amount of [125I]APNEA bound is on the ordinate. Assays are described under “Experimental Procedures.” This experiment is representative of four similar assays.
FIG. 5. Representative experiment demonstrating R-PIA-mediated inhibition of forskolin-stimulated adenylylcyclase activity in membranes prepared from CHO cells stably expressing bovine brain A1AR cDNA.
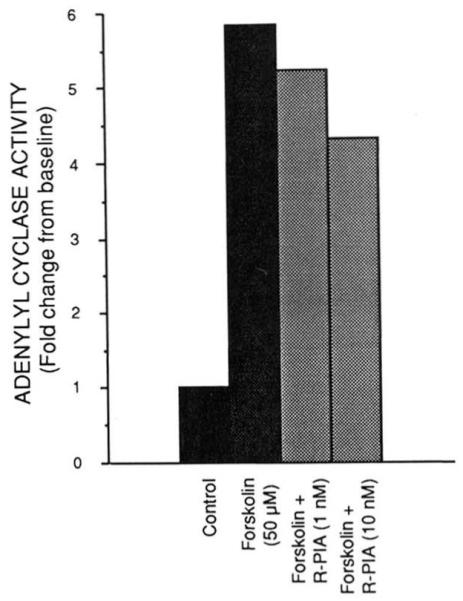
Assays were performed as described under “Experimental Procedures” using ~100 μg of protein per assay tube. Forskolin was present at 50 μM where indicated and the concentration of R-PIA was as shown. This experiment was performed four times with similar results.
The distribution of the A1AR mRNA in bovine tissues by Northern analysis is presented in Fig. 6 and is in agreement with the findings from previous ligand binding and functional studies (31). Two transcripts of 3.4 and 5.7 kb are abundant in brain with a significant amount also in heart. In the kidney, a 3.4-kb transcript was detected. The thyroid contained only a 5.5-kb message. No transcript was detected in bovine spleen, lung, or liver. DDT1 MF-2 cells, a cell line known to contain A1AR, also contained two transcripts of 3.4 and 5.6 kb.
FIG. 6. Northern blot analysis of tissue distribution of A1AR mRNA.
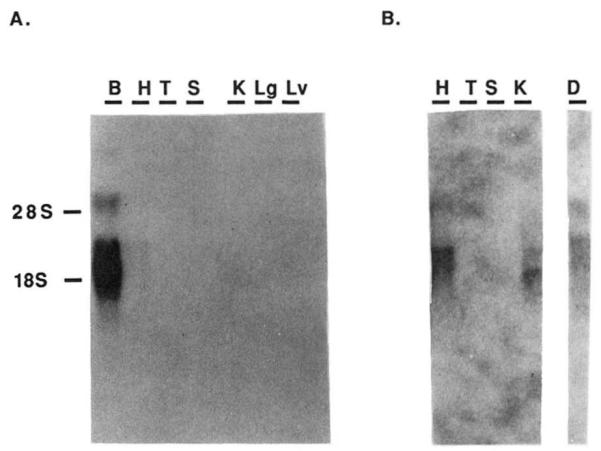
Poly(A)+ RNA (3–5 μg) was run on agarose/formaldehyde gels, transferred to nylon membranes, and probed with 32P-labeled full-length bovine A1AR cDNA. Washing conditions were described under “Experimental Procedures.” Tissues analyzed were bovine brain (B), heart (H), thyroid (T), spleen (S), kidney (K), lung (Lg), and liver (Lv). A is a photograph of film exposedt o theb lot for 12 h. B is a selected panel from the same blot exposed to the film for 48 h to allow visualization of transcripts present in heart, thyroid, and kidney. For comparison, an analysis is shown of transcript levels in DDT1 MF-2 smooth muscle cells (D). Position of the 18 and 28 S ribosomal RNA is shown to the left of A.
As mentioned in the Introduction, biochemical studies have suggested that histidine residues may be important for adenosine binding (17). Indicated in Fig. 2 are 2 histidines within the putative transmembrane domains (one each in transmembrane domains 6 and 7). In order to assess if these amino acids are important for agonist or antagonist binding, sitedirected mutagenesis was utilized to individually change the histidines to leucines as described under “Experimental Procedures.” The mutagenized receptors were then expressed in COS7 cells, radioligand binding was performed, and the results were compared to the wild-type receptor. Antagonist ([3H]XAC) and agonist ([125I]APNEA) binding to the His-278 → Leu-278 (seventh domain) mutant were both dramatically decreased compared to wild-type receptor (specific binding < 10%). This mutant was, therefore, not evaluated further. Much different results were observed for the His-251 → Leu-251 mutant. In [3H]XAC saturation assays (Fig. 7), a 74% decrease in antagonist binding (Bmax) was observed and the Kd changed increased from 0.17 ± 0.02 to 0.65 ± 0.15 nM in mutant as compared to wild-type A1AR (n = 4). Agonist binding as assessed by [125I]APNEA saturation assays (Fig. 8) decreased by 74%, while the Kd was not affected (1.68 ± 0.4 and 1.23 ± 0.14 nM in wild-type and His-251 → Leu-251, respectively, n = 3). Full R-PIA competition curves versus [3H]XAC were also constructed to completely examine the effect of the histidine substitution on agonist binding in that [125I]APNEA saturation assays may only provide information regarding high affinity binding. In these experiments, the IC50 for R-PIA was 56.6 ± 17 nM (n = 4) and 50.7 ± 14.3 nM (n = 6) for wild-type and His-251 → Leu-251, respectively. It should be pointed out that R-PIA affinity for the A1AR when competing with [3H]XAC rather than [125I]APNEA is always lower even in native bovine membranes. Therefore, the point mutation had no apparent effects on agonist affinity. The His-251 → Leu-251 also bound agonist ligands in the potency order noted above for wild-type A1AR, again indicating agonist recognition remained intact.
FIG. 7. Representative [3H]XAC saturation assay in membranes prepared from COS7 cells transiently expressing wild-type and mutagenized (His-251 → Leu-251) A1AR cDNA.
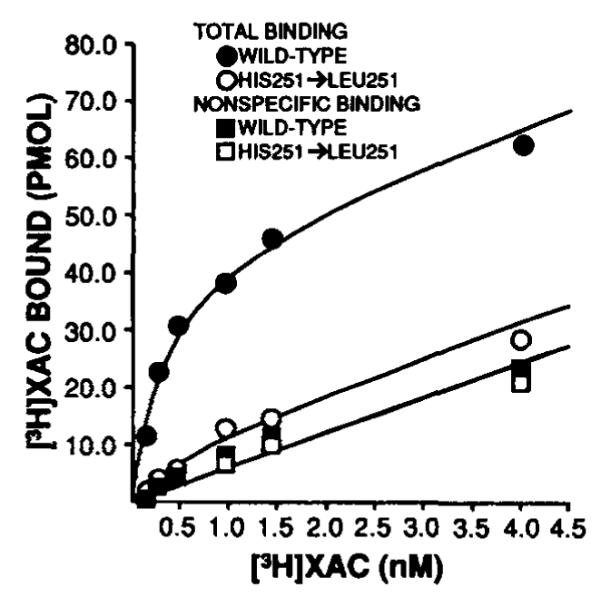
Nonspecific binding was defined with 10 mM theophylline. The bestfit lines for nonspecific binding for both groups were superimposable. Approximately equivalent amounts (~3.5 μg) of protein were used per assay tube for both groups. Other experimental details are described under “Experimental Procedures.” Similar results were obtained in three other experiments.
FIG. 8. Representative [125I]APNEA saturation assay in membranes prepared from COS7 cells transiently expressing wild-type and mutagenized (His-251 → Leu-251) A1AR cDNA.
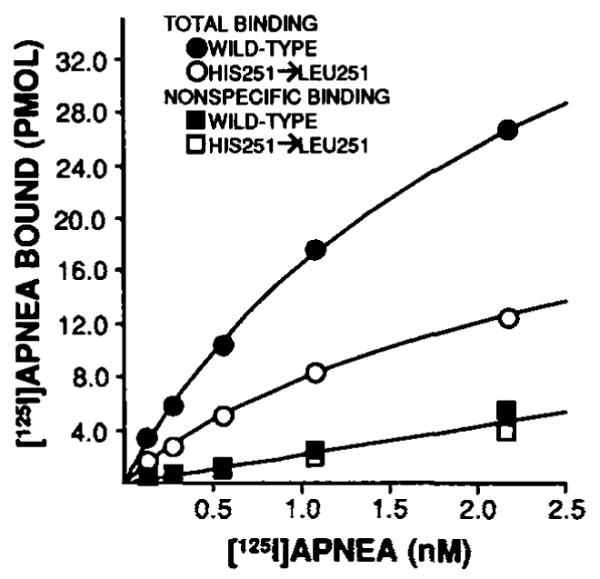
Nonspecific binding was defined with 10 mM theophylline. The best-fit lines for nonspecific binding for both groups were superimposable. Approximately equivalent amounts (~3.5 μg) of protein were used per assay tube for both groups. Other experimental details are described under “Experimental Procedures.” This experiment was repeated three times.
DISCUSSION
The bovine brain A1AR has long been recognized as a distinct form of the A1AR in that it binds agonists with a different potency series and has an ~10-fold higher affinity for both agonists and antagonists compared to rat or other A1ARs (8–11). In this paper we describe the cloning and characterization of the cDNA for the bovine brain A1AR and through site-directed mutagenesis studies have begun to probe the binding pocket of the receptor.
Radioligand binding studies demonstrate that in COS7 cells, the cDNA codes for an adenosine receptor that displays a pharmacologic potency series which is identical to that reported for the bovine brain A1AR in both itsm embrane (11) and purified (10) forms. This potency series of R-PIA > S-PIA > NECA is unique for bovine cortex as it differs from that reported for A1ARs obtained from tissues of several other species in which NECA possesses a significantly greater affinity than S-PIA. The expressed receptor has all the pharmacological characteristics expected including high affinity agonist and antagonist binding and selective and stereospecific binding. In addition, the expressed receptor functionally couples to its G protein and can inhibit adenylylcyclase activity.
The bovine brain A1AR has also been considered unique based on the binding of the radiolabeled antagonist, [3H]XAC. The affinity of this compound for bovine brain A1AR is several-fold greater than that reported for several other A1ARs including those present in rat fat (5) and brain (9) and DDT1 MF-2 smooth muscle cells (25). COS7 cells transfected with the bovine brain A1AR cDNA bind [3H]XAC with a Kd of ~0.4 nM in agreement with that displayed for the native receptor. This would indicate that the greater affinity of this receptor for the antagonist is due to distinct intrinsic properties of the bovine A1AR protein and not merely differences provided by the milieu of the membrane in its nativet issue.
Recently, the cloning of A1ARs from canine thyroid (12) and rat brain (13, 14) cDNA libraries has been reported. Mahan et al. (13) observed the typical potency order of R-PIA > S-PIA > NECA for displacement of [3H]DPCPX from binding sites in A9-L cells transiently expressing the rat brain A1AR cDNA. Reppert and coworkers (14) reported this same potency order following the expression of a rat brain A1AR cDNA in COS-6 M cells. This agonist series was not used in the characterization of the canine A1AR clone, RDC7, expressed in COS7 cells (12). However, this study reported a Ki value of 4.55 nM for NECA in competition assays with [3H] CHA. This value is similar to that reported for the high affinity agonist state of NECA (Ki = 4.3 nM) for the rat brain A1AR (13) and several fold lower than that found in the present study for bovine brain A1AR
This tissue distribution of A1AR mRNA in bovine tissues as determined by Northern analysis corresponds with the results of radioligand binding and functional studies. The A1AR is abundant in brain while heart, thyroid and kidney also contain appreciable amounts of message. Northern analysis in rat (13, 14) and canine (12) tissues also demonstrates A1AR mRNA in brain and heart. However, unlike the bovine tissue, neither canine nor rat kidney contained detectable A1AR mRNA. Also, rat spleen appeared to possess A1AR transcripts while no message is present in bovine spleen.
Based on the unique ligand binding properties of the bovine A1AR and apparent differences in tissue distribution of the mRNA for this receptor, we propose that the bovine A1AR is a distinct subclass of this receptor. The existence of distinct subtypes of A1ARs is in keeping with what has been found in other G protein-coupled receptors. The presence of multiplereceptor subtypes has recently been appreciated in almost all receptor families. Cloning techniques have allowed for the recognition of multiple α1 (32, 33) and α2 (34) adrenergic receptors as well as D1 and D2 dopamine (35), muscarinic (36–40), and serotonergic (41–43) receptors. Such receptor subtypes are often demonstrated to be distinct either pharmacologically, functionally, or on the basis of tissue distribution.
The differences in pharmacology described above exist despite there markable amino acid homology between the bovine brain A1AR and those recently cloned. The encoded proteins are nearly identical in molecular mass with 326 amino acids constituting all of the cloned A1ARs. The three A1ARs now cloned are 92% identical overall and in the putative transmembrane regions the bovine A1AR differs from the rat and canine receptors by 7 and 8 amino acid substitutions, respectively. In comparison, only 3 amino acid differences exist in the transmembrane regions between the rat and canine A1ARs. It is generally believed that the composition of the transmembrane domains of the G protein-coupled receptors determine their ligand binding characteristics and, therefore, confer the specificity of receptor subtype activation (26, 27). It is possible that very few or perhaps only a single amino acid difference in these regions is (are) responsible for the differences in bovine brain A1AR pharmacology relative to other A1ARs. Suryanarayana et al. (44) recently demonstrated that a single point mutation (Phe to Asn) in the seventh transmembrane domain of the α2 adrenergic receptor dramatically reduced the receptor’s affinity for selective agonists while increasing its affinity for a β-adrenergic receptor antagonist 3000-fold.
Site-directed mutagenesis provides a powerful technique for assessing the importance and role of individual or groups of amino acids in the functioning of proteins. Klotz et al. (17) have previously documented that diethylpyrocarbonate, a histidine-specific agent, was capable of specifically decreasing both agonist and antagonist binding to the A1AR of rat brain membranes. Diethylpyrocarbonate treatment decreased the number but did not alter the affinity of the receptors for antagonists. This suggests a modification of the ligand binding site itself. The fact that there was a selective protective effect of antagonist binding by antagonists but not agonists and similarly a protective effect of agonist binding by agonists but not antagonists suggested that there were at least 2 histidine residues near the binding pocket.
In this study we have now been able to directly study the role of the 2 histidine residues in transmembrane domains 6 and 7. Our data demonstrate that the His-278 (seventh domain) appears to be extremely important for the binding of both agonists and antagonists. Substituting leucine for this histidine leads to the almost but not quite complete loss of agonist and antagonist binding. In contrast, His-251 is important for both agonist and antagonist binding in that agonist and antagonist bindings are both decreased by substitution of Leu for His, but not to the same extent as thaotf His-278 → Leu-278. At the present time, no antibodies to the A1AR are available for immunodetection of the receptor protein. Therefore, agonist and antagonist radioligand binding studies are the most suitable methods available to quantitate membrane A1ARs. It is possible, therefore, that the reduction in receptor number as assessed by [125I]APNEA and [3H]XAC binding may indicate a decline in the actual quantity of A1AR protein rather than the loss of a specific ligand-receptor interaction. Such a decline could result if the process by which the mutant A1AR cDNA is transcribed, processed, and translated in the COS7 cells is different from that for the wild-type cDNA. In addition, the mutated protein may not undergo the proper folding necessary for transport to and insertion in the membrane in the appropriate orientation. This latter possibility would be difficult to detect, for even immunologic techniques would not likely be able to distinguish between A1ARs differing only in their three-dimensional structure. To minimize the occurrence of the possibilities described above, leucine, which is an aliphatic and relatively nonbulky amino acid that is prevalent in protein transmembrane domains, was selected to substitute for the histidine. However, even with all these caveats we believe that the mutant studies provide new insights into A1AR structure and function.
In addition to the effects on A1AR Bmax, the His-251 → Leu-251 substitution has differential effects on agonist and antagonist binding affinity. As judged from Kd values for [3H]XAC, the mutated A1AR demonstrates approximately a 4-fold decline in antagonist affinity. However, both [125I]APNEA and R-PIA versus [3H]XAC competition curves indicate that the His-251 → Leu-251 substitution produces no effect on agonist affinity. The ability of His-251 → Leu-251 to bind agonists with high affinity demonstrates that the functional properties of the ligand binding pocket remain intact, and, furthermore, the mutated receptor does couple to a G protein, presumably Gi. The latter finding is reasonable based on previous analysis of structure and function of G proteincoupled receptors which indicates that transmembrane domain regions are involved in ligand binding, whereas intracellular regions are critical for G protein activation (25, 26).
The differential effects of the His-251 → Leu-251 substitution on agonist/antagonist binding are in agreement with previous studies on ligand-receptor interactions of the A1AR. These findings extend those of Klotz et al. (17) by specifically examining the histidine residues involved. Not only has sequencing allowed the location of the histidines to be determined but site-directed mutagenesis permits their individual contributions to be studied. The histidine at position 251 appears to be directly involved in antagonist binding since antagonist affinity is significantly altered. Mutation at this site does not alter agonist affinity, only receptor number. Changes in Bmax are much more complicated in this system (A1AR) than changes in Kd since it is known that the quantity of receptor detected by both agonist and antagonist radioligands is dependent on receptor-G protein coupling, ion concentration, and the presence or absence of guanine nucleotides (45). Interestingly, Barrington et al. (46), using proteolytic digestions of agonist and antagonist occupied A1AR in native bovine brain membranes, demonstrated that although these ligands may share the same binding pocket they induce unique conformational changes of the A1AR. The conformational changes induced will of necessity determine not only the apparent affinity for a given ligand but also its coupling to the G protein and hence the number of receptors detected.
In summary, this paper provides evidence for the existence of a subtype of the A1 adenosine receptor which is in agreement with data obtained with the native bovine receptor. Apparently, minor variations in amino acid sequence may account for the differences in pharmacologic properties of the bovine A1AR as compared to the canine and rat A1ARs previously described. Furthermore, this study significantly extends previous findings regarding A1AR-ligand binding interactions. His-278 in transmembrane domain 7 is apparently crucial for ligand recognition. His-251 in transmembrane domain 6, though involved in part ina gonist binding, plays a much more important role in A1AR-antagonist interaction. Future studies probing other domains of the A1AR should provide a more detailed description of its ligand binding pocket.
Acknowledgments
We thank Linda Scherich for her assistance in the preparation of this manuscript and Mary Pound for technical assistance. The helpful discussions with Drs. Keith Jarvie, Mario Tiberi, and Donna Coulter-Karis are also appreciated.
Footnotes
The work was supported in part by National Institutes of Health Postdoctoral Fellowship 1F32-GM-13713-01 from the National Institute of General Medical Sciences (to M. E. O.), by NHLBI Grant R01-HL35134 (and Supplement) (to G. L. S.), and by Grant-in-Aid 91008200 from the American Heart Association (to G. L. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The nucleotide sequencefs) reported in this paper has been submitted to the GenBank™/EMBL Data Bank with accession number(s) M86261.
The abbreviations used are: G protein, guanine nucleotide regulatory protein; Gi protein, inhibitory guanine nucleotide regulatory protein; A1AR, A1 adenosine receptor; R-PIA, N6-R-phenylisopropyladenosine; S-PIA, N6-S-phenylisopropyladenosine; NECA, 5′-N-ethylcarboxamidoadenosine; XAC, xanthine amine congener; APNEA, N6-2-(4-aminophenyl)ethyladenosine; SDS, sodium dodecyl sulfate; kb, kilobase(s); CHO, Chinese hamster ovary; bp, base pair(s); CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
REFERENCES
- 1.Olsson RA, Pearson JD. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 2.Stiles GL. Clin. Res. 1990;38:10–18. [PubMed] [Google Scholar]
- 3.Stiles GL. News Physiol. Sci. 1991;6:161–165. [Google Scholar]
- 4.Parsons WJ, Stiles GL. J. Biol. Chem. 1987;262:841–847. [PubMed] [Google Scholar]
- 5.Longabaugh JP, Didsbury J, Spiegel A, Stiles GL. Mol. Phurmacol. 1989;36:681–688. [PubMed] [Google Scholar]
- 6.Ramkumar V, Bumgarner JR, Jacobson KA, Stiles GL. J. Clin. Invest. 1988;82:242–247. doi: 10.1172/JCI113577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rapiejko PJ, Malbon CC. Biochem. J. 1987;341:765–771. doi: 10.1042/bj2410765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy KMM, Snyder SH. Mol. Pharmacol. 1982;22:250–257. [PubMed] [Google Scholar]
- 9.Ukena D, Jacobson KA, Padgett WL, Ayala C, Shamin MT, Kirk KL, Olsson RO, Daly JW. FEBS Lett. 1986;209:122–128. doi: 10.1016/0014-5793(86)81096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olah ME, Jacobson KA, Stiles GL. Arch. Biochem. Biophys. 1990;283:440–446. doi: 10.1016/0003-9861(90)90665-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stiles GL, Jacobson KA. Mol. Phurmacol. 1987;32:184–188. [PMC free article] [PubMed] [Google Scholar]
- 12.Libert F, Schiffman SN, Lefort A, Parmentier M, Gerard C, Dumont JE, Vanderhoeghen J-J, Vassart G. EMBO J. 1991;10:1677–1682. doi: 10.1002/j.1460-2075.1991.tb07691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahan LC, McVittie LD, Smyk-Randall EM, Nakata H, Monsma FJ, Jr., Gerfen CR, Sibley DR. Mol. Pharmacol. 1991;40:1–7. [PubMed] [Google Scholar]
- 14.Reppert SM, Weaver DR, Stehle JH, Rivkees SA. Mol. Endocrinol. 1991;5:1037–1048. doi: 10.1210/mend-5-8-1037. [DOI] [PubMed] [Google Scholar]
- 15.Stiles GL. J. Biol. Chem. 1986;261:10839–10843. [PubMed] [Google Scholar]
- 16.Yeung S-MH, Green RD. J. Biol. Chem. 1983;258:2334–2339. [PubMed] [Google Scholar]
- 17.Klotz K-N, Lohse MJ, Schwabe U. J. Biol. Chem. 1988;263:17522–17526. [PubMed] [Google Scholar]
- 18.Stiles GL, Daly D, Olsson RA. J. Biol. Chem. 1985;260:10806–10811. [PubMed] [Google Scholar]
- 19.Libert F, Parmentier M, Lefort A, Dinsart C, Van Sande J, Maenhaut C, Simons MJ, Dumont JE, Vassart G. Science. 1989;244:569–572. doi: 10.1126/science.2541503. [DOI] [PubMed] [Google Scholar]
- 20.Cullen BR. Methods Enyzmol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- 21.Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 22.Nelson RM, Long GL. Anal. Biochem. 1989;180:147–151. doi: 10.1016/0003-2697(89)90103-6. [DOI] [PubMed] [Google Scholar]
- 23.De Lean A, Hancock AA, Lefkowitz RJ. Mol. Pharmacol. 1982;21:5–16. [PubMed] [Google Scholar]
- 24.Bradford MM. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Ramkumar V, Barrington WW, Jacobson KA, Stiles GL. Mol. Pharmacol. 1990;37:149–156. [PMC free article] [PubMed] [Google Scholar]
- 26.Strader CD, Sigal IS, Dixon RAF. FASEB J. 1989;3:1825–1832. doi: 10.1096/fasebj.3.7.2541037. [DOI] [PubMed] [Google Scholar]
- 27.O’Dowd BF, Lefkowitz RJ, Caron MG. Annu. Rev. Neurosci. 1989;12:67–83. doi: 10.1146/annurev.ne.12.030189.000435. [DOI] [PubMed] [Google Scholar]
- 28.Strosberg AD. Eur. J. Biochem. 1991;196:1–10. doi: 10.1111/j.1432-1033.1991.tb15778.x. [DOI] [PubMed] [Google Scholar]
- 29.Ramkumar V, Olah ME, Jacobson KA, Stiles GL. Mol. Phurmacol. 1991;40:639–647. [PMC free article] [PubMed] [Google Scholar]
- 30.O’Dowd BF, Hnatowich M, Caron MG, Lefkowitz RJ, Bouvier M. J. Biol. Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 31.Daly JW. In: Advances in Cyclic Nucleotide and Protein Phosphorylation Research. Cooper DMF, Seamon KB, editors. Vol. 19. Raven Press; New York: 1985. pp. 29–46. [PubMed] [Google Scholar]
- 32.Cotecchia S, Schwinn DA, Randall RR, Lefkowitz RJ, Caron MG, Kobilka BK. Proc. Natl. Acad. Sci. U. S. A. 1988;85:7159–7163. doi: 10.1073/pnas.85.19.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schwinn DA, Lomasney JW, Lorenz W, Szklut PJ, Fremeau RT, Yang-Feng TL, Caron MG, Lefkowitz RJ, Cotecchia S. J. Biol. Chem. 1990;265:8183–8189. [PubMed] [Google Scholar]
- 34.Lorenz W, Lomasney JW, Collins S, Regan JW, Caron MG, Lefkowitz RJ. Mol. Pharmacol. 1990;38:599–603. [PubMed] [Google Scholar]
- 35.Andersen PH, Gingrich JA, Bates MD, Dearry A, Falardeau P, Senogles SE, Caron MG. Trends Pharmacol. Sci. 1990;11:231–236. doi: 10.1016/0165-6147(90)90249-8. [DOI] [PubMed] [Google Scholar]
- 36.Kubo T, Fukuda K, Mikami A, Maeda A, Takahashi H, Mishina M, Haga T, Haga K, Ichiyama A, Kangawa K, Kojima M, Matsuo H, Hirose T, Numa S. Nature. 1986;323:411–416. doi: 10.1038/323411a0. [DOI] [PubMed] [Google Scholar]
- 37.Kubo T, Maeda A, Sugimoto K, Akiba I, Mikami A, Takahashi H, Haga T, Haga K, Ichiyama A, Kangawa K, Matsuo H, Hirose T, Numa S. FEBS Lett. 1986;209:367–372. doi: 10.1016/0014-5793(86)81144-9. [DOI] [PubMed] [Google Scholar]
- 38.Peralta EG, Winslow JW, Peterson GL, Smith DH, Ashkenazi A, Ramachandran J, Schimerlik MI, Capon DJ. Science. 1987;236:600–605. doi: 10.1126/science.3107123. [DOI] [PubMed] [Google Scholar]
- 39.Bonner TI, Buckley NJ, Young AC, Brann MR. Science. 1987;237:527–532. doi: 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- 40.Liao C-F, Themmen APN, Joho R, Barberis C, Birnbaumer M, Birnbaumer L. J. Biol. Chem. 1989;264:7328–7337. [PubMed] [Google Scholar]
- 41.Fargin A, Raymond JR, Lohse MJ, Kobilka BK, Caron MG, Lefkowitz RJ. Nature. 1988;335:358–360. doi: 10.1038/335358a0. [DOI] [PubMed] [Google Scholar]
- 42.Julius D, MacDermott AB, Axel R, Jessel TM. Science. 1988;241:558–564. doi: 10.1126/science.3399891. [DOI] [PubMed] [Google Scholar]
- 43.Pritchett DB, Bach AWJ, Wozny M, Taleb O, Dal Toso R, Shih JC, Seeburg PH. EMBO J. 1988;7:4135–4140. doi: 10.1002/j.1460-2075.1988.tb03308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suryanarayana S, Daunt DA, Von Zastrow M, Kobilka BK. J. Biol. Chem. 1991;266:15488–15492. [PubMed] [Google Scholar]
- 45.Stiles GL. J. Neurochem. 1988;51:1592–1598. doi: 10.1111/j.1471-4159.1988.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 46.Barrington WW, Jacobson KA, Stiles GL. J. Biol. Chem. 1989;264:13157–13164. [PMC free article] [PubMed] [Google Scholar]