

Abstract
Summary: Peptide identification algorithm is a major bottleneck for mass spectrometry based chemical cross-linking experiments. Our lab recently developed an intensity-incorporated peptide identification algorithm, and here we implemented this scheme for cross-linked peptide discovery. Our program, SQID-XLink, searches all regular, dead-end, intra and inter cross-linked peptides simultaneously, and its effectiveness is validated by testing a published dataset. This new algorithm provides an alternative approach for high confidence cross-linking identification.
Availability: SQID-XLink program is freely available for download from http://quiz2.chem.arizona.edu/wysocki/bioinformatics.htm
Supplementary information: Supplementary data are available at Bioinformatics online.
Contact: vwysocki@email.arizona.edu
1 introduction
Chemical cross-linking coupled with mass spectrometry is a powerful approach to analyze protein structures and interactions (Rinner et al., 2008; Sinz et al., 2003). In such an experiment, spatially adjacent amino acid residues from one or more proteins are covalently linked by chemical reagents. The cross-linked proteins are then enzymatically digested and the resulting cross-linked peptide pairs can be detected by mass spectrometry. Identification of these cross-linked peptides enables researchers to define the interaction sites of proteins in a complex in their native states and to build or confirm structural models. Compared with traditional techniques such as NMR and X-ray crystallography, mass spectrometry-based chemical cross-linking does not require a large quantity of sample (Lee, 2008). Moreover, recent development of high-resolution and high-throughput mass spectrometers such as the Orbitrap have offered increased efficiency and sensitivity required for the identification of cross-linked peptides.
Although the chemical cross-linking approach is promising, the data analysis for chemical cross-linking forms a major limitation for this technique (Lee, 2008). This is mainly because cross-linked sequences are non-linear, thus traditional protein database search algorithms such as Sequest (Eng et al., 1994) and X!Tandem (Craig and Beavis, 2004) cannot be directly employed. The development of new database searching algorithms enables more and more protein identification from a single shot-gun proteomics experiment; however, these novel approaches have seldom implemented a cross-linking search function to increase the number of identified cross-linked peptides. Moreover, many current cross-linking algorithms are slow, lack graphical user interfaces and need extensive manual data interpretation before and after the search. These shortcomings motivated us to make a powerful and user-friendly tool to identify cross-linked peptides.
Here we report the implementation of our recently developed peptide identification algorithm, SQID (Li et al., 2011), to cross-linked peptide identification (SQID-XLink). Our algorithm features an intensity incorporated scoring function: when a strong peak in a spectrum agrees with the statistical value, the confidence will be boosted. For example, if a CID-induced cleavage occurs N-terminal to proline or C-terminal to glutamic acid or aspartic acid, the score will be higher. This is similar to manually checking a spectrum to confirm if the strong peaks are from cleavages expected to lead to abundant peaks. Due to the low abundance of cross-linked peptides and corresponding poorer spectral quality, incorporating intensity into cross-linking search algorithms will be potentially very beneficial. In addition, SQID-XLink searches all regular, dead-end (cross-linked at only one reactive site of the cross-linker), intra-peptide (cross-linked at two locations within a single peptide) and inter-peptide cross-links simultaneously with the same scoring function, so that the probability of false identification can be minimized.
2 IMPLEMENTATION
SQID-XLink is a modified version of SQID which is specifically designed for cross-linking searches. It is written in C language with a user-friendly interface from visual basic 6.0. It has been tested in Windows XP and Windows 7 operating systems. Currently, the program supports BS2G-d0/d4 (Bis[Sulfosuccinimidyl] glutarate), BS3-d0/d4 (Bis[Sulfosuccinimidyl] suberate) and EDC(1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide) cross-linkers.
SQID-XLink processes a fasta database by generating regular peptides, and peptides with a variable modification of the mass of dead-end or intra-peptide cross-linker. Peptides containing cross-linkable residues are extracted and paired through combination of any two peptides. During the search, the two peptides in a cross-linkable pair are linearized into two sequences by putting one sequence before the other (AB and vice versa BA) and searched respectively, as reported by Maiolica et al. (2007). Ions generated by cleaving the sequences between the cross-linkable locations are excluded and the search results of the two linearized sequences are combined as the final result of for cross-linked peptide pair. The final SQID-XLink score is calculated as:
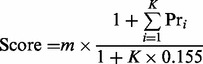 |
(1) |
where m is the number of matched peaks, Pr is the probability for a certain amino acid pair to have strong peaks (stored in a table) and K is the number of most intense peaks used to calculate the intensity score ΣPr [K depends on the mass of peptide, and equals the integer portion of (2 + mass/330)]. The term (1+ ΣPr)/(1 + 0.155K) measures whether the observed intensity (the numerator) is better than the expected value (the denominator).The function is similar to the SQID scoring function except that consecutive ion series are not used. This is because consecutive ion series tend to greatly increase the confidence when a part of the whole peptide sequence is matched, but cross-linked peptides involve two independent sequences. The Pr table and a more detailed explanation of Equation (1) can be found in Li et al. (2011).
3 results
A published EDC cross-linking dataset (McIlwain et al., 2010; Singh et al., 2008) of human cytochrome P450 2E1 (P450) and cytochrome b5 (B5) was used to test the program. The dataset contains 3314 spectra, and was collected using an LTQ-Orbitrap with high resolution for both the precursor and MS2 masses. The search was performed with three missed cleavages and a 50 ppm precursor and 20 ppm fragment m/z tolerance against both the target and decoy version of the database. The decoy database was built with reverse sequences of the two proteins plus twice the number of randomized sequences, with 5.4 times larger search space compared with the target database. Figure 1a shows a plot of score versus precursor m/z error. The majority of high score hits observed have a precursor mass error within −5 to 20 ppm, and decoy hits have a maximum score of 3.22. As a result, using −5 to 20 ppm and a score of 3.22 as a threshold should give a false discovery rate (FDR) close to 0. With these parameters, we discovered 163 high confidence peptide-spectrum matches, with 140 from non-cross-linked tryptic peptides, 22 from cross-linked peptides and 1 from intra-peptide cross-links. The minimum score for matched cross-linked peptides was 4.45, which is far above the threshold used. Figure 1b summarizes the unique cross-linked peptides that are assigned by SQID-XLink, and comparison with a popular cross-linking search engine, xQuest (Rinner et al., 2008), as well as comparison with previously published results from Crux (Table 2, McIlwain et al., 2010) and Popitam (Table 1, Singh et al., 2008). XQuest was searched using the same parameters as SQID-XLink and the FDR was determined with the same target-decoy database search strategy. We use published Popitam and Crux results directly instead of using our own search results because Popitam needs an additional algorithm to pre-filter the data and needs extensive manual interpretation to associate the modification mass with peptide sequence, while Crux relies on its own FDR estimation system which needs the optimization of many parameters. The published data were already optimized by the authors and manually verified, so they represent the best performance of the two algorithms. Our results show that SQID-XLink can identify a larger number of cross-linked peptides at high confidence. The two intra-protein cross-linked products (GTVVVPTLDSVLYDNQEFPDPEK, FKPEHFLNENGK) and (LYTMDGITVTVADLFFAGTETTSTTLR, YGLLILMKYPEIEEK) in Table 2 of the reference McIlwain et al. (2010) are matched to linear peptides with missed cleavages by both SQID-XLink and xQuest. The spectra of these products as well as a complete list of identified peptides by SQID-XLink, Crux (from our own searches) and xQuest can be found in the Supplementary Material. In terms of speed, the total search time including database processing was only 2.3 min for SQID-XLink and 6.5 min for Crux, on a 64-bit computer with Intel Xeon 2.4 GHz cpu (Crux only works on 64-bit computer), while it took 36–127 min for the xQuest webserver, depending on the server load.
Fig. 1.
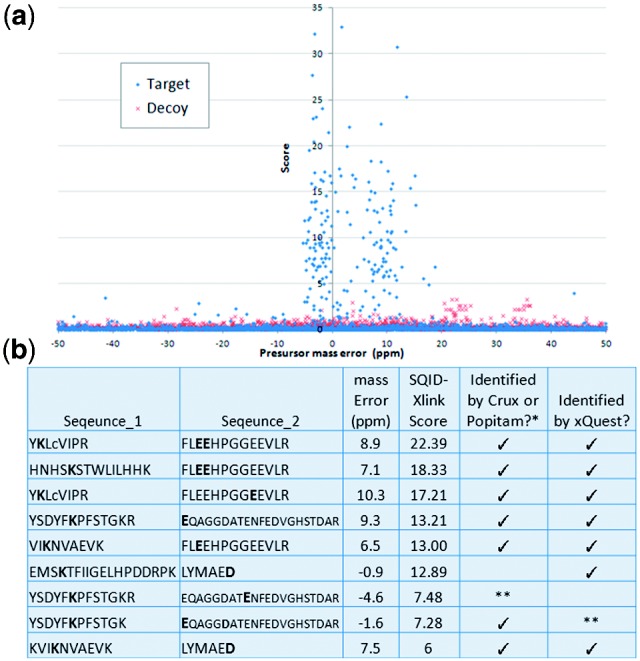
(a) Score distribution versus precursor m/z error. Blue spots represent the hits by searching with cytochrome P450 2E1 and cytochrome b5 sequences, whereas red spots are by searching with decoy sequences. (b) Unique cross-linked peptides identified by SQID-XLink, and a comparison with xQuest and Crux/Popitam. Only high confident matches (FDR < 1%) are considered. Bold font indicates the location of cross-linking. *Data from Table 2, McIlwain et al. (2010). **These two peptides can be identified with a higher FDR by Crux (3%) or xQuest (5%)
4 CONCLUSION
We have introduced SQID-XLink, an open source program for cross-linked peptide identification. By testing it with a published dataset and comparing it with the results of existing algorithms, SQID-XLink demonstrated its ability to identify more cross-linked peptides at high confidence. In addition, SQID-XLink is fast and has an easy-to-use graphical user interface. More cross-linker support and better visualization of the results will be added in the near future.
Funding: National Institutes of Health (2R01GM051387 to V.H.W.).
Conflict of Interest: none declared.
Supplementary Material
References
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Eng JK, et al. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Lee YJ. Mass spectrometric analysis of cross-linking sites for the structure of proteins and protein complexes. Mol. BioSyst. 2008;4:816–823. doi: 10.1039/b801810c. [DOI] [PubMed] [Google Scholar]
- Li W, et al. SQID: an intensity-incorporated protein identification algorithm for tandem mass spectrometry. J. Proteome Res. 2011;10:1593–1602. doi: 10.1021/pr100959y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolica A, et al. Structural analysis of multiprotein complexes by cross-linking, mass spectrometry, and database searching. Mol Cell Proteomics. 2007;6:2200–2211. doi: 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- McIlwain S, et al. Detecting cross-linked peptides by searching against a database of cross-linked peptide pairs. J Proteome Res. 2010;9:2488–2495. doi: 10.1021/pr901163d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinner O, et al. Identification of cross-linked peptides from large sequence databases. Nat. Methods. 2008;5:315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, et al. Characterization of protein cross-links via mass spectrometry and an open-modification search strategy. Anal. Chem. 2008;80:8799–8806. doi: 10.1021/ac801646f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A, et al. Chemical cross-linking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J. Mass Spectrom. 2003;38:1225–1237. doi: 10.1002/jms.559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.